
Abstract
Despite the advent of antiretroviral therapy, people living with HIV suffer from a range of infectious and noninfectious pulmonary complications. HIV impairs antioxidant defenses and innate immune function of the alveolar macrophage by diminishing granulocyte macrophage-colony stimulating factor (GM-CSF) signaling. Since GM-CSF may be linked to mitochondria, we sought to determine the effects of HIV on GM-CSF receptor expression and alveolar macrophage mitochondrial function. At an academic medical center, studies were completed on alveolar macrophages isolated from both wild-type and HIV transgenic (HIV Tg) rats and human subjects with and without HIV. Primary macrophages were plated and evaluated for expression of GM-CSF receptor beta, phagocytic index, and mitochondrial function in the presence and absence of GM-CSF treatment. GM-CSF receptor expression and mitochondrial function were impaired in macrophages isolated from HIV Tg rats, and treatment with GM-CSF restored GM-CSF receptor expression and mitochondrial function. GM-CSF treatment of HIV Tg rats also increased alveolar macrophage levels of the mitochondrial proteins voltage-dependent anion-selective channel 1 (VDAC) and glucose-regulated protein 75 (Grp75). Similar to the HIV Tg rat model, impairments in mitochondrial bioenergetics were confirmed in alveolar macrophages isolated from human subjects with HIV. HIV-associated impairments in alveolar macrophage mitochondrial bioenergetics likely contribute to innate immune dysfunction in HIV infection, and GM-CSF treatment may offer a novel therapeutic strategy for mitigating these deleterious effects.
Introduction
The advent of antiretroviral therapy (ART) greatly increased the life expectancy of people living with HIV. 1 However, individuals on ART with undetectable viral loads and normal CD4+ T cell counts are still at high risk for pneumonia 2 and other noninfectious pulmonary complications, 3 which account for the majority of hospital admissions in people with HIV. 4 Novel strategies are needed to improve lung health in this vulnerable population.
The alveolar macrophage is one of the key resident innate immune effectors of the lung and plays a critical role in recognizing pulmonary pathogens, initiating immune responses, and recruiting the adaptive immune system. However, alveolar macrophage function is also impaired among people with HIV. These deficits include essential macrophage functions such as phagocytosis and antioxidant defense and have been found to persist despite immune reconstitution with ART. Persistent impairment of immune function may be driven by the ongoing presence of the virus itself within alveolar macrophages. 5 HIV transgenic (HIV Tg) rats serve as a noninfectious animal model of HIV infection that express HIV proteins such as glycoprotein-120 (gp120) and trans-activator of transcription (Tat). We have previously demonstrated that alveolar macrophages from HIV Tg rats recapitulate many of the hallmarks of chronic HIV infection and exhibit functional defects in a wide range of key signaling pathways, including decreased antioxidant capacity and phagocytosis. 6 Others have also recently reported that gp120, which persists in the alveolar space of individuals with well-controlled HIV infection, is sufficient to inhibit pneumococcal killing. 7
Granulocyte macrophage-colony stimulating factor (GM-CSF) is secreted by alveolar epithelial cells and directs the maturation of myeloid-lineage cells in the lung. It retains an important role in mature alveolar macrophages by promoting phagocytosis, pathogen killing, surfactant clearance, and macrophage polarization. 8 Many of the infectious complications in people with HIV, including their increased risk of pneumonia and opportunistic infections from atypical pathogens such as Pneumocystis jirovecii and Nocardia, have also been seen in the setting of GM-CSF impairments. 9 We previously demonstrated that exposure to HIV proteins decreases alveolar macrophage expression of the β-subunit of the receptor for granulocyte/macrophage-colony stimulating factor (GM-CSFRβ). 10 GM-CSF has also been found to be a key mediator of macrophage metabolism and mitochondrial bioenergetics. 11 This discovery of a link between GM-CSF and alveolar macrophage immunometabolism, alongside growing literature supporting global perturbations in cellular metabolism in HIV, 12 suggests that impaired GM-CSF signaling may drive mitochondrial impairments and thereby contribute to the wide range of defects that we and others have previously described. We therefore hypothesized that alveolar macrophages exposed to HIV proteins would demonstrate impaired mitochondrial bioenergetics and that treatment with GM-CSF would improve their energy utilization from oxidative phosphorylation. These studies identify GM-CSF as a novel modulator of alveolar macrophage mitochondrial function in HIV.
