
Abstract
Understanding the evolutionary dynamics of the viruses within an individual at or near the moment of transmission can provide critical inputs for the design of an effective vaccine for HIV infection. In this study, high-throughput sequencing technology was employed to analyze the evolutionary rate in viruses obtained at a single time point from drug-naive recently infected infants and adults in the chronic stage of disease. Gene-wise nonsynonymous (pN) and synonymous (pS) mutation rates were estimated and compared between the two groups. Significant differences were observed in the evolutionary rates between viruses in the early and late stages of infection. Higher rates of adaptive mutations in the HIV-1 envelope gene (env) were found in the chronic viruses as compared with those in the early stages of HIV infection. Conversely, percentage of nonsynonymous substitutions in env was found to be higher in recently transmitted viruses. In addition, a positive correlation was found between mutation and the evolutionary rate, and infectivity titer in recent infection. Despite the small sample size, the study identified useful information about viral evolution on transmission-associated bottlenecks. The effect of intraindividual HIV-1 evolution at the population level was highly contemporary, and the higher percentage of nonsynonymous substitutions seen in env during recent HIV-1 infection has suggested a pattern of convergent evolution leading to a positive selection for survival fitness and disease progression.
Introduction
The HIV-1 surface glycoprotein (Env) is a principal target of the humoral immune response in HIV-1-infected individuals. Genetic diversity of HIV-1 envelope (env) has been associated with the development of broadly neutralizing antibodies (bNAbs) 1 and is a major challenge for HIV vaccine design. Envelope-specific antibodies emerge in HIV-infected individuals within a few weeks of infection, but these fail to neutralize the virus. 2,3 HIV-1 has developed several mechanisms to evade neutralizing antibodies, such as inaccessibility of relevant epitopes resulting from the conformational flexibility of the trimeric Env structure, increased density of glycosylation, and varying lengths of variable loops. 4 Extensive research on the HIV-1 envelope has enabled us to understand some of the mechanisms involved in viral/host interaction, viral replication kinetics, and disease progression. A better understanding of the characteristics of early transmitted viruses will help discover Env vulnerabilities that can be targeted by vaccines designed to elicit sustainable protective antibodies against HIV-1. 5
The HIV-1 genome comprises of structural, regulatory, and accessory genes that code for different proteins involved in establishing HIV infection and promoting viral replication. The structural genes play an important role in the establishment of HIV infection and quasispecies dynamics in the human host. Infection by HIV-1 is a stepwise process starting with the fusion of gp120 to the CD4 cells with the help of one of its two coreceptors that permits viral entry. 6 Soon after fusion, the virus releases its nucleocapsid into the host cell cytoplasm where the enzyme reverse transcriptase converts RNA into cDNA. 7 Successful viral entry into the host cell is crucial and important for establishing HIV-1 infection. 8 The highly adaptive nature of the HIV-1 envelope enables viral escape to evade the host immune response, particularly, CD8+ cytotoxic T lymphocyte (CTL) activity, which the virus encounters during the early and chronic stages of infection. 8,9
Host immune pressure is unevenly distributed across the HIV-1 genome with clear gene-specific differences. The continuous processes of mutant generation, insertion, or deletion within the gene, result in dominance of one or several genomes with a higher fitness divergence. In addition, the loss of some unique patterns such as potential N-linked glycosylation site (PNGs) and coreceptor recognition motifs exhibit failure to establish infection or cause slower disease progression. 10 It is important to examine minor genetic variations that occur within the population to characterize the host adaptation of the viral quasispecies and its evolution toward drug resistance and immune evasion. 11 It is often difficult to distinguish minor mutations that exist in the viral population through Sanger sequencing. On the contrary, high-throughput sequencing 12 technology provides enough throughput data and sensitivity to detect very rare viral mutations for diagnosis, surveillance, and epidemiology. 13
Numerous studies have examined the structural aspects of the HIV-1 envelope and host cell receptor/coreceptor interactions. 14 However, studies describing the frequent escape mutations that the virus chooses to adapt and evolve within the human host, are limited. Viral escape mutations have frequently been incorporated by HIV, without affecting its biological properties, for better fitness and disease progression. 15 Evolution rate is described as the elevation in the rate of nonsynonymous substitutions (pN) over the rate of synonymous substitutions (pS) under host immune pressure. In this study, the evolutionary dynamics of HIV-1 within and among individuals with recent and chronic infections have been investigated in an attempt to understand the pN and pS contributions toward the viral evolutionary rate using HTS data. An enhanced knowledge on the selection of escape mutation by the virus is one of the several elements that could throw light on the design of both preventive and therapeutic vaccines to achieve sustainable protection and better control of HIV-1 infection.
