
Abstract
Investigation of disease and intervention in populations of men having sex with men (MSM) has garnered attention globally, a primary reason being the rapid increase in the prevalence of human immunodeficiency virus (HIV)-1 among MSM. The purpose of this study was to understand the current HIV-1 molecular characteristics and characterize HIV-1 transmission networks in the MSM population. Nine hundred and fourteen newly diagnosed HIV-positive MSM were included in this study. The HIV-1 pol gene region was amplified and sequenced. A maximum likelihood phylogenetic tree was constructed, and transmission clusters were identified using 1.5% distance and 0.9 bootstrap values. In total, 767 sequences were successfully obtained, with CRF01_AE being the major genotype (43.3%, 332/767), followed by CRF07_BC (31.3%, 240/767), CRF67_01B (7.2%, 55/767), and URF (6.4%, 49/767). The transmitted HIV drug resistance rate was 4.0% (31/767), and the most common mutations were E138G (n = 4) and G190A (n = 4). A total of 182 (23.7%) sequences were included in the HIV-1 transmission networks, forming 79 clusters. Four clusters were identified as fast-growing, and the proportion of young MSM was higher than that of non-MSM (51.6% vs. 31.8%). The genetic diversity of HIV-1 in Jiangsu was complex, and cross-region transmission might exist for CRF67_01B. Transmission among young MSM within networks was greater than the other age groups; thus, they could be essential in the control of the HIV epidemic in Jiangsu. This study was approved by the ethical review board of the National Center for AIDS/STD Control and Prevention (Project No. X140617334).
Introduction
Although the incidence of human immunodeficiency virus (HIV) infections/acquired immunodeficiency syndrome (AIDS) in all age groups is decreasing globally, 1 HIV/AIDS remains a serious problem in China. In 2017, 134,512 new HIV-1 infections 2 were reported by the National Center for AIDS/STD Control and Prevention, Chinese Center for Disease Control and Prevention (CDC), signifying a 7.99% (9,957 patients) increase compared with the proportion reported in 2016. Most of these new infections (95.1%) were transmitted through sexual contact in 2017, making sexual transmission the most common mode of HIV-1 transmission in China. 2 In some developed areas and large cities, the population of men having sex with men (MSM) constitutes >50% of the local people living with HIV/AIDS (PLWHA): 53.4% (3461/6486) in Shenzhen, 3 54.3% (150/276) in Zhejiang province, 4 and 55.6% (240/432) in Shanghai. 5 The increasing number of new infections among MSM poses a severe challenge for HIV control. Therefore, it is necessary to assess and understand any new changes in the HIV-1 epidemic among MSM in China.
Chinese MSM generally have multiple sexual partners and low rates of condom use. A meta-analysis study reported that the HIV-1 infection rate among MSM was 5%, and the major risk factors were anal sex without the use of condom, commercial sex behavior, and multiple sexual partners. 6 Another Chinese study suggested that HIV-1-positive MSM with multiple sexual partners are common, and these partners constitute a “continuous transmission network.” 7 Therefore, elucidating the MSM transmission network is key to restricting HIV-1 transmission from MSM networks to members of the general population.
HIV drug resistance affects the ability of drugs to block replication of the virus. 8 Once patients experience first-line antiretroviral treatment failure, drug-resistance mutations can be transmitted to others. Recent studies have reported an increase in the prevalence of transmitted HIV drug resistance (TDR) among MSM in China. In Shanghai, the prevalence of TDR increased from 2.9% in 2012 to 8.0% in 20149; in Tianjin, the prevalence of TDR had increased from 3.9% in 2014 to 12.2% in 2017. 10 Clearly, monitoring the prevalence of TDR is very important.
Molecular epidemiology approaches have been widely used to elucidate HIV-related characteristics and risk factors. Oster et al. 11 used HIV-1 pol sequences obtained from drug resistance surveillance projects conducted by the US CDC to obtain rapidly growing clusters, given that genetic sequence data allow a visible understanding of molecular transmission network analyses. 12
In Jiangsu, one of the most developed provinces in eastern China, 50% of the population of people living with HIV/AIDS (PLWHA) consists of MSM (since 2014), and homosexual transmission is a major means of HIV-1 transmission in the province. A recent study showed that 71.3% of newly diagnosed HIV-1 infection cases in Nanjing, which is the provincial capital of Jiangsu, occurred through homosexual transmission. 13 It is, therefore, crucial to understand the characteristics of HIV-1 transmission networks among MSM to control the HIV/AIDS epidemic in Jiangsu. A recently established and ambitious goal of the Jiangsu provincial CDC is to construct a complete serum bank for newly diagnosed (treatment-naive) HIV-1-positive patients, and sample collection from MSM clients began in January 2017. Using these samples, we aimed to characterize the molecular epidemiology and TDR of HIV-1 and elucidate the transmission pattern among MSM, which could contribute to the development of targeted interventions.
