
Abstract
Iron is a key factor at various stages of HIV life cycle and determines the progression of HIV infection. Data about cellular labile iron pool (LIP) in the settings of contemporary antiretroviral therapy (cART) are lacking. Yet LIP is directly related to the generation of reactive oxygen species, and may contribute to immune activation, dysfunction, and exhaustion. Using multiparameter flow cytometry, we evaluated LIP in CD4 and CD8 T cells from HIV+ patients with sustained viral suppression (SVS) as a result of continuous long-term cART. Based on the recovery of CD4/CD8 ratio, two patients' subgroups were defined: A (n = 26), CD4/CD8 > 0.9, and B (n = 37), CD4/CD8 < 0.9, with significantly differing CD4 absolute count (AC) (mean 752 vs. 571 cells/μL, p < .05). Although hemoglobin and serum iron had recovered in all patients, CD4 T cell LIP and CD8 T cell LIP were significantly higher than that of controls, both in the subgroup with complete (A) and with incomplete (B) immune recovery [mean CD4 mean fluorescence intensity (ΔMFI) 318.7 and 777.8 vs. 157.6; mean CD8 ΔMFI 359.5 and 628.7 vs. 179.2, analysis of variance p < .05 for both]. CD4 LIP correlated inversely with CD4 AC (R = −0.4, p < .01), and both CD4 LIP and CD8 LIP—with CD4/CD8 ratio (R = −0.4, p < .01). Thus, increased CD4 T cell LIP and CD8 T cell LIP in the settings of SVS and immune recovery are a sensitive marker of residual immune activation and may predict immune exhaustion in long-term cART-treated patients.
Although contemporary antiretroviral therapy (cART) efficiently suppresses HIV replication, and promotes CD4 T cell reconstitution, persistence of viral reservoirs is a well-established fact. The proliferation of latently infected cells, occasional viral replication, and low-level immune activation may compromise the long-term prognosis of people living with HIV. Highly sensitive biomarkers of residual viral activity are warranted to improve the treatment and follow-up of ART-treated patients.
Iron is essential for immune functions and plays a key role at various stages of HIV life cycle. Increased iron stores were shown to correlate with faster progression of HIV-1 infection in the settings of thalassemia major, haptoglobin 2-2 polymorphism, or oral iron substitution. 1 In contrast, iron chelation can prevent HIV-1 reactivation. 2 Systemic and cellular iron are complexly regulated. Essentially all extracellular is transferrin bound iron (TBI), and >95% of intracellular iron is also protein bound. The term “labile iron pool” (LIP) refers to the transitory (i.e., exchangeable and chelatable) redox-active form of cell iron important for cell metabolism. It results from a balanced uptake of circulating TBI, its utilization in mitochondria, and storage of unutilized cell iron in ferritin complexes. 3 LIP is regulated by ferroportin-mediated transmembrane export, and hepcidin-mediated inhibition of ferroportin receptors. Enhanced hepcidin synthesis during inflammation results in increased level of LIP. 4 Poorly liganded ferrous iron is related to production of reactive oxygen species (ROS), with deleterious role in case of compromised iron balance. 3,5
The few studies on systemic iron in cART-treated HIV+ patients are inconclusive as to the independent effects of cART on iron homeostasis. Chang et al. showed that HIV induces a significant increase in serum iron (sFe) level, persisting in adequately ART-treated patients. 2 No data are available on LIP at the level of T cell subsets in HIV+ subjects, before or in the course of cART. We hypothesized that in the settings of HIV infection, CD4 T cell LIP and CD8 T cell LIP are related to viral activity changes in the course of treatment. Modifying a published method, 6 we measured LIP directly in CD4 and CD8 T cells from HIV+ patients with sustained viral suppression (SVS) and different extent of immune recovery in response to cART.