Materials and Methods
Study population
Two milliliters of venous blood was obtained from eight infants (<1 year of age) who were infected with HIV-1 through mother-to-child transmission, and from two adults who acquired HIV infection through horizontal transfer. All the subjects were drug naive with the viral load >3,000 copies/mL (range: 83,400–47,23,382 RNA copies/mL of plasma) at the time of sample collection. Their HIV status was confirmed by HIV-1 DNA-PCR and ELISA, respectively. The detailed demographic profile, including viral load and CD4 count is shown in Table 1. Ethics clearance for the study was obtained from the Institutional Ethics Committee of the National Institute for Research in Tuberculosis (NIRT IEC No.: 2009009).
Clinical and Demographic Profile of Study Participants
F, female; M, male; MOT, mode of transmission; HET, heterosexual route; MTCT, mother-to-child transmission; A1, acute HIV infection; A2, chronic HIV infection; NA, not available.
HIV-1 near full-length genome amplification and high-throughput sequencing
Near full-length HIV-1 genome was sequenced based on the protocol described previously. 16 In brief, viral RNA was isolated from recently infected infant plasma samples having the viral load >3,000 copies/mL. cDNA was synthesized using gene-specific and oligo d(T)18 primers for two fragments (F1-Gag-to-Vpu and F2-Tat-to-3′LTR). First-round amplification was performed with 0682F and 6352R primers for f1 fragment and 5550F and 9555R primers for f2 fragment, respectively. Second-round amplification was performed with 0776F and 6231R primers for f1 fragment and 5861F and 9555R primers for f2 fragment, respectively. All PCRs were performed using KAPA HiFi Taq DNA polymerase following the manufacturer's protocol. The amplified fragments (F1-Gag-to-Vpu and F2-Tat-to-3′LTR) were purified using the PureLink Quick Gel Extraction Kit (Invitrogen) and pooled together in equal molar concentrations (10 nM each). The mixture was then fragmented on the Covaris S200 at 300 bp for 75 s with the peak power 50 and the cycle/burst 200. The library was prepared using the NEBNext® UltraTM DNA Library Prep Kit for Illumina® (New England Biolab) with multiplexed NEB next adaptors. The samples were then mixed along with other unrelated nonviral indexed libraries. Paired-end sequencing of length 250 bp was carried out on the Illumina HiSeq2500 platform. All the samples were resequenced to define reproducibility.
Genome assembly, base calling, and detection of single nucleotide polymorphisms
The de novo genome assembly was performed using IVA (Iterative Virus Assembler). 17 Any sample for which IVA was not able to yield a single contig of length ∼9 kb, VICUNA was used. 18 The base calling script, bustard, removed the instrument noise from each cluster, along with a confidence level for each nucleotide base. After these corrections, the base with the highest intensity was chosen. 19 Then, the filter aligned sequences were realigned against individual near full-length genomes (NFLG) to evaluate the “position-specific base count,” which was performed in integrative genomics viewer. 20 Consensus sequences were generated from the base count data with the occurrence of a particular nucleotide in >50% of the reads or the first most common nucleotide occurring at a given position. 16 Adaptive mutations (single nucleotide polymorphism, SNP) at a particular position were identified based on the base count range 10% to 50% or the second most common nucleotide occurring at a given population. Pseudo sequence to the consensus sequence was generated by replacing the existing nucleotide in the consensus sequence with the SNP nucleotide. Both the full-length nucleotide sequences were then spliced into individual gene and amino acid sequences for downstream analysis. Pairwise alignment of consensus and pseudo sequence from each individual was carried out to identify the nonsynonymous amino acid substitutions. Positions of nonsynonymous substitutions were numbered according to the HXB2 reference sequence position.
Estimation of pS and pN rates
The evolutionary rate of HIV-1 viruses within each individual infant was estimated separately using a Perl-based version of SNAP v2.1.1 tool available in the HIV database with necessary modifications needed to analyze the pN/pS ratio for each pair of sequence. This program is based on the method of Nei and Gojobori, 21 and incorporates a statistics developed by Ota and Nei. 22 SNAP calculated synonymous and nonsynonymous substitution rates based on a set of codon-aligned nucleotide sequences. In brief, in-frame nucleotide sequences of each gene of the consensus and variant sequences from each individual were codon aligned based on the HXB2 reference sequence position and were analyzed both individually (one individual at a time), and jointly (all individuals together) employing the NJ tree-based distance profile. The ratio of pN to pS (pN/pS) remains one of the most common and reliable measures of selective evolutionary pressure on protein coding regions. Much of its popularity branches from the simple, spontaneous interpretation of pN/pS <1 as a faithful indication of negative selection, pN/pS = 1 as neutrality, and pN/pS >1 as strong positive selection, 23 respectively, where, pN is the number of nonsynonymous changes per nonsynonymous site, pS is the number of synonymous changes per synonymous site, and pN/pS is the evolutionary pressure often quantified by the ratio of substitution rates at nonsynonymous (pN) and synonymous sites (pS) within species. 24 In addition, the amino acid distance for nonsynonymous amino acid substitutions was estimated in each gene based on the formula derived by Grantham, (1974). 25
Estimation of infectious titer
To estimate the infectious potential of the virus, the recombinant virus was generated as described previously. 3 In brief, gp120 of the env was amplified and cloned into the pMN-K7-Luc-IRESs-NefΔgp120 plasmid. Twenty-five clones from each individual were transfected into 293T cells and the viral supernatant was harvested after 48 h and clarified by centrifugation. The infectivity of the recombinant virus was determined by infecting TZM-bl cells. Luciferase expression from TZM-bl cells was measured as relative light units at 48 h postinfection, using the Bright-Glo luciferase assay system (Promega) on an Infinite M200Pro plate reader (Tecan, Switzerland). Sixty-five out of 250 clones were found to be infectious. The number of infectious clones obtained from each individual is provided in Table 1.
Definition of HIV-1 individual genes
HIV-1 NFLG nucleotide sequence was spliced into individual genes based on the HXB2 (K03455) reference sequence position. The viral structural genes (5′gag-pol-env3′) were defined based on the corresponding sequences in the HXB2 as nucleotides 790-2292, 2085–5096, and 6225-8795, respectively. The essential regulatory genes (tat-rev-nef) were identified to correspond to the nucleotides (tat exon 1: 5831-6045, tat exon 2: 8379-8469), (rev exon 1: 5970-6045, rev exon 2: 8379-8653), and 8797-9417 in the HXB2 sequence, respectively. The accessory regulatory genes (vif-vpr-vpu) were defined to span nucleotides corresponding to the positions 5041-5619, 5559-5850, and 6062-6310, respectively, in the HXB2 sequence.
Results
Participant characteristics
HIV-1 NFLG (5′gag to 3′LTR: position 0711-9555) were sequenced using the Illumina HiSeq2500 platform from the HIV-1-infected infants who acquired infection through mother-to-child transmission (n = 8) and the adults who got infected through heterosexual transmission (n = 2) (Table 1). The chosen sample size was 10 and the sampling period was from 2013 to 2016. Of the eight infants, four (50%) were males. The median peak viral load was 5.7 log10 viral RNA copies/mL of plasma (interquartile range, IQR: 5.1–6.4 log10); the median CD4+ T cell count was 392 cells/μL (IQR: 250–647 cells/μL). Due to insufficient sample availability, CD4 count could not be done for a couple of samples. As expected, the phylogenetic analysis showed a tight clustering of the sampled sequences with the subtype C reference sequences (data not shown). Sequences of full-length HIV-1 genomes were submitted to GenBank with the accession nos. KX069219—KX069228. The data available for the study participants are provided in Table 1.
Estimation of genetic diversity
The genetic variation/nucleotide substitution in terms of SNPs identified in each sequence is represented in Figure 1. SNPs were calculated based on the variations in nucleotide observed at a single position. The true variants with set point of >10% reads in the HTS data were examined at each genomic site to call SNPs that occurred more often than expected. The number of SNPs in recently infected individuals was in the range of 18 (CON_IN09) to 104 (CON_IN03) with a median value of 57.5. As expected, this number was significantly higher in chronic infection, —206 (CON_IN01) and 254 (CON_IN02). Interestingly, no diversity was found in the gag, pol, vif, and vpr genes in one of the individuals with chronic infection (CON_IN01) (Fig. 1). The number of SNPs (Supplementary Table S1) identified in the env was found to be higher with 40.9% (30.5% in recent infection and 51.7% in chronic infection), followed by pol and nef with 18.2% [24.2% (A1) and 11.9% (A2)] and 11.7% [10.7% (A1) and 12.8% (A2)], respectively. There was very little diversity in the rest of the genes that is, <10% [gag (7.3%); vif (4.6%); vpr (2.0%); vpu (4.9%); tat (5.1%); and rev (5.2%)].

Average nucleotide coverage and genetic diversity. SNPs at each genomic site were plotted using the highlighter tool available in the HIV Database with default parameters. The true variant with a sensitivity of up to 1/10,000 was achieved for SNP calling at a coverage of around 5,000 to 15,000. Number 1–8425 represents the start of gag and end of nef and does not correspond to the position of sequence in the full-length genome. SNP, single nucleotide polymorphism. Color images are available online.
Nonsynonymous amino acid substitutions
The percentage of nonsynonymous amino acid substitutions in the individual genes from all the individuals are illustrated in Figure 2a. The percentage substitution in the env (p value <.0538) was found to be higher than in any other gene; although this value is not statistically significant, it may be considered as biologically significant with an average value of 60.8%. In contrast to the percentage diversity, percentage nonsynonymous amino acid substitution for the env was found to be significantly higher in strains from recent infection (68.3%) as compared with 56.3% in those from chronic infection. It was intriguing to note that the percentage of nonsynonymous amino acid substitution in the pol region was much lower (24.1%) than in all the other genes [gag (32.4%); vif (46.5%); vpr (42.1%); vpu (41.3%); nef (43.6%); tat (39.6%); and rev (51.0%)]. Taken together, the overall estimation indicated that the percentage of nonsynonymous amino acid substitution per individual in recent infection was comparatively higher (except regulatory genes: tat and rev) than that seen in chronic infection (Fig. 2b).
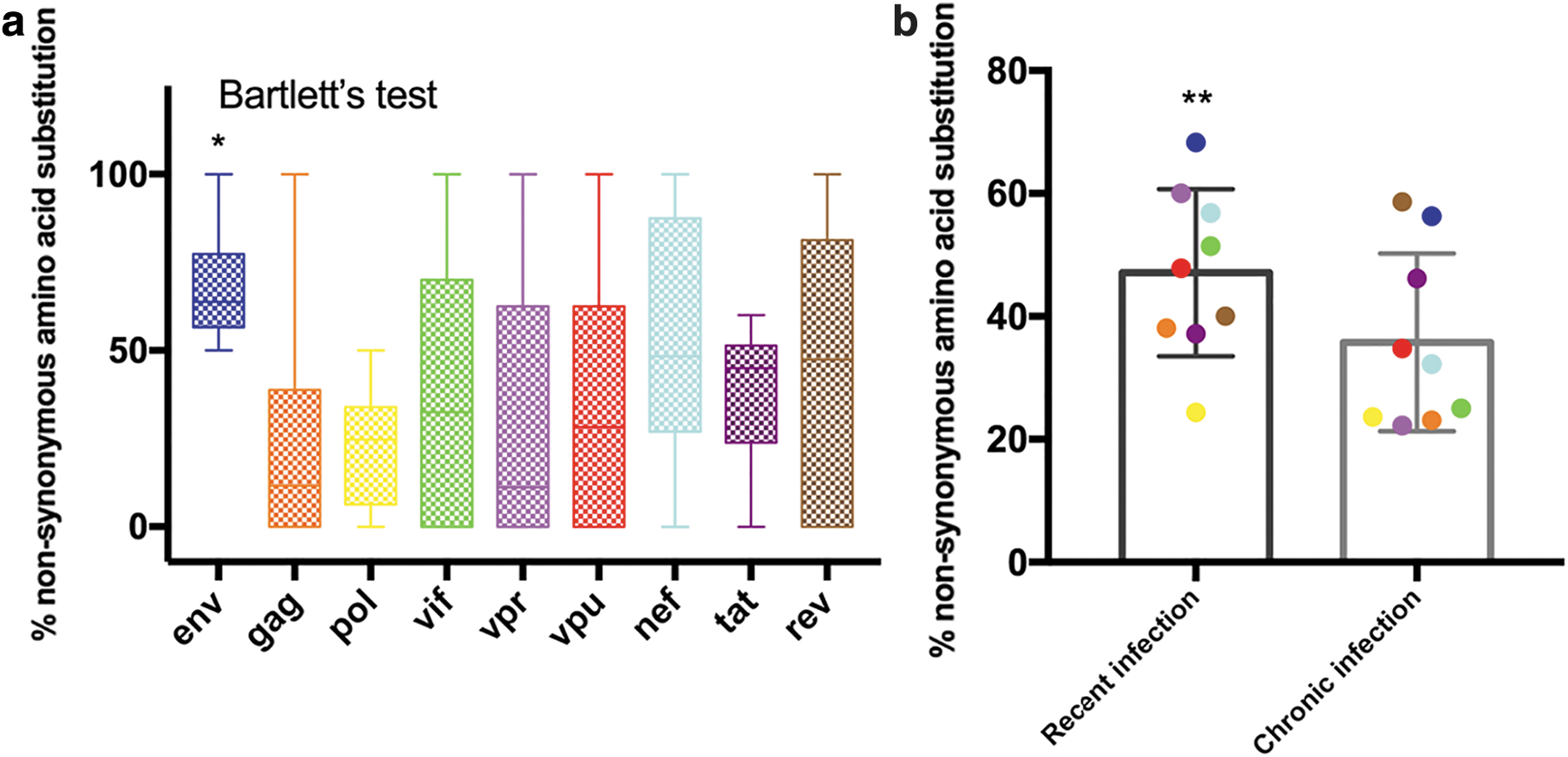
Percentage of nonsynonymous substitutions observed in different HIV-1 genes of variants obtained from recent and chronic infection. Box-and-whiskers plot showing percentage variation of pN values per gene per site
pN and pS substitution rate
pS and pN rates of individual genes of the full-length HIV-1 genomes sequenced in this study were determined. Gene-wise nonsynonymous amino acid substitutions with their corresponding positions are listed in Supplementary Tables S1 and S2. The rate of divergence at synonymous sites differed substantially between the genomic regions. Not surprisingly, the divergence rate at nonsynonymous sites was found to be much higher in the env than in pol (Fig. 3 and Supplementary Table S3). A distinct divergence was observed between the envs of recent and chronic isolates at both synonymous and nonsynonymous sites. However, selection of divergence in the env at synonymous sites was strikingly lower in strains from recently infected individuals as compared with those from chronically infected individuals. This could probably be attributed to the humoral immune pressure of the host that resulted in positive selection of escape mutations, which would rapidly spread through the population. This possibly led to a much faster accumulation of nonsynonymous substitutions in the env region.

Comparison of synonymous (pS) and nonsynonymous (pN) substitution rates in different genes of HIV from recent and chronic infection, where pS is the number of synonymous changes per synonymous site and pN is the number of nonsynonymous changes per nonsynonymous site. Color images are available online.
Estimation of intrapatient evolution rate (pN/pS)
Taken together, the average evolutionary rate was significantly higher for env (0.58 with IQR 0.32–0.71, p value <.05) followed by nef (0.48 with IQR 0.07–0.81) (Fig. 4a). When comparing the two groups, the average evolutionary rate of env was significantly higher in recent infection (0.65) as compared to the chronic stages of infection (0.36) (Fig. 4b). In contrast, the substitution rate in the env for nonsynonymous substitution in chronic infection was found to be higher than in recent infection with mean values of 0.036 and 0.006, respectively (Fig. 4c). This difference was not observed between the two groups for the rest of the genes. The patient-specific evolutionary rate across the env was quite high, followed by nef, than all the other genes in recent infection. In an individual estimation, positive selection (pN/pS >1) was not found in certain genes in three recently infected individuals (vif in CON_IN06; env and nef in CON_IN07; nef in CON_IN08). None of the genes of chronic isolates exhibited positive selection. In short, the analysis of evolutionary rate indicated a moderate purifying selection across env and nef (0.65 and 0.51, respectively) in recently infected individuals. As expected, the average amino acid distance in the env for nonsynonymous substitution in recent infection (79.1) was found to be significantly higher than that of chronic viruses (63.7) (Fig. 4d).
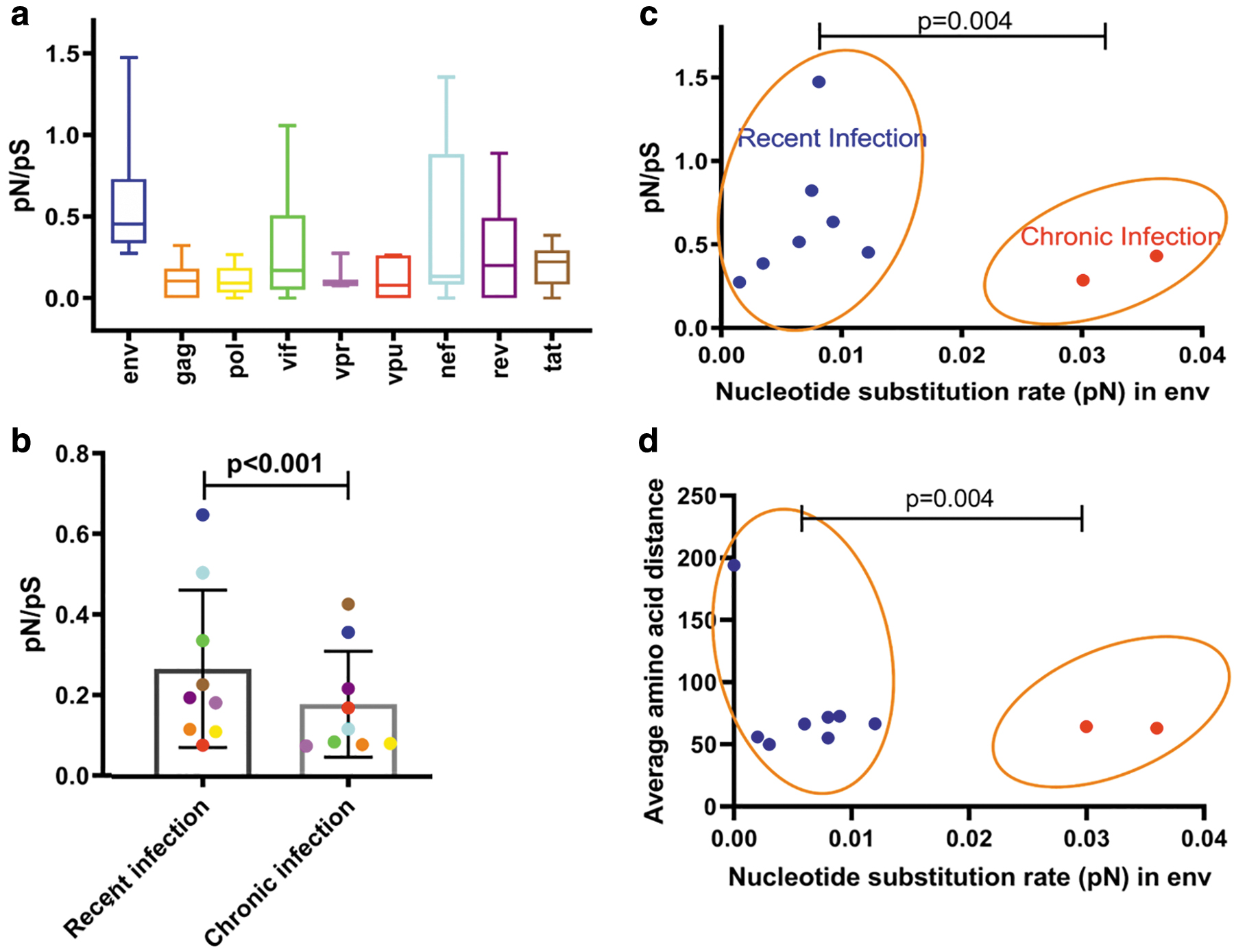
Evolution rates across genes and individuals with recent and chronic infection; and its impact on infectivity. Box-and-whisker plot showing evolution rate per gene
Impact of nonsynonymous substitutions in the env on infectivity
Next, the impact of mutation and/or amino acid substitution rate on infectivity of the recently transmitted and chronic viruses, was estimated. A positive correlation was found between mutation and evolutionary rate, and infectivity titer in recent infection (Fig. 5). Viruses from all recently infected individuals exhibited higher infectivity rates as well as a higher degree of evolution. On the contrary, the viruses present during the chronic stage of infection had a very low infectivity titer as well as evolutionary rate, in spite of the higher mutation rate accumulating over time, as compared with the recent infection. Unfortunately, in one of the two chronic cases, no infectious virus was obtained despite testing 25 molecular clones. These observations suggested that the viral quasispecies present very near the moment of transmission had incorporated nonsynonymous escape mutations for the purpose of immune evasion and enhanced its infectivity for successful homeostasis maintenance. There is strong evidence to support the impact of viral evolution on infectivity; it has been reported that even a single genetic mutation can influence establishment of infection. 26 At the same time, the large number of synonymous mutations identified in the chronic stage of infection might help the virus to survive under strong immune pressure.

Correlation analysis between mutations (SNPs), evolutionary rate, and infectivity titer in recent (left) and chronic (right) infection. Color images are available online.
Impact of nonsynonymous substitutions on host neutralizing antibody response and antiviral drug resistance
We noticed an enormously high substitution rate in the variable loops of the env gene. We had previously also reported that envs with shorter V1V2 loops were associated with increased neutralization resistance among recently transmitted HIV-1 strains. 3 We found a similar pattern in the present study as well. We hypothesize that the increase in neutralization resistance was due to the increased number of PNGs in the V1V2 loops that form a tight glycan shield and hide the neutralizing antibody epitopes in the env from the envelope-specific neutralizing antibodies. We also noticed a few minor drug resistance mutations to the NRTI and NNRTI class of drug in the pol gene of four of the eight infants who acquired HIV-1 infection from the mother. 27 It is likely that the mutations could have been transmitted from the mother to the child as the infants were breastfed for 6 months after delivery, although this could not be verified since the treatment history and drug resistance profile of the HIV-1-infected mothers were not available.
Discussion
In general, it is well known that RNA viruses have a high mutation rate and a fast evolution rate within the host. In particular, ss (+) RNA viruses (e.g., HCV) and retroviruses (e.g., HIV) often evolve faster than other viruses, with 10–4 to 10−2 substitutions per nucleotide per year. 28 In accordance with the literature, this study also found 10−2 and 10−3 substitutions per synonymous and nonsynonymous site, respectively. The substitution rate per nonsynonymous site was found to be very high (10−2) in the env. The tendency for higher substitution might reflect the adaptation of the virus to maintain replicative fitness during the early event of transmission. Improvement of knowledge on mother-to-child transmission, responsible for most pediatric HIV infections, would throw light on the dynamics of early viral evolution in the host environment to which the virus has partially adapted. 2 Uneven patterns of divergence and infectivity have been found among MTCT. Intrapatient genetic diversity at individual sites has been examined in the recently infected full-length HIV-1 viral genomes sequenced through HTS. A positive correlation between evolutionary rate and disease progression has been demonstrated by several studies in HIV-1-infected individuals. 29 –31 Positive selection (pN/pS >1) was observed in four of the infant samples, indicating a higher rate of viral replication and viral evolution. 32
The sample size used in this study was not significant enough to correlate the evolutionary rates between the recent and chronic stages of infection. However, useful informations about viral evolution on transmission-associated bottlenecks were obtained. Furthermore, the study provided a unique opportunity to examine the impact of immune pressure on evolutionary rates at/very near the moment of HIV transmission. Before analyzing the sequences for evolutionary rates, the quality of the sequence reads was checked in two independent runs and tight clustering of the same samples together with 100 bootstrap supports, was identified. Several studies have investigated the evolutionary rates at different time points within an individual and between individuals. Conversely, there is limited data on differences in the evolutionary rates between the viruses present in an individual at a given time point. Most studies have found lower evolutionary rates within the hosts than between hosts. 33 –35 However, an in-depth analysis of within-host and between-host evolutionary rates of HIV-1 genomes from HTS data has provided insights into distinct patterns of convergent and positive selection of amino acids under immune pressure in recent and chronic stages of infection.