Materials and Methods
Study subjects
All the study subjects were newly diagnosed with HIV-1 infections between January and December 2017, as confirmed by municipal CDC data submitted to the Jiangsu provincial CDC. We collected client demographic information (including age, marital status, education, number of sex partners, history of sexually transmitted diseases, and CD4 count) through face-to-face interviews administered by staff of the local CDCs. All subjects were male, treatment-naive, and infected through homosexual sex. This study was approved by the ethical review board of the National Center for AIDS/STD Control and Prevention (Project No. X140617334). All participants provided verbal informed consent.
RNA extraction, amplification, and sequencing
Whole blood samples collected at room temperature were sent within 4 h to the local city institutions for CD4+ T cell count testing, and the plasma of each sample was stored at −80°C until use. Viral RNA was extracted from 200 μL of plasma using the Abbott Sample Preparation System (Abbott Molecular, Inc., Des Plaines, IL) according to the manufacturer's instructions. The obtained RNA samples were amplified using reverse transcription polymerase chain reaction (PCR) and nested PCR for the pol region (HXB2 coordinates: nt 2253-3312) consisting of protease (PR)- and partial reverse transcriptase (RT)-coding sequences. The sequencing protocol has been described previously, 14 and the primers and cycling conditions are described in the Supplementary Material.
Sequence analysis
We assembled the original sequence files for the pol gene and aligned them using the ChormasPro 1.6 program and the Gene Cutter online tool (
HIV-1 transmission network analysis
Cluster Picker v1.2.5 was used to extract potential transmission clusters from the phylogenetic tree, with bootstraps support values ≥90% and maximum pairwise genetic distance ≤1.5% nucleotide substitutions per site. For all sequences extracted, Tamura-Nei 93 pairwise genetic distances were calculated using HyPhy 2.2.4; (3), and clusters included at least two sequences. Tamura-Nei 93 Cytoscape v3.7.0 was used to illustrate the transmission network.
A link implies a potential transmission relationship between subjects connected by lines. Degree refers to the number of links each sequence has, and higher degree values in the HIV-1 transmission network suggest a higher probability of virus transmission. The information of all the obtained sequences was divided into three groups: subjects with no link to others (0 link), subjects linked to one other subject (1 link), and subjects linked to two or more other subjects (≥2 links).
To obtain information to guide HIV intervention strategies, we defined a ≥5 sequence increase in the number of network sequences within a 1-year sequence as fast-growing networks. 15 The basic transmission networks were constructed using samples collected in the first 3 months of the study.
Statistical analysis
The variables of demographic information, CD4 count, and virus subtypes were compared among the three groups, and the differences were considered statistically significant at p < .05. Analyses were performed using SPSS 21.0.
Results
Information on study samples
Among the 914 newly confirmed HIV-1-positive subjects included in our study, the mean age was 33.8 ± 12.3 years (range 15–79 years), and 98.6% (901/914) were of Han ethnicity. Most of them were single (56.2%, 514/914), and 29.5% (270/914) were married to women. Participants within the sexually active age group (25–49 years) represented 61.2% (559/914) of the study group, and 41.8% (382/914) had received a college-level or higher education. All subjects were treatment-naive and verbally agreed to their blood samples being used for this study.
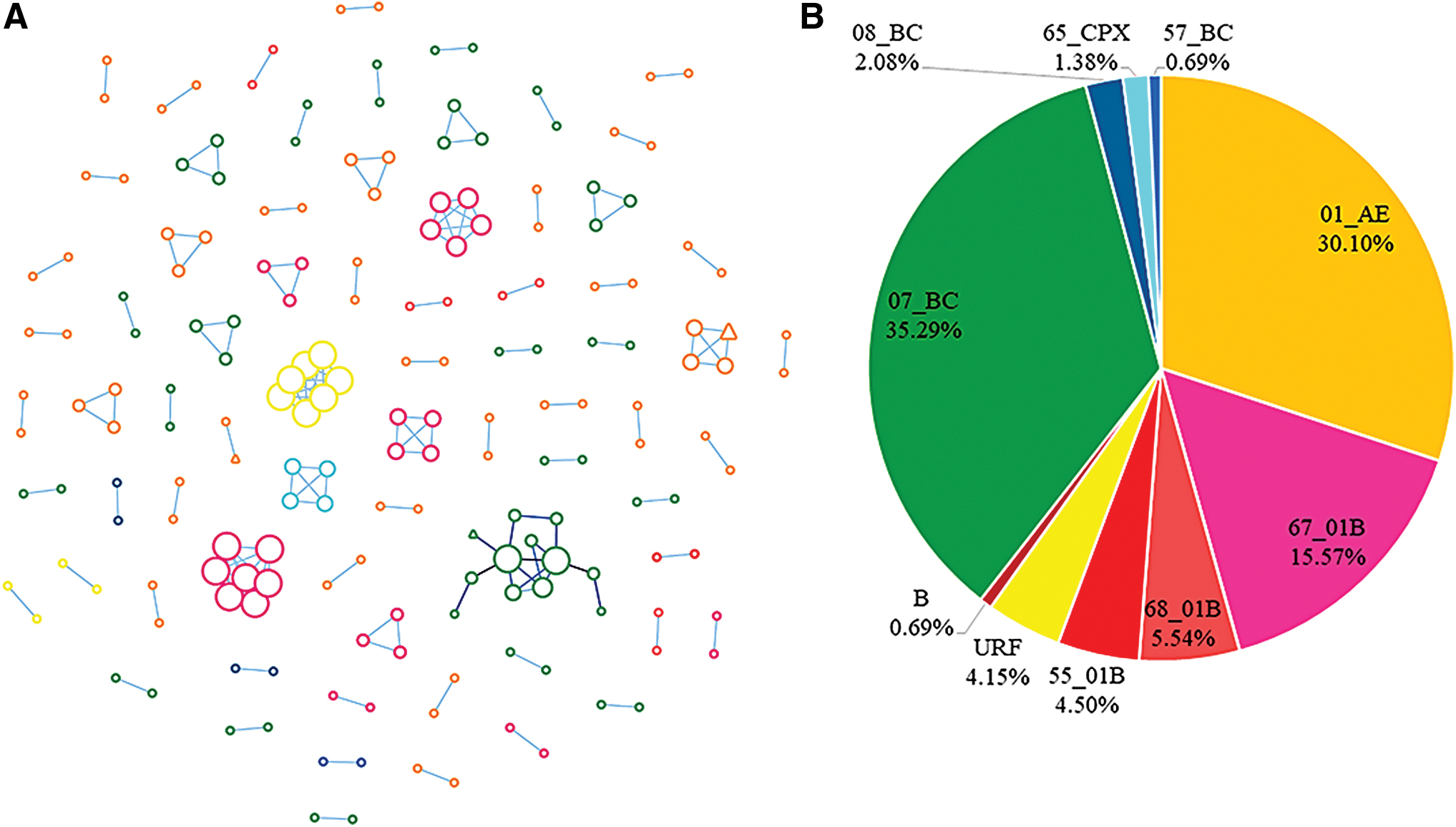
Of the 914 samples, 767 were (83.9%, 767/914) amplified successfully and the pol sequences were obtained using laboratory tests. The final length of the partial pol gene region of 1030 bp (HXB2:2253-3283) was used to construct the ML tree (Fig. 1A). We detected 11 HIV-1 genotypes, with CRF01_AE being the major genotype (43.3%, 332/767), followed by CRF07_BC (31.3%, 240/767); the other genotypes were CRF67_01B (7.2%, 55/767), URF (6.4%, 49/767), CRF68_01B (3.3%, 25/767), B (3.0%, 23/767), CRF55_01B (2.7%, 21/767), CRF08_BC (2.0%, 15/767), CRF65_cpx (0.5%, 4/767), CRF57_BC (0.3%, 2/767), and CRF59_01B (0.1%, 1/767) (Fig. 1B).

ML tree of pol (protease and partial reverse transcriptase) sequences from Jiangsu and globally sampled reference data set of sequences with known HIV-1 subtypes. The distribution of different subtypes is illustrated using different colors. The fast-growing clusters (clusters A, B, C, and D in Fig. 3) detected in this study are also shown. ML, maximum likelihood; HIV, human immunodeficiency virus. Color images are available online.
Prevalence of TDR
According to the Surveillance Drug Resistance Mutations (SDRMs) list published in 2009, 16 the proportion of 767 pol sequences with SDRMs was 2.74% (n = 21), including 6 samples with NRTI mutations (M41L, M184I, T215S), 13 samples with NNRTI mutations (K101E, K103S, K103N, V106A, V106M, Y181C, G190A, G190S), and 4 samples with PIs mutations (M46I, G73S, N88S). Drug resistance analysis showed that more than half of the participants had high or medium resistance to Nevirapine (NVP) and Efavurebz (EFV). Detailed information is shown in Supplementary Table S1.
Characteristics of HIV-1 transmission networks
All 767 MSM sequences were used in the transmission network analysis. A total of 74 transmission networks were identified, involving 182/767 (23.7%) samples. Network sizes ranged from 2 to 12, with the majority (79.7%, 59/74) made up of two subjects. Comparing the sequences included to those not included in the transmission networks, marital status (χ 2 = 16.060, p < .001), age (χ 2 = 16.669, p < .001), and sample genetic subtypes (χ 2 = 12.509, p < .006) were significantly different (Supplementary Table S2).
Different subtypes can reveal different transmission models (Fig. 2A), and the distribution of subtypes in the network was different from that of all the obtained sequences (Fig. 2B). CRF01_AE (36.8%, 67/182) was the most common genotype in the transmission networks, followed by CRF07_BC (30.8%, 56/182), CRF67_01B (15.4%, 28/182), URFs (6.0%, 11/182), CRF68_01B (4.4%, 8/182), CRF08_BC (2.2%, 4/182), CRF65cpx (2.2%, 4/182), CRF55_01B (1.1%, 2/182), and CRF57_01B (1.1%, 2/182). Sequences of subtype B and CRF59_01B were not included in the networks.
The average degree values also varied, as sequences with URFs had the highest degree (each sequence with URFs linked to 4.2 sequences) in the transmission networks, followed by CRF67_01B (linked to 3.3 sequences) and CRF65_cpx (linked to 3.0 sequences). Each sequence was separated into three groups based on the number of links (i.e., sequence not connected to other sequences, sequence connected to another sequence, and sequence connected to at least two other sequences) to understand the potential transmission risks. As shown in Table 1, the three groups statistically differed according to marital status (χ 2 = 18.535, p = .001), age (χ 2 = 16.680, p = .002), location of sample collection (χ 2 = 9.968, p = .041), and HIV-1 genotype of sequences (χ 2 = 38.705, p < .001).
The Demographic Information Between Three Groups with Different Links
1. subtypes: AE (CRF01_AE), BC (CRF07_BC, CRF08_BC, CRF57_BC), 01B (CRF67_01B, CRF68_01B, CRF55_01B, CRF59_01B), Others (URF, B, CRF65_cpx).
To elucidate the temporal trends, we constructed an HIV-1 transmission network for every 3 months in 2017 (Fig. 3). As seen in the set of figures, four clusters were observed to grow faster than others within 1 year and were categorized as fast-growing. The fast-growing clusters included 31 sequences with a mean age of 31.1 ± 11.5 years. The proportion of <25 infections in the fast-growing group (51.6%, 16/31) was higher than that in other age groups (33.8%, 51/151). Detailed information is listed in Supplementary Table S3.
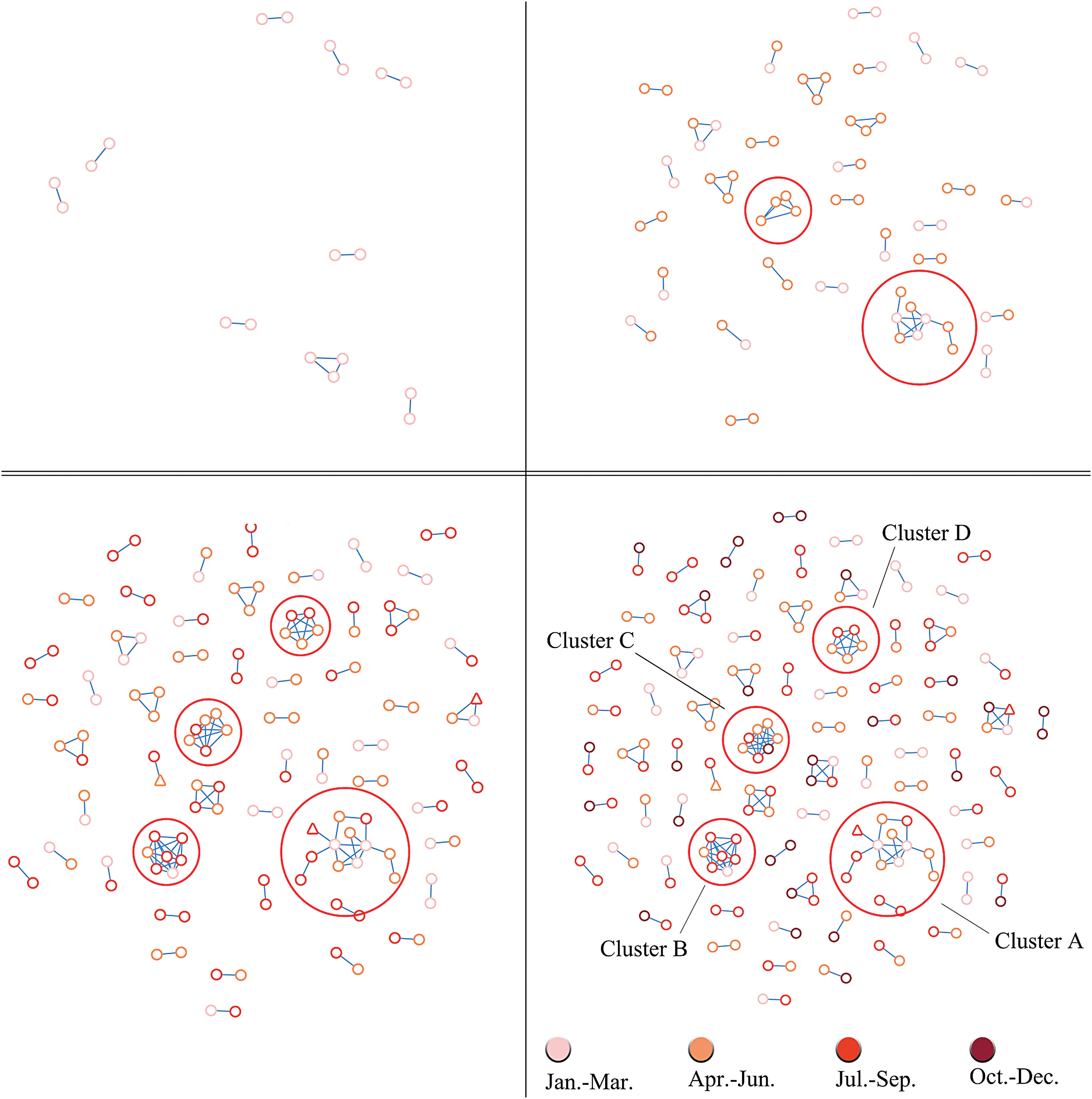
The temporal trends of HIV-1 transmission network, separated by every season in 2017. Color images are available online.
Discussion
In this study, a total of 914 MSM with newly diagnosed HIV-1 infection were included. From 767 available sequences, 11 HIV-1 subtypes and 74 transmission networks were identified. The TDR rate was 2.74%, and the distribution of the HIV subtypes was different from that in previous related research in Jiangsu.
The earliest HIV-1-positive patient detected in the Jiangsu province was in 1991. According to the provincial scale molecular epidemiology surveys, the main HIV-1 epidemic subtypes in Jiangsu have been changing over time. The main HIV-1 subtypes transformed from C (40.5%) and B’ (38.1%) in the period 1991–200217 to B (35.7%), CRF01_AE (35.7%), and CRF07_BC (28.6%) in the period 2006–2007. 18 This pattern subsequently changed to CRF01_AE (60.1%) and CRF07_BC (22.3%) from 2012 to 201519 and to CRF01_AE (43.3%) and CRF07_BC (31.3%) by 2017, per our study findings. Notably, CRF67_01B was rarely detected or reported in Jiangsu in recent years, 20 –22 but the proportion of CRF67_01B (7.2%) in our study was followed by those of CRF01_AE and CRF07_BC in that order. CRF67_01B was recombined by CRF01_AE and subtype B, it was initially reported among MSM in Anhui province in 2013. 23 However, soon after, a near full-length sequence of CRF67_01B was described among MSM in Jiangsu province in 2014. 24 However, this is not surprising, as Anhui and Jiangsu are neighboring provinces sharing almost 850 km of provincial boundary. In addition, a previous study showed that CRF67_01B from Jiangsu and Anhui may have a genetic relationship, 25 and thus cross-regional virus transmission might exist between the two provinces. Therefore, cross-regional prevention and monitoring of HIV-1 transmission would be of benefit for epidemic control.
The prevalence of HIV-1 TDR was lower in Jiangsu province in comparison with previous trends reported in the HIV Drug Resistance Threshold Survey in 2009–201126 and 2015. 27 In our study, the TDR was 2.74%, showing no obvious difference from previous study findings, which suggests that current treatment strategies can exert curative effects in Jiangsu province.
Identifying and responding to groups of HIV-infected persons who have an epidemiological connection related to HIV transmission is a critical step toward the goal of recording no new infections. To achieve this goal, the United States CDC published guides 28 and conducted routine analyses in each quarter. In addition, the British Columbia (Canada) CDC detected a fast-growing cluster for a period of 3 months using a monitoring system. 29 In our study, we found four fast-growing clusters within 1 year, with a clear change every 3 months. Accordingly, we believe that high-density sampling together with high-frequency analysis can be an effective method to discover fast-growing clusters.
In this study, almost a quarter of all sequences (23.7%, 182/767) were successfully extracted, constituting 74 molecular transmission networks. Single young patients seem more likely to be included in a network. Different subtypes have different transmission modes. The transmission networks of CRF01_AE were small and dispersed. There were 31 clusters with CRF01_AE, of which 27 (87.1%) contained two sequences each. The largest cluster contained four sequences, and no large cluster with CRF01_AE was formed. In contrast, the construction of CRF07_BC transmission networks differed from that of CRF01_AE. There were 21 clusters with CRF07_BC in the network, with one cluster (cluster A) containing 12 sequences, of which one sequence had drug-resistance mutations. In a similar study in Beijing, a large cluster with CRF07_BC was described, which suggests that the prevalence of the CRF07_BC subtype could have an increasing trend nationwide and may have easily formed larger transmission networks compared with other subtypes. It is worth nothing that approximately half of the CRF67_01B sequences were included in the transmission network, and two networks (clusters B and D) were identified as especially fast-growing. Considering the growing proportions of CRF07_BC and CRF67_01B, local CDCs should continue monitoring the molecular changes in newly diagnosed MSM to conduct targeted interventions and control epidemics in a timely manner.
Limitations
Our study had three major limitations. First, the samples in this study were not from wholly PLWHA populations in Jiangsu province. Therefore, there might be key transmission points we did not detect. This is because although we sequenced nearly 40% HIV-1—positive MSM in 2017, higher than the percentage of minimum sampling, 30 there were still many PLWHA who we could not find or were unwilling to participate in this research. This might have impacted the results of our study. Second, our research includes only information among MSM, and we did not assess transmission among other populations. Finally, with limited time and funds, we only sequenced the pol gene, which may not holistically represent the epidemic, as there might be other HIV-1 genotypes in existence in Jiangsu.
Conclusions
The present subtypes of HIV-1 among MSM are complex and diverse; therefore, cross-regional prevention and monitoring are necessary. The TDR rate was low among MSM in Jiangsu province, which implies that good treatment approaches should be maintained. Regular surveys and follow-up studies should be continued to understand the potential risk factors of HIV-1 transmission.
Sequence Data
All the nucleotide sequences obtained in this study have been submitted to GenBank. MT986065-MT986829.
Footnotes
Acknowledgment
We thank Editage for English language editing.
Author Disclosure Statement
The authors declare no competing interests.
Funding Information
This study was supported by the Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX17_0184) and molecular network analysis and social network exploration of HIV-1 infection transmission among young students in Jiangsu Province (0701-184160070478).
Supplementary Material
Supplementary Data
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.