Peripheral blood samples from 63 HIV+ men, aged 41 ± 11 years [mean ± standard deviation (SD)], on long-term cART (mean therapy duration, 5.8 ± 4.6 years), with SVS [HIV viral load (VL) <1.6 logHIV RNA copies/mL for at least 1 year), were compared with those of 16 HIV(−) age- and sex-matched controls. The study was conducted during follow-up of patients according to European ethical standards. Hemoglobin (Hb) and sFe were determined by routine laboratory methods. T cell subset absolute counts were determined in TRUCount standard tubes (BD Biosciences). Samples were incubated with calcein acetoxymethyl ester (CA-AM: BioLegend) and T-subset markers followed or not by deferiprone. The difference between the mean fluorescence intensity (ΔMFI) of chelator treated and untreated cells was used to evaluate the amount of LIP in CD4+ and CD8+ T. Statistical analysis included Student's paired t-test and analysis of variance (ANOVA) with post hoc Bonferroni test, and Pearson's correlation test (SPSS version 23). All data are expressed in mean ± SD.
Virally suppressed patients had different immunological responses to cART: CD4 absolute count (AC) varied from 219 to 1,130 cells/μL, and CD4/CD8 ratio varied from 0.17 to 3.8. Therefore, we defined subgroups based on CD4/CD8 ratio as a sensitive marker of immune recovery: A, n = 26, CD4/CD8 > 0.9 (mean 1.34 ± 0.61) and B, n = 37, CD4/CD8 < 0.9 (mean 0.55 ± 0.17). CD4 AC differed significantly between the subgroups: 752 ± 227 vs. 571 ± 196, (p < .01), unlike the age (44 ± 10 vs. 39 ± 11, years), time without cART (18 ± 27 vs. 22 ± 31, months), treatment duration (6.7 ± 5.0 vs. 5.0 ± 4.0, years), baseline CD4 AC (493 ± 311 vs. 409 ± 372), baseline CD4/CD8 ratio (0.49 ± 0.37 vs. 0.36 ± 0.26), or baseline HIV VL (4.5 ± 1.26 vs. 4.6 ± 1.22 logHIV RNA copies/mL), p > .05 for all. Hb and sFe recovered to normal in all patients with SVS (143.9 ± 16.7 vs. 149.6 ± 7.6 g/L and 21.2 ± 8.2 vs. 25.4 ± 6.2 μmol/L, p > .05 for both). Despite some contradictory data, 2 these results corroborate with the fact that replicating HIV, together with the generalized immune activation in the absence of cART are major Fe consumers. However, CD4 T cell LIP and CD8 T cell LIP were significantly higher in patients than in HIV(−) controls (CD4 ΔMFI 604 ± 509 vs. 158 ± 114, and CD8 ΔMFI 527 ± 446 vs. 179 ± 178, p < .001). Importantly, this was valid both for the subgroup with complete (A) and with incomplete (B) immune recovery (CD4 ΔMFI 319 ± 236 and 778 ± 540 vs. 158 ± 114; CD8 ΔMFI 360 ± 240 and 629 ± 505 vs. 179 ± 178, ANOVA, p < .05), Figure 1A. In addition, a significant inverse correlation existed between CD4 LIP and CD4 AC (R = −0.4, p < .01) as well as between CD4 LIP (data not shown), CD8 LIP, and CD4/CD8 ratio (R = −0.4, p < .01; Fig. 1B, C).