Materials and Methods
Cell culture and treatments
Cells were plated in F12K media +5% fetal bovine serum (FBS). Cells were incubated with 10 ng/mL GM-CSF (PeproTech, Rocky Hill, NJ), 10 ng/mL of Tat (ImmunoDiagnostics, Inc., Woburn, MA), or 10 ng/mL gp120 (ImmunoDiagnostics, Inc.). 10
Primary rat macrophages
Primary alveolar macrophages were obtained by bronchoalveolar lavage from ∼10-month-old Fischer 344/NHsd HIV Tg rats or their wild-type littermate controls (n = 3–4/group). This rat model lacks the gag and pol genes necessary for functional virion formation, but otherwise produces the full spectrum of HIV-related proteins, including Tat, rev, Nef, and gp120. 13 All procedures were approved by the Institutional Animal Care and Use Committee at the Atlanta Veterans Affairs Health Care System and Emory University.
Phagocytosis assay
Primary alveolar macrophages isolated from either HIV Tg rats or littermate controls were seeded at 80,000–100,000 per well in 16-well tissue culture chamber-slides. Based on our prior work on HIV, cells were treated with or without a low concentration of GM-CSF (10 ng/mL), and phagocytosis index was assessed as previously described. 6
Cytoimmunofluorescence
Primary rat alveolar macrophages were isolated and plated as previously described 14 before incubation with polyclonal rabbit antibodies to GM-CSFRβ (Santa Cruz Biotechnology, Santa Cruz, CA), glucose-regulated protein 75 (Grp75; Abcam, Cambridge, MA) or voltage-dependent anion-selective channel 1 (VDAC; Abcam) overnight at 4°C. Wells were then incubated with anti-rabbit secondary antibodies (Jackson ImmunoResearch, West Grove, PA) and DAPI (Vector Laboratories, Burlingame, CA) for nuclear staining. Images of 80–100 alveolar macrophages/experimental group were acquired as previously described 15 and analyzed using a binary image map to capture cell borders with automated background correction.
Mitochondrial bioenergetics
The rate of change in oxygen consumption rate (OCR) was examined in alveolar macrophages isolated from rats (n = 4/group) and human subjects (n = 5/group) with the Seahorse XFp extracellular flux analyzer (Agilent Seahorse Bioscience, Inc., Billerica, MA) using a Cell Mito Stress Test as previously described. 16 OCR values were normalized to the protein concentration of each sample.
Human alveolar macrophages
Five subjects with HIV and five without HIV were recruited from the Grady Health System in Atlanta, GA. Subjects with HIV were well-controlled on ART with CD4 cell counts >350 cells/μL and undetectable HIV viral loads. Additional details, including inclusion and exclusion criteria, are available in Table 1. Written informed consent was obtained from all subjects. The informed consent form and study protocol were approved by the Emory University Institutional Review Board (IRB00088993). Alveolar macrophages were obtained by bronchoalveolar lavage as previously described. 5 Cells were washed and then plated in RPMI-1640 (ATCC) with 10% FBS and gentamycin/amphotericin (Thermo Fisher) on a Seahorse XFp cell culture miniplate (Agilent Seahorse Bioscience, Inc.).
Inclusion and Exclusion Criteria for Human Subjects With and Without HIV
ART, antiretroviral therapy; COPD, chronic obstructive pulmonary disease; ESRD, end-stage renal disease; HD, hemodialysis; TB, tuberculosis.
Statistical analyses
Student's t-test was used for single comparisons (Microsoft Excel, Redmond, WA). In studies with more than two groups, statistical significance was calculated using one-way ANOVA followed by Tukey-Kramer post hoc tests to detect differences between individual groups (GraphPad Prism version 5, San Diego, CA). Data are presented as mean ± standard error of the mean. Significance was accepted at p < .05.
Results
Exposure to HIV-related proteins suppresses rat alveolar macrophage GM-CSF receptor β expression, and exogenous treatment with GM-CSF restores both receptor expression and phagocytic function
Because alveolar macrophages from HIV Tg rats demonstrated decreased expression of GM-CSFRβ, 10 we assessed whether GM-CSF treatment could stimulate GM-CSFRβ expression by treating alveolar macrophages isolated from HIV Tg rats with GM-CSF for 48 h ex vivo. As shown in Figure 1, GM-CSF treatment restored GM-CSFRβ expression to levels seen in alveolar macrophages from wild-type littermates. Given the importance of GM-CSF for macrophage innate immune function 17 and our prior work demonstrating that HIV transgene expression impairs phagocytic function to ∼50% of the wild-type baseline, 6 we next determined the effects of GM-CSF treatment on the phagocytic function of HIV Tg macrophages against Staphylococcus aureus. GM-CSF treatment markedly improved phagocytic capacity of alveolar macrophages from HIV Tg rats (Fig. 2), thereby demonstrating a restoration of this key macrophage immune function.
