
Abstract
In this study, we sequenced the full-length HIV type 2 (HIV-2) long terminal repeat region from the proviral DNA of 23 HIV-2-infected individuals from the southern parts of India. We identified two different promoter variant strains circulating in this region along with the globally circulating common promoter variant. Seven sequences had an additional nuclear factor kappa-light chain enhancer of activated B cells (NF-κB) binding motif and the sequence from another subject showed one NF-κB and one RBE-III binding site. Phylogenetic and subtyping analyses revealed that the circulating strains comprised HIV-2 subtype A. The occurrence of two NF-κB binding sites in ∼30% of the sequences analyzed in our study prompts us to hypothesize that as in the case of HIV-1 subtype C viruses that possess additional κB sites, the two NF-κB HIV-2 variants might possess superior replication fitness because of the increased magnitude of transcription, thus leading to the expansion of these variants in the country.
HIV type 2 (HIV-2) infections are not only endemic in West Africa, but are also present in India, Brazil, and Europe, particularly Portugal, Spain, Germany, France, Sweden, and the United Kingdom. 1 In general, HIV-2-infected individuals survive longer than HIV-1-infected persons. The first case of HIV-2 was reported from India in 19912 and since then various studies have reported the prevalence of subtype A infections in the country. 2,3 Like HIV-1, HIV-2 uses the host's signaling pathways and viral regulatory proteins synergistically for transcriptional regulation.
The HIV-2 long terminal repeat (LTR) sequence, present at both ends of the viral genome is 850 base pairs long and comprises the U3, R, and U5 regions. The U3 region contains the promoter, enhancer, and modulatory sequences. The R region contains sequences specific for tat binding called the TAR (Trans-activation response) element. The U5 region contains transcription factor binding sites. The HIV-2 LTR shows an overall similarity of 40% with the HIV-1 LTR at the genomic level, but the conserved regions within the LTR show up to 50% similarity. Numerous transcription factors and regulatory elements actively interact with the HIV LTR and regulate its activity.
The modulatory region in the U3 segment of the HIV-2 LTR contains binding sites for transcription factors such as PuB1 and PuB2 (purine-rich site majorly used for T cell receptor binding), peri-ets sequence (Pets) or enhancer-like purine boxes, CCAAT/enhancer binding protein (C/EBP) site, activating transcription factor/cyclic AMP response element binding (ATF/CREB) site, lymphocyte enhancer factor (LEF-1) binding site, and the nuclear factor of activated T cells (NF-AT) binding site. The enhancer element upstream of the core region is defined by the presence of a 10 bp binding site for nuclear factor kappa-light chain enhancer of activated B cells (NF-κB). The core region is primarily known for the presence of the TATAA box and GC-rich binding sites for the specificity protein (Sp) family of transcription factors.
The TATAA box is the regulatory element present 25 to 35 bases upstream of the initiation site of transcription that binds the TATAA binding protein, and aids in effective transcription at the transcription initiation site. Three tandem binding sites for Sp factors, rich in G + C content, are positioned immediately upstream of the TATAA box and are majorly involved in the cell differentiation process by promoting transcription. They also have other functions such as responding to DNA damage and immune responses. The three isoforms of the Sp site are referred to as Sp1, Sp2, and Sp3. All the three sites are required for efficient replication of HIV. The transcription factor NF-κB plays a pivotal role in the host innate immune and antiviral response and is an important host factor that promotes HIV-1 replication during acute infection. 4,5 The HIV LTRs contain typically one NF-κB binding site in HIV-2 and two to four in HIV-1. 6,7
With regard to the HIV infection landscape in India, subtype C accounts for >95% of HIV-1 infections, and ∼95% of HIV-2 infections occurring in Asia are from India. 1 In India, since 1991, HIV-2 infections have been reported in many states. Yet, reliable and up-to-date information on the HIV-2 epidemic in India is still lacking. Various seroprevalence studies carried out in different parts of the country have reported HIV-2 prevalence ranging from 0% to 7%. 8 Given the complexity of the HIV epidemic in the country, accurate genetic characterization and identification of various elements of the genome including the architecture of the LTR sequences in the circulating HIV-1 and 2 strains in the country is important for population-based molecular epidemiology studies. The availability of full-length genomic sequences is critical to address the evolving HIV epidemic.