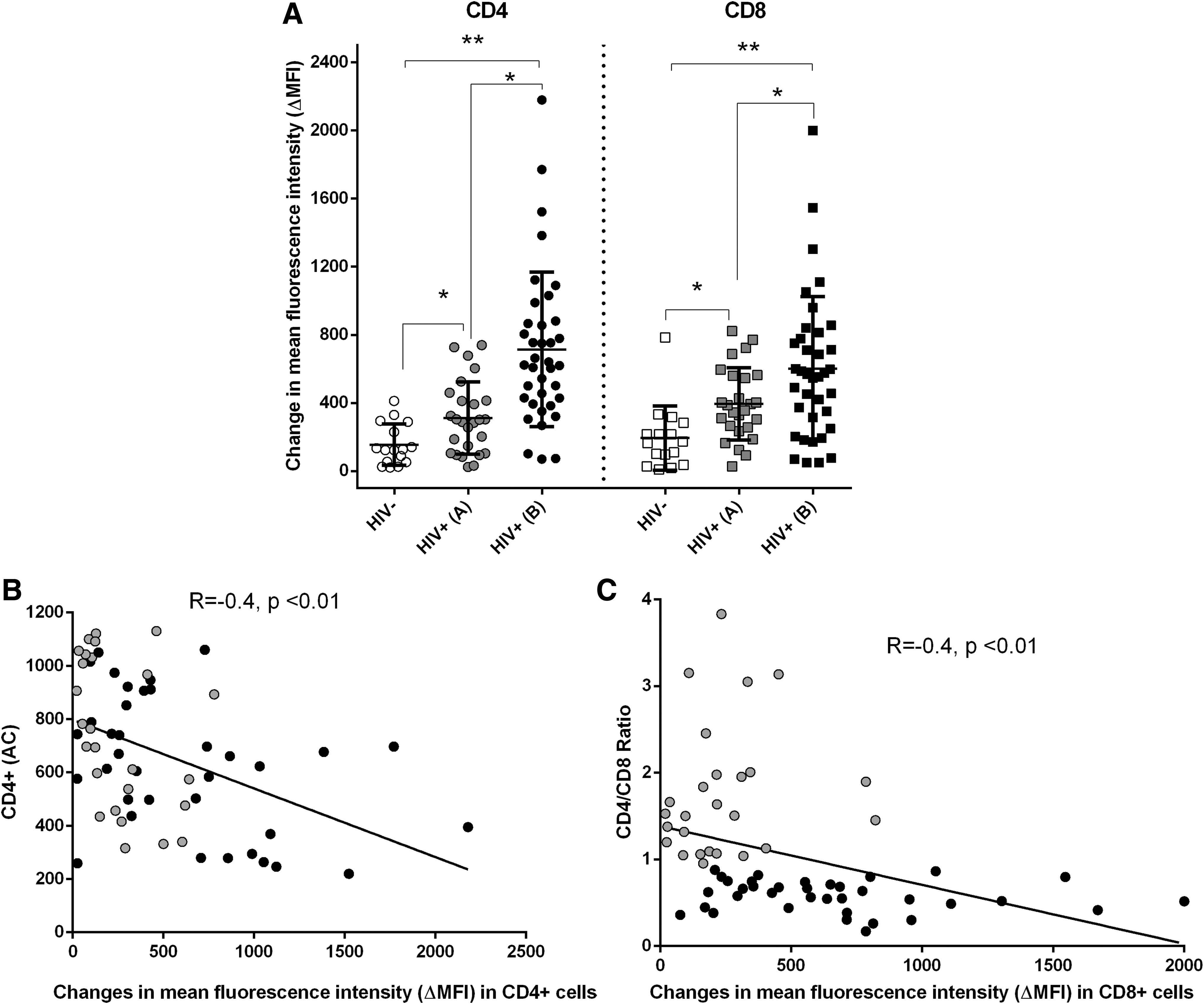
Elevated LIP levels in CD4+ and CD8+ T cell subsets of cART+ HIV+ patients despite SVS. Freshly isolated peripheral blood mononuclear cells were incubated simultaneously with 0.5 μM green fluorescent CA-AM (BioLegend) and the combination of monoclonal antibodies CD45-PerCP/CD8-APC/CD4-PB/CD3-AmCyan (all purchased from BD Biosciences) for 20 min at 37 RT, followed by washing and 60 min. incubation with 100 μM deferiprone (Sigma-Aldrich). At least 5,000 events were acquired in the lymphocyte gate defined on CD45/SSC plots, using an FACSCanto II instrument Diva 6.1.2 software (BD Biosciences). LIP was calculated as the difference between the ΔMFI of chelator-treated and untreated cells.