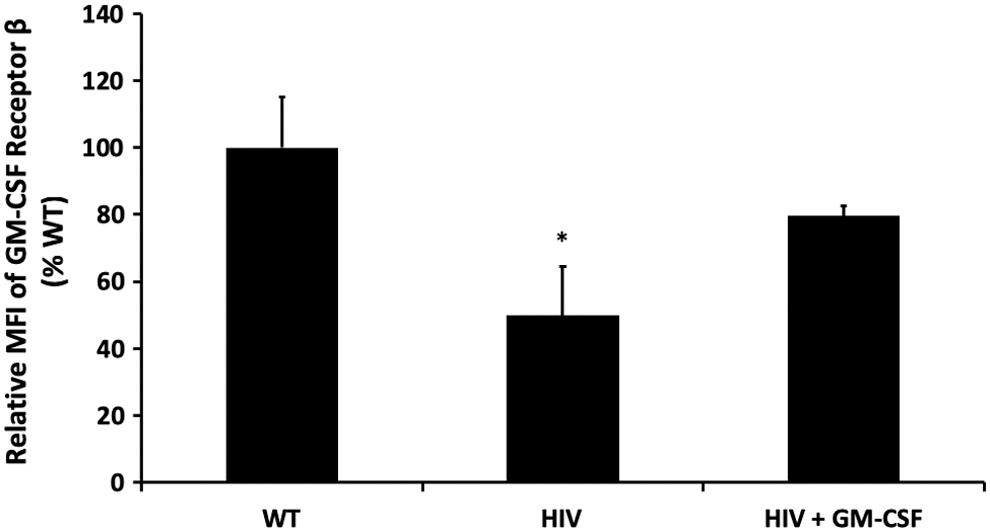
GM-CSF treatment restores protein expression of GM-CSFRβ. Alveolar macrophages from HIV-1 transgenic rats were cultured ± GM-CSF in parallel with their littermate controls. Protein expression was quantified by cytoimmunofluorescence after 48 h of treatment with GM-CSF. HIV-1 transgene expression significantly decreased GM-CSFRβ expression relative to wild-type littermate controls, which was restored with GM-CSF treatment (*p < .05 relative to wild-type littermate controls). Data presented as a ratio of the fold change (mean ± SEM, n = 3 animals and 100–150 cells/group). GM-CSF, granulocyte macrophage-colony stimulating factor; GM-CSFRβ, β-subunit of the receptor for GM-CSF; SEM, standard error of the mean.
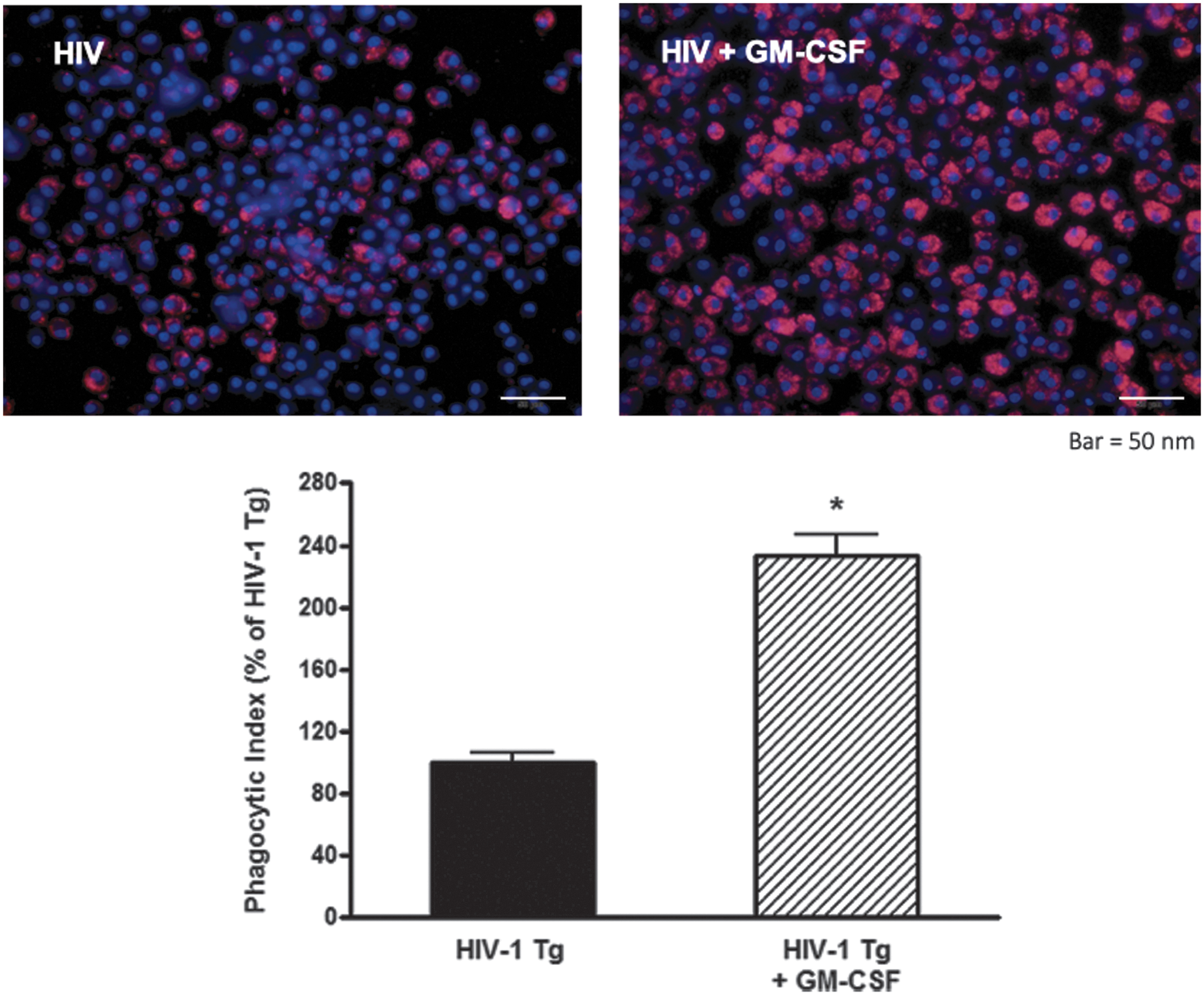
GM-CSF treatment of HIV-1 Tg rat macrophages restores phagocytic function. Alveolar macrophages were treated ex vivo with 10 ng/mL of GM-CSF for 24 h before incubation with pHrodo-labeled Staphylococcus aureus for 2 h. Cells were analyzed by mean fluorescence intensity with a representative image shown here. An over twofold increase in phagocytosis is present in the HIV macrophages treated with GM-CSF, suggesting a restoration of innate immune effector function. *p < .05 compared to HIV-1 Tg. Data presented as phagocytic index ± SEM, n = 3 animals and 100–150 cells/group. HIV-1 Tg, HIV-1 transgenic.
GM-CSF treatment improves mitochondrial bioenergetics in HIV Tg rat alveolar macrophages
Having identified a key role for GM-CSF in the maintenance of macrophage innate immunity in the setting of HIV-related protein exposure, we next investigated the mechanism by which GM-CSF produces its salutary effects. GM-CSF has been linked to cell energy utilization, 11 which may be critical for immunologic functions in alveolar macrophages such as phagocytosis. Therefore, we sought to determine whether HIV impairs mitochondrial function and whether GM-CSF treatment could mitigate this effect. OCR, representing the overall mitochondrial bioenergetic profile, were diminished in primary alveolar macrophages isolated from HIV Tg rats compared with wild-type littermates (Fig. 3A). Specifically, basal respiration (Fig. 3B), ATP-linked respiration (Fig. 3C), maximal respiration (Fig. 3D), and spare respiratory capacity (Fig. 3E) were decreased in HIV Tg rat alveolar macrophages. Treatment with GM-CSF improved the OCR and mitochondrial bioenergetics of HIV Tg rat alveolar macrophages across all of these parameters.

Chronic exposure to HIV-related proteins impairs mitochondrial bioenergetics and the effects are mitigated by treatment with GM-CSF. The rate of change in OCR in alveolar macrophages isolated from HIV-1 Tg or littermate control rats treated with or without GM-CSF was assessed (n = 3 group/condition). During the experiment, injections 2 μM oligomycin (Oligo, mitochondrial complex V inhibitor) and 0.5 μM of FCCP (ATP synthase inhibitor and uncoupler), and 0.5 μM of rotenone/antimycin-A/rotenone (R/A, mitochondrial complex III/complex I inhibitor, respectively) were applied in sequence. Rate of change in OCR was normalized to protein concentrations in the same samples.
GM-CSF treatment improves expression of the mitochondrial proteins Grp75 and VDAC in HIV Tg rat alveolar macrophages
Grp75 is important for the maintenance of intracellular calcium homeostasis, 18 and VDAC is important for maintaining flux across the outer mitochondrial membrane. 19 To determine how GM-CSF improves mitochondrial function, we studied the effects of GM-CSF treatment on these key mitochondrial proteins. As shown in Figure 4, alveolar macrophages isolated from HIV Tg rats exhibited reduced baseline levels of VDAC but did not alter Grp75 levels. However, in HIV Tg macrophages (but not WT macrophages), GM-CSF treatment significantly increased levels of both VDAC and Grp75 above baseline wild-type expression. These data suggest Grp75 and VDAC regulation as possible mechanisms by which GM-CSF treatment may improve HIV Tg rat alveolar macrophage mitochondrial derangements.

GM-CSF treatment improves levels of key mitochondrial proteins in alveolar macrophages from HIV Tg rats. Alveolar macrophages from HIV Tg rats and their littermate controls were cultured with or without GM-CSF before being stained for Grp75 and VDAC. HIV transgene expression reduced VDAC but did not alter Grp75 levels. GM-CSