
Abstract
Dynamic recombination is the driving force in the genetic diversity of human immunodeficiency virus type 1 (HIV-1). When multiple subtypes are circulating in the same area of a population, new HIV-1 strains are likely to be generated through recombination. In this study, we report a novel recombinant strain (2018GXQZLSHET001) of HIV-1, isolated from a HIV-1-positive heterosexual individual infected in Guangdong province, who recently lived in Guangxi province, China. Phylogenetic analysis of the near full-length genome suggested that 2018GXQZLSHET001 was a recombinant of strains CRF55_01B and subtype B. Similarity plotting and bootscaning showed that a subtype B segment was inserted into the CRF55_01B genome with one breakpoint in the nef and 3′ long terminal repeat regions. Further subregion phylogenetic analysis demonstrated that the CRF55_01B segment originated from Guangdong. The subtype B segment was similar to a Thai B lineage. This indicated that the strain might be a novel recombinant, comprising sequences of both CRF55_01B and B. The emergence of this unique recombinant strain illustrated the complexity of the HIV-1 epidemic, and the need to strengthen molecular epidemiological surveillance and measures to reduce its spread.
China is a country with a high prevalence of human immunodeficiency virus type 1 (HIV-1) recombinant strains, with HIV subtype circulating recombinant forms (CRFs) being responsible for >90% of HIV-1 infections. 1 Despite the epidemic of different subtypes and recombinant forms in China, the dominant HIV-1 genotypes are strains CRF01_AE, CRF07_BC, CRF08_BC, and subtype B/B′ (Thailand variant of subtype B). 2 Subtype B was originally found in the United States and Europe, and was first isolated in China among injecting drug users in1989. Subtype B′ was also first detected in this same risk group in the Golden Triangle 1 year earlier, and was epidemic in Yunnan, China, by the early 1990s. 3 –6 CRF01_AE was identified in Thailand in the 1980s, and was later confirmed to have originated in Central Africa in the 1970s, rapidly expanding in China around 1999–2000 through sexual transmission. 7,8 In recent years, the proportion of HIV-1 infections caused by CRF01_AE has increased significantly, whereas cases due to subtype B/B′ have shown a steady decline; however, recombinants of subtype B/B′ and CRF01_AE account for more than three-fourth of cases in some regions of China. 9 A total of five CRF strains (CRF55_01B, CRF59_01B, CRF67_01B, CRF68_01B, and CRF101_01B) and diversified unique recombinant form (URF) strains, CRF01_AE/B and B′, have been confirmed to exist in mainland China, with most being identified in groups who partake in high-risk sexual activities. 10
Guangxi has one of the highest incidences of HIV/AIDS in China. It is located in southern of China, bordered by Vietnam in the southwest and Guangdong to the east. Guangdong was the first province to open up to the outside world. Its postmodern environment has attracted many people, especially those from the neighboring province of Guangxi. As such, Guangdong and Guangxi share many similarities in terms of the AIDS epidemic. For 2008–2009, the number of people infected by HIV through sexual transmission was greater than through intravenous drug use, with the predominate HIV genotypes being CRF01_AE, CRF08_BC, CRF07_BC, and CRF55_01B in Guangxi and Guangdong province. 11 –13 Dramatic changes in transmission patterns, dynamic changes in subtypes, and the cocirculation of multiple subtypes in the same region have led to the emergence of recombinant strains with increasing frequency in these two provinces.
In this study, we identified a new HIV-1 strain, 2018GXQZLSHET001, formed by recombination between subtypes CRF55_01B and B.
After obtaining informed consent, a blood sample was collected from an individual taking antiretroviral medication, who was from Guangxi, China, as of December 19, 2018. The patient was a 42-year-old married male with junior high school education. He was first diagnosed as being HIV positive in July 2006, and believed he was infected through heterosexual contact in Guangdong province. His most recent CD4+T count was 530 cells/μL in March 2018.
The proviral DNA was extracted using an Aidlab DNA kit (Aidlab Biotechnologies Co, Ltd., Beijing, China). The near full-length genome (NFLG) was amplified by a nested polymerase chain reaction (PCR). PCR was carried out with the same cycling conditions in both rounds: preheating at 94°C for 2 min; followed by 35 cycles at 94°C for 15 s, 60°C for 30 s, and 68°C for 4 min; a final extension at 68°C for 10 min; and a holding temperature of 8°C. PCR products were separated by agarose gel electrophoresis, and excised bands were sent to Tianyi Huiyuan Biological Technology Co., Ltd. (Beijing, China) for purification and nucleotide sequencing. Primary sequence was trimmed, spliced, cleaned, and mixed bases with Sequencher 5.0.
Finally, a contig of 9,005 base pairs (bp) was obtained, which on alignment with HXB2 corresponded to nucleotides (nt) 790 to 9,606. The NFLG sequence was subjected to online HIV-1 Sequence Quality Analysis (
Phylogenetic tree analysis was implemented by FastTree and displayed by FigTree, using the approximately maximum-likelihood method with the generalized time-reversible (GTR) model for the NFLG alignment. Branches with bootstrap values >0.9 were considered to be phylogenetic clusters. 8 As shown in Figure 1, strain of 2018GXQZLSHET001 was clustered with JF340054 (bootstrap values = 1) and distinct from other clusters of CRF55_01B.
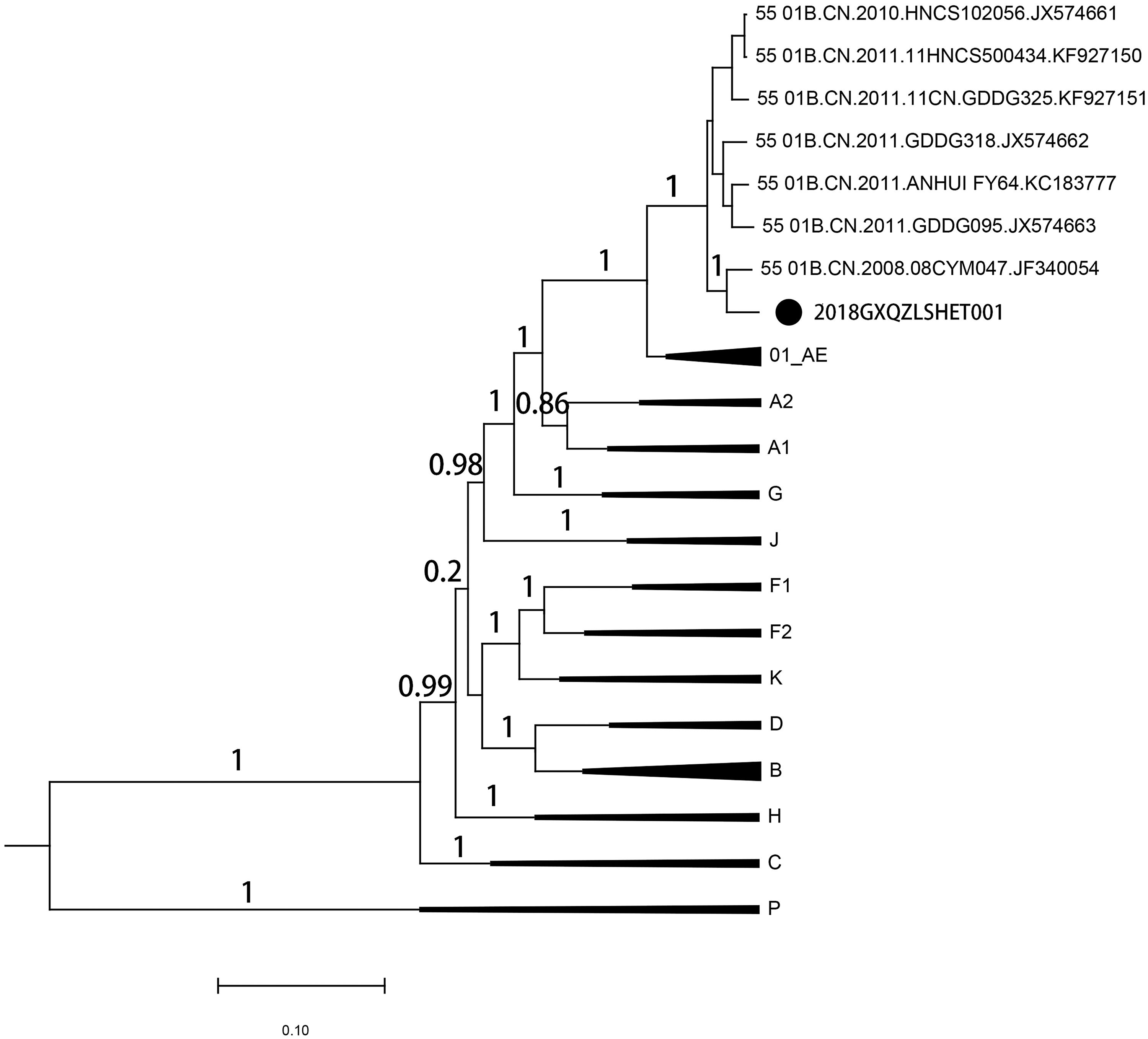
Phylogenetic analysis of the NFLG sequence of 2018GXQZLSHET001. The approximately maximum-likelihood phylogenetic tree was constructed with the GTR model. Three to four HIV-1 sequences in each subtypes of group M, group P, and several CRFs (CRF01_AE and CRF55_01B) were used as reference sequences. Bootstrap values >70% were shown at the nodes of the tree. The bar scale was set as 0.10. The branch of 2018GXQZLSHET001 was displayed as a straight line with black solid circle. CRFs, circulating recombinant forms; HIV-1, human immunodeficiency virus type 1; NFLG, near full-length genome.
Simplot version 3.5.1 was used to determine recombinant structure. The similarity plot (Fig. 2A) showed that strain 2018GXQZLSHET001 consisted of subtypes B and CRF55_01B. Bootscan (Fig. 2B) further verified that the backbone of the 2018GXQZLSHET001 sequence was CRF55_01B, with one subtype B segment inserted into the nef and 3′ LTR regions.

Recombinant analysis of 2018GXQZLSHET001 strain.

Subsequently, subregion phylogenetic trees (Fig. 3) were constructed to further describe the subtype and to explore the likely origin of each portion of the genome. As expected, phylogenetic analysis showed that the subtype origins of two genome segments were the same as those identified by the similarity plot and bootscan, all with high bootstrap values (>0.9). The results indicated that region I was clustered together with the CRF55_01B isolates, whereas the likely parental origin of region II was associated with the Thailand variant of subtype B.
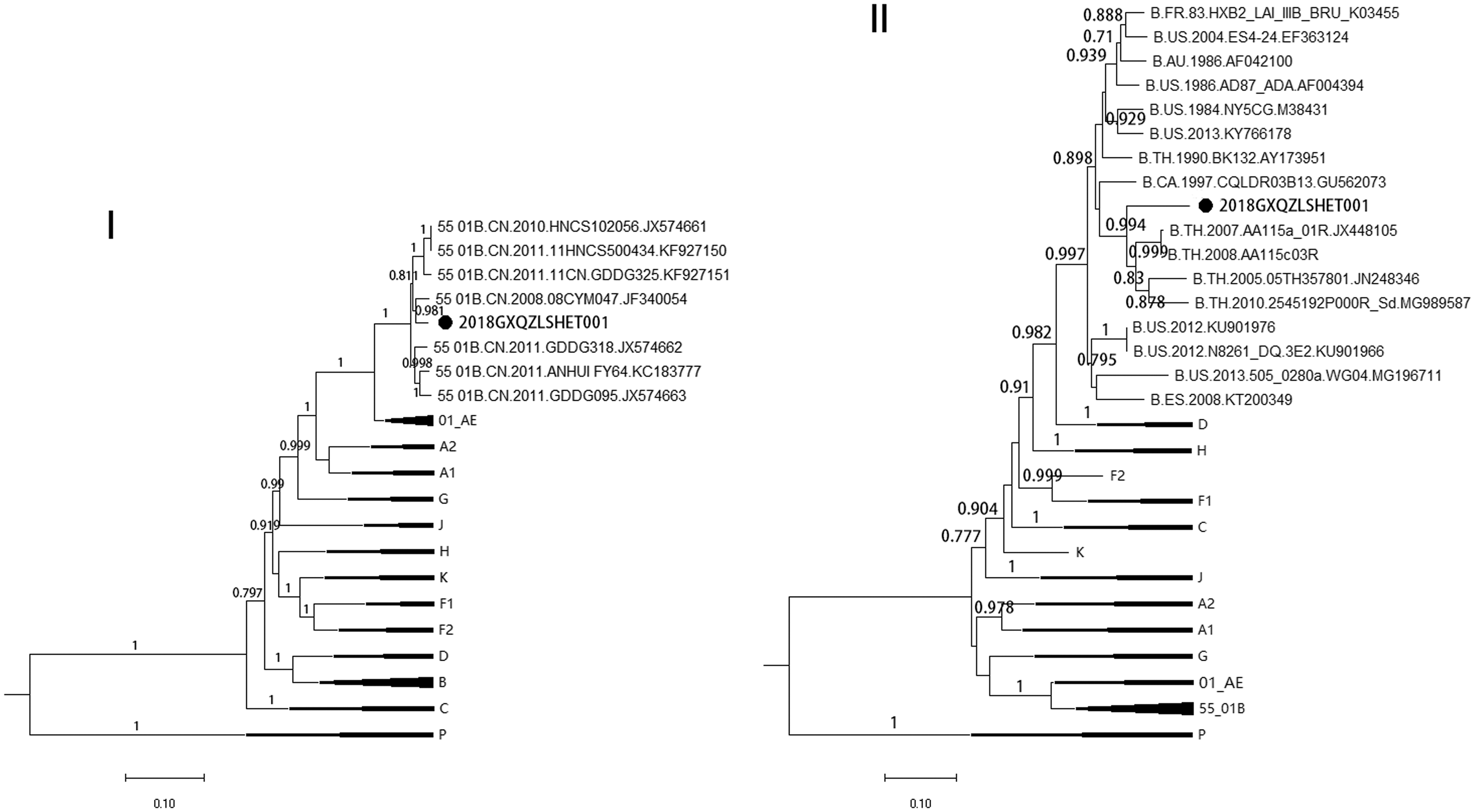
Subregion tree analysis of 2018GXQZLSHET001 strain. The phylogenetic trees of the two segments (region
To provide a more intuitive view and comparison between 2018GXQZLSHET001 and the typical CRF55_01B strain, mosaic structures for these genomes were drawn with the recombinant HIV-1 drawing tool available at the Los Alamos HIV sequences database (
With the extension of the epidemic from high-risk groups to the general population, the distribution of a particular HIV-1 subtype is not localized to a specific population anymore. 16 Therefore, coinfection of multiple subtypes of HIV-1 has become increasingly common, resulting in elevated number of CRFs and URFs.
As mentioned before, a variety of recombination forms of CRF01_AE and B have been found in China, where they are thought to spread by sexual transmission, particularly among heterosexuals. Therefore, it was not surprising to identify a new recombination of CRF55_01B and B in this study. Notably, CRF55_01B was initially identified in the MSM population, 17 with subsequent outbreaks in parts of Guangdong, and ultimately spreading to MSM populations in numerous regions of the country. 9,18 However, this strain has also spread rapidly through heterosexual populations, 19 likely contributing to the prevailing complexity of HIV-1. According to a previous study in Guangdong, 21.6% of MSM had been married, and 24% had engaged in heterosexual behaviors in the prior 6 months. 12 Sexual activities between different high-risk groups were likely to promote the emergence of new recombinant forms and affect the distribution of HIV-1 subtypes. This could explain why 2018GXQZLSHET001 likely formed by recombination of CRF55_01B and subtype B, whereas epidemiological data indicated he was infected with HIV through heterosexual contact in Guangdong.
With the convenient transportation and the huge economic gap between the two provinces, a large number of people from Guangxi work in Guangdong. A variety of transmission routes and multiple HIV-1 subtypes coexist in both these two regions, which usually indicate active cross-infection, particularly for dominant strains, such as CRF01_AE and subtype B, which were recombinant as CRF55_01B. In this study, the patient stated that he was diagnosed with HIV infection after sex with prostitutes in Guangdong, suggesting that the infection originated in that province. Our analysis indicated that the infecting virus was a recombinant strain containing portions of CRF55_01B and subtype Thai-B. The Thai-B has also been reported to be prevalent among heterosexual populations in Guangdong. 12 Therefore, the recombination pattern of 2018GXQZLSHET001 was consistent with the current prevalence in Guangdong of CRF55_01B and Thai-B in MSM and heterosexual populations, respectively.
In conclusion, the NFLG sequence of strain 2018GXQZLSHET001 was characterized, and shown to be a novel second-generation recombinant form. Although there is no evidence that this recombinant strain was circulating in the region from which it was isolated, its presence demonstrated that the continuously increasing prevalence of subtype B and CRF55_01B will promote the emergence of more recombinant forms. As such, it is a warning that molecular epidemiological surveillance of HIV-1 needs to be vigorously strengthened, to assess the dynamic changes of HIV-1 genotypes and their distribution in real time.
Sequence Data
The near full-length genomic sequence of 2018GXQZLSHET001 has been submitted to GenBank with accession number: MN067522.
Footnotes
Authors' Contributions
L.Y. conceived and designed the study. B.Y.L. and Q.Y.W. conducted the data analysis, literature review, and drafted the article. Y.Y., J.L., and Y.Y. were involved in data collection and interpretation. J.M.C., R.F.C. and H.L. assisted with data management and data analysis. All authors contributed to the revision of the article and approved the final version.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the National Key Science and Technology Project of China (grant no. 2018ZX10101002-001-006), National Natural Science Foundation of China (grant no. 81560326), Guangxi Natural Science Foundation (grant no. 2018GXNSFAA138070), Science Foundation for The Excellent Young Scholars of Guangxi Collaborative Innovation Center for Biomedicine (grant no. GCICB-TC-2017015), Key Program of Guangxi Collaborative Innovation Center for Biomedicine (grant no. GCICB-SR-2017007).