It was found that the substitution rate varied between individuals infected recently through MTCT, with higher degree of substitution rates in both env and nef. Conversely, pol had a lower substitution rate. This was in agreement with the study that demonstrated a lower substitution rate in 5′ half of the HIV-1 genome than 3′ half. 36 However, substitution rates were influenced by both pathogenicity of the virus and selective immune pressure from the host, with the earlier reflected in the pS rate and the latter in the pN rate. 37 pS rates for all the genes, except env, were found to be higher than pN although not statistically significant. Nonetheless, the pS and pN rates for env were found to be similar. This reflects selective immune pressure exerted during the early event of infection. Collectively for all the genes, a significantly higher pS rate was observed in chronic infection than in recent infection. This observation was further confirmed through physicochemical analysis of the amino acid distance between the replaced and replacing amino acids, 25 which gave similar results. It is speculated that the higher pS rate in the former is associated with a higher replicative fitness as compared to the latter.
Surprisingly, a considerable number of SNPs and nonsynonymous amino acid substitutions were evident in recent infection. Particularly in the env, it was evident from the analysis that viruses from recently infected individuals undergo enormous numbers of nonsynonymous mutations for establishing successful infection. When compared with chronic infection, the number of SNPs in env was found to be lower in recent infection but the conversion of nonsynonymous mutations was found to be higher in recent infection. The higher percentage of nonsynonymous substitutions seen in the env might have resulted from a pattern of convergent evolution leading to a positive selection for survival fitness and disease progression. 38 The intraindividual HIV-1 evolution in the env in recent infection could reflect viral escape from the host immune responses. This is in evidence with the study that describes how an RNA virus mediates autoregulation mechanism (replicative hemostasis), which establishes stable, but highly reactive, equilibria that expands quasispecies and generates escape mutation. 39 This has been frequently reported to be under strong selection pressure, mediated naturally by emerging neutralizing antibodies, 40 T-helper cells, or cytotoxic T Lymphocytes. 41
The estimation of evolutionary rate across the genome, revealed a significant difference in the env and nef, as compared with the rest of the genes. The mean evolutionary rate was approximately twofold higher in recent infection than in chronic infection. However, this rate was fourfold higher in the case of nef in recent infection. This could reflect the differences in the evolutionary rate between the recent and chronic stages of infection, or differences between infant and adult infections. For example, higher viral loads in infants might influence evolutionary rates. 42 Viral loads at the later stages of infection were found to be more suppressed than the very early stages of infection although direct comparison between the two groups was difficult due to the difference in the sample number. In agreement with the existing literature, 35 nef, a myristilated protein that influences HIV replication, enhances infectivity of viral particles and downregulates CD4 and HLA on target cells, was found to evolve at the highest rate in recent infection. In contrast, the evolutionary rate for rev, which plays a role in regulating the export of nonspliced and partially spliced viral mRNA, was found to be higher in chronic infection.
Limitation
This present study had a couple of limitations that need to be mentioned. First, the sample size was too small to compare the substitution rate between the individuals in the two groups. Second, the study analyzed samples from a single time point only. Subsequent samples were not collected from the study participants since treatment was started after first sample collection, and treatment initiation could alter the immunological pressure and affect the natural selection of viral mutations.
Conclusions
The current study reconciles the findings that the dynamics of nonsynonymous over synonymous substitution rate in recent HIV-1 infection, particularly in the env, is likely to be the driving force of the humoral immune response, and the viral variants, which are resistant to neutralization by maternal antibodies, and drug resistance mutations acquired from mother, are preferentially transmitted to the infants. As per the neutral theory of molecular evolution, synonymous substitutions will be tolerated but nonsynonymous substitutions will be removed by purifying selection. Consequently, nonsynonymous substitutions will be fewer than the synonymous substitutions. The effect of intraindividual HIV-1 evolution at the population level is highly contemporary, and the selection of nonsynonymous amino acid substitutions, could likely reflect an advantage for the virus to survive against host immune pressure.
Footnotes
Author Contributions
Conceptualization: M.A., S.P.T., and L.E.H.; Data Curation: M.A., U.N., and L.E.H.; Formal Analysis: M.A., S.P., and U.N.; Investigation: M.A. and L.E.H.; Methodology: M.A. and S.P.; Project Administration: L.E.H.; Resources: U.N. and L.E.H.; Software: M.A. and S.P.; Supervision: L.E.H.; Validation: M.A., S.P.T., and L.E.H.; Visualization: M.A. and L.E.H.; Writing – Original Article: M.A. and L.E.H.; Writing – Review & Editing: M.A., S.P., S.P.T., U.N., and L.E.H.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.