Our results clearly show that chronic HIV infection affects the regulation of iron turnover leading to increased LIP in the settings of SVS, independently from the age, baseline CD4 AC, CD4/CD8 and HIV VL, and cART duration. This effect was probably associated with HIV activity since CD4 LIP correlated inversely with CD4 AC, but also—with the general activation since both CD4 LIP and CD8 LIP correlated negatively with CD4/CD8 ratio—a sensitive marker of residual immune inflammation in the virally suppressed. 7 The problem of HIV persistence and accelerated immune aging of long-term treated HIV+ patients is well recognized. A number of studies have focused on the functional and metabolic state of immune cells “beyond the undetectable viral load” looking for sensitive markers of HIV reactivation and/or immune inflammation. Very few though have looked at LIP in the settings of long-term cART.
In vitro studies have linked iron with HIV progression: higher LIP and oxidative stress were associated with HIV reactivation in infected macrophages, with increased HIV infection and replication in primary CD4+ T cell cultures, whereas iron chelators decreased viral replication. 2 It was further shown in vitro that HIV infection results in cellular iron overload through increasing iron uptake, and several ART drugs (tenofovir, old-generation protease inhibitors) may increase cellular iron independently of HIV. 2 In our study, specific cART effects were negligible since regimens were either darunavir or INSTI based, and the number of patients on tenofovir was comparable between subgroups A and B. To our knowledge, this study is the first to show lasting effects of HIV infection and cART on LIP ex vivo, in the settings of controlled viral replication and immune recovery.
As LIP plays a key role in cell metabolism, the balance between iron uptake and deposition into ferritin is tightly regulated. Since all studied patients had normal sFe levels, increased CD4 LIP and CD8 LIP were most probably associated with inefficient ferroportin-mediated export, in the settings of inflammation-enhanced hepcidin expression.
At pathological levels, LIP catalyzes ROS formation that can surpass cellular antioxidant capacities and seriously damage its constituents. 3,5 Recent studies have linked oxidative stress and dysregulated T cell metabolism and functions in some cancers and chronic infections. In particular, high ROS content in HIV-specific CD8 T was associated with signs of exhaustion (e.g., high PD1 and low CD127 expression, impaired proliferative capacity), 8 as well as induction of apoptosis, and depletion of CD4+ T. 5 In vitro findings suggested involvement of ROS in the activation of NF-κB, which potently increases the production of HIV virions in latently infected cells. 5
Although no other data are available on LIP at the subset level, studies similar to ours evaluated ROS production in CD4+ and CD8+ T cells from virologically suppressed HIV+ individuals, with contradictory results. Although Masson et al. found no significant differences between HIV(−) and HIV+/cART (>6 years) participants, 9 Yu et al. established increased level of ROS in CD4+ but not in CD8+ T of HIV+/cART(<3 years) subjects. The discrepancies were attributed to different cART duration and/or different methods for ROS quantification. 10 In our hands, LIP levels were similarly increased in both CD4 and CD8 T cells and correlated with each other (R = 0.7, p < .0001), regardless of cART duration. These results suggest that the mechanisms of HIV-related mitochondrial toxicity may differ in CD4 and CD8 T cells, and that increased LIP (whether leading to increased ROS or not) is the more straightforward marker of perturbed cellular metabolism.
In conclusion, we report for the first time continuously elevated LIP levels in CD4 and CD8 T cells from successfully cART-treated HIV+ patients. Since elevated LIP promotes HIV replication, and is associated with T cell dysfunction, exhaustion, and apoptosis, it may serve as a sensitive marker for clinical follow-up. A larger prospective study is warranted to assess the prognostic significance of LIP, independently from other markers of mitochondrial disturbance, and the undesirable effects of certain cART components on iron homeostasis and mitochondrial metabolism.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by Research Grant DM13/6/20.12.2017, Bulgarian National Science Fund, “The role of iron homeostasis for chronic inflammation in HIV infection.”