Although there are increasingly large numbers of HIV-1 genome sequences, there are only two full-length HIV-2 sequences (CRIK-147 and NNVA) available from India and these were collected and sequenced >10 years ago (CRIK-147 isolated in 1995 and NNVA in the year 2007). Given the rapidly evolving HIV epidemic in India, a periodic sequence-level characterization of circulating strains is important to understand viral evolution. In this study, we describe the sequencing and molecular characterization of full-length HIV-2 LTR sequences derived from 23 subjects from South India. The study identified viral variants with uncommon features in the promoter region that are being reported for the first time.
The study was approved by the Institutional Ethical Review Board of the National Institute for Research in Tuberculosis (NIRT), Chennai, India. Written informed consent was obtained from the study participants before blood collection. The demographic and clinical details of the participants and the deduced subtype of the viruses are given in Table 1. The sampling period was between 2012 and 2016. In brief, proviral DNA was extracted from whole blood using the QIAamp DNA mini kit (Qiagen, Valencia, CA). Full-length HIV-2 LTR was amplified by nested PCR with a set of outer and inner sense and antisense primers that were designed in-house (Table 2).
Demographic Details and Virus Subtype of the HIV Type 2-Infected Individuals
F, female; M, male.
List of Primers Used for the Amplification and Sequencing of the HIV Type 2 Long Terminal Repeat Region
Reference sequences representing the LTR region of the HIV-2 genome of multiple subtypes were downloaded from the Los Alamos National Laboratory HIV database (
Full-length LTR contigs were edited based on the purity of the peaks in the electropherogram and assembled manually with the HIV-2 BEN (M30502) sequence using the Seqscape assembly program, version 2.5, with prescribed default parameters. We took BEN as reference sequence for this study because the other reference sequence, HIV-2 ROD, was obtained from viral RNA. The 23 full-length LTR sequences were subjected to extensive sequence and phylogenetic analysis. The virus subtype was analyzed using three different web-based HIV genotyping tools (NCBI HIV genotyping, COMET, and REGA HIV subtyping tools).
Multiple sequence alignment of the isolated HIV-2 LTR sequences revealed many unique substitutions and a few deletions. The alignment was generated by a HMMER model with HIV-2 BEN sequence (accession no. M30502) as reference. Phylogenetic and cluster analyses were performed using online phylogeny software program
Detailed molecular analysis of the 23 full-length HIV-2 LTR sequences from this study identified the presence of all previously reported potential transcription factor binding sites. The LTR in 16 of the 23 sequences contained one NF-κB site, typical of HIV-2 subtype A. In the other seven cases (NIRT01, NIRT06, NIRT08, NIRT09, NIRT11, NIRT18, and NIRT24), the LTR contained two NF-κB sites (Fig. 1), a molecular feature quite uncommon for HIV-2 strains. Tamhane et al. 12 had previously reported an additional NF-κB site from one Indian HIV-2 isolate (HIV2CRIK147).

Multiple sequence alignment of the Indian type-2 HIV 5′ LTR sequences showing three classes of promoter variant strains: (a). Those strains having two NF-κB sites (the commonly observed NF-κB site marked as NF-κB-I and the additional NF-κB site marked with *). (b). The strain having one RBE-III site and one NF-κB site. (c). Those strains having one NF-κB site, the commonly observed NF-κB site. LTR, long terminal repeat; NF-κB, nuclear factor kappa-light chain enhancer of activated B cells. Figure can be viewed in greater detail online.
The original NF-κB site (5′ AGGGACTTTCCA), which is present in all subtypes of HIV-2, was found to be present and highly conserved in 21 of 23 (91%) viral sequences, suggesting its critical role. The NF-κB site in one viral sequence (NIRT29) had a single point mutation (G to A substitution) at the fourth position. Yet another sequence (NIRT19) contained mixed bases (R and S) in the first and sixth position, respectively. In addition to the two NF-κB binding sites, we identified at least five other cis-acting enhancer elements in the study sequences: two purine-rich sequences (PUB-1 and PUB-2), a peri-ets sequence (P-ets), a monocyte-specific peri-kb sequence, and Sp elements that have already been reported in literature (Fig. 2). Another interesting and unusual feature we observed was the presence of an RBE-III binding site in one of the LTR sequences (NIRT07).
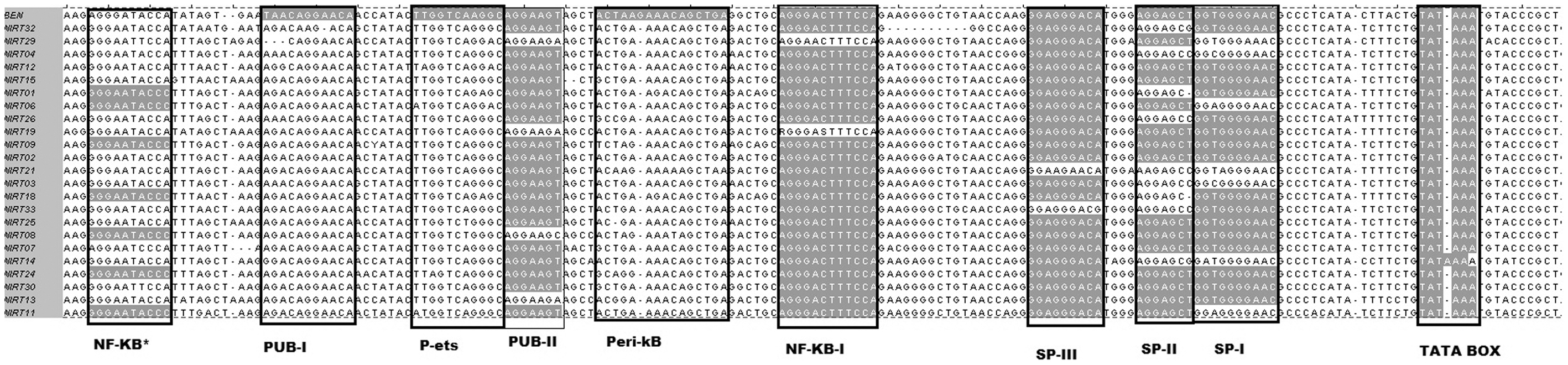
Diagrammatic representation of the sequence alignment of the 5' LTR nucleotide sequences of the 23 HIV-2 isolates from south Indian population. The sequences were aligned with the HIV-2 BEN sequence (accession no: M30502). Different TFBS previously reported in literature are marked in gray and their names are shown in the boxes. TFBS, transcription factor binding sites. Figure can be viewed in greater detail online.
We next examined the degree of conservation in each of the transcription factor binding sites (TFBS). As given in Figure 3, the mean mutant frequency was calculated by dividing the number of mutations with respect to the reference BEN sequence by the total number of nucleotides in each TFBS. We observed relatively higher levels of nucleotide diversity in the PUB1, Peri-kb, and P-ets sites, than in the PUB-2, NF-κB, and SP binding sites.

Mean nucleotide distance of different TFBS between the BEN sequence and the present study isolates from South India. Mutation frequency was calculated by dividing the number of mutations with respect to the reference BEN sequence by the total number of nucleotides sequenced in each TFBS as described. *The mean mutation frequency was calculated only for the commonly observed NF-κB site and not the additional κB site reported in this study.
Phylogenetic analysis was performed with the study sequences and 27 full-length HIV-2 subtype A sequences that were downloaded from the Los Alamos Database representing different continents and different countries. One subtype B sequence and two previously reported Indian subtype A sequences (CRIK and NNVA). All the isolates sequenced in this study were found to cluster tightly with the subtype A reference sequences (Fig. 4). Within the subtype A cluster, majority of the study samples were found to be closely associated with the CRIK and NNVA sequences.
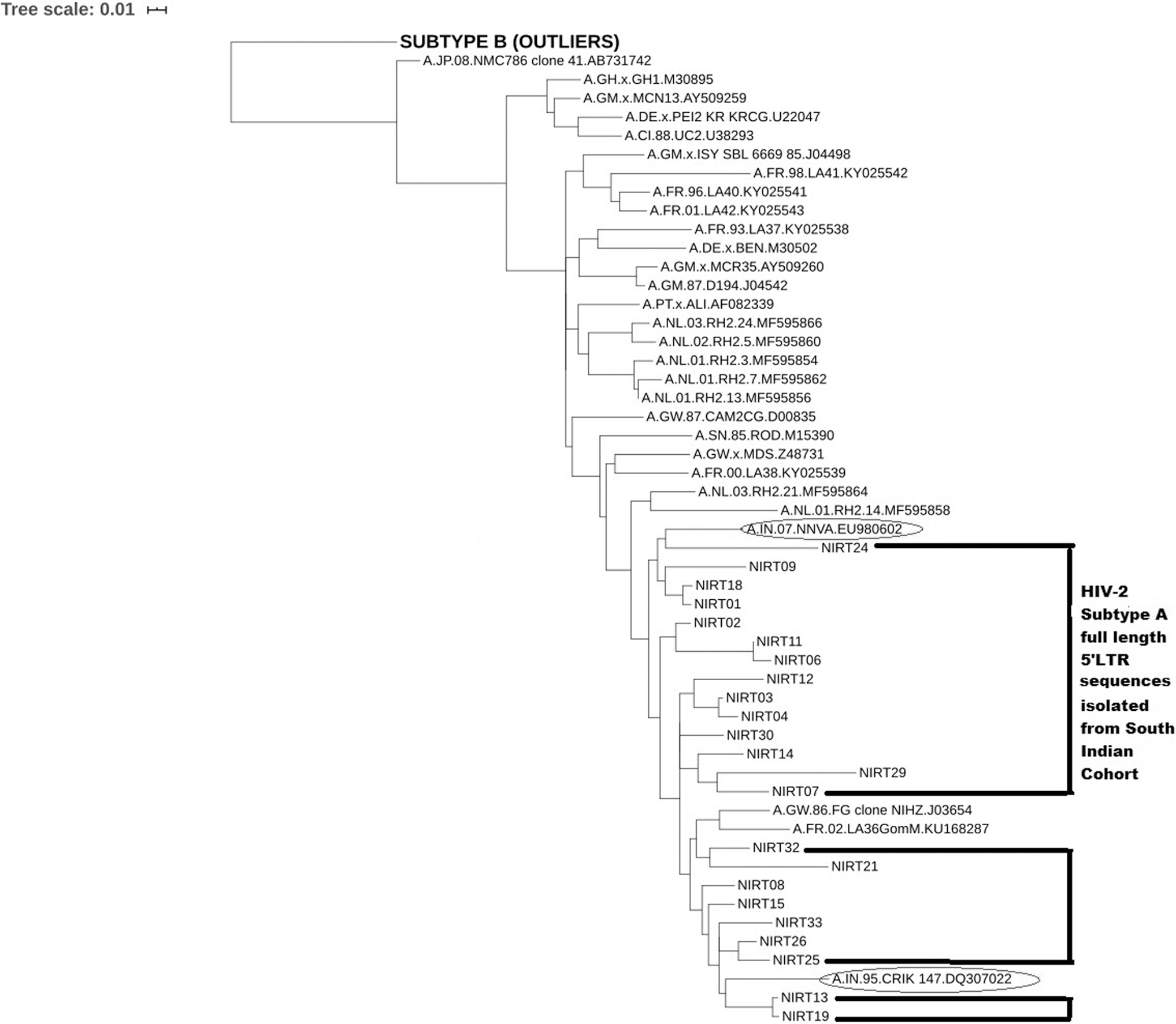
Phylogenetic analysis of full-length 5′ HIV-2 LTR sequences. Phylogenetic analysis was performed using the full-length 5′ LTR sequences isolated from south Indian HIV-2-infected individuals and the database derived sequences representing HIV isolates from different countries including Indian NNVA and CRIK 147 isolates and a subtype B isolate as an outlier. Sequences that belong to the subjects of this study are indicated within the box-type black lines and the previously reported Indian isolates are marked in oval-shaped circles. The number of bootstrap replications was set as 100.
We also analyzed the full-length HIV-2 LTR sequences obtained from the HIV Los Alamos database (accessed on November 7, 2019) to understand the molecular heterogeneity in the promoter region. To this end, a total of 35 full-length HIV-2 subtype A, 8 subtype B, 1 subtype G, 5 subtype H2-AB, and 1 MAC reference sequence were downloaded from the Los Alamos database and subjected to multiple sequence alignment and comparative analysis. We found that only three HIV-2 sequences, two subtype A sequences (Z48731 and CRIK), and one subtype B sequence (KYO25545) contained an additional NF-κB site (data not shown).
Similarly, the RBE-III site was found only in two subtype A strains (KYO25543 and MH681610). These data collectively suggest that the presence of 2-κB and RBE-III site in HIV-2 viruses is an emerging phenomenon that is also seen in Indian HIV-2 strains. The occurrence of 2-κB sites in ∼30% of the sequences in our dataset clearly reflects expansion of this promotor variant in the Indian population.
Investigators have noticed a similar phenomenon in HIV-1 subtype C viruses and have reported rapid emergence of viral variants with duplicated NF-κB and RBE-III binding sites. 13 The emergence of additional NF-κB binding motifs in HIV-1 has been implicated as a potential advantage to the virus in many ways. This prompts us to speculate that as in the case of HIV-1 subtype C, the presence of additional NF-κB site in the HIV-2 LTR confers a definite advantage to these viral variants in terms of superior magnitude of transcription, replicative fitness, and enhanced viral predominance. This relatively new phenomenon of acquiring additional NF-κB sites and/or RBE III motifs in emerging circulating viral strains is associated with a higher magnitude of genetic variation not previously reported in HIV-2 viruses. Whether these emerging promotor variant strains of HIV-2 will alter the landscape of the Indian HIV demographics and prevalence in the country remains to be seen.
The data presented in this study have the following merits and limitations. Although the meritorious side of the study is that the analyzed LTR sequences are all full/near full length and were obtained from proviral DNA, the limitation is that no infectivity assay was performed to confirm the effect of various mutations in the transcription factor binding sites on viral infectivity and replicative fitness. Most of the bioinformatics analyses performed in this study were based on the sequences primarily generated through this study and sequences available from extant databases. Given this limitation, our results could be considered only as inferential evidence suggesting a positive evolutionary selection of variant viral strains. In addition, the inference on the potential advantage of the identified molecular features to the HIV-2 virus should be confirmed using validated functional assays.
In summary, we demonstrate for the first time that at least two different promotor variants of HIV-2 subtype A are present in significant proportions in India. These viral strains contain an additional NF-κB, or RBEIII site, in addition to the original NF-κB site. Although it is tempting to speculate that the differences in the regulatory motifs are very likely to influence viral fitness by acting at the transcription level, the potential effect of the second NF-κB site on the replicative capacity of the virus and rate of disease progression in HIV-2-infected individuals remain to be established. The present findings prompt us to suggest that HIV-2 exploits a small window of opportunity for relatively superior magnitude of transcription and replication fitness. Phylogenetic analysis of the HIV-2 sequences obtained from the 23 HIV-infected individuals showed that all individuals harbored HIV-2 subtype A strains that were closely linked to the previously reported Indian strains. As molecular surveillance is the backbone for finding effective interventions, characterization of more full-length sequences of HIV-2 viruses would contribute to a better understanding of the epidemiology of the infection in the Indian subcontinent.
Sequence Data
The nucleotide sequences of the 5′ LTR of the 23 HIV-2 sequences generated in this study have been submitted to GenBank (accession nos. MN942007–MN942029).
Footnotes
Acknowledgments
The authors thank the Indian Council of Medical Research (ICMR) and the individuals who participated in the study.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
There was no separate funding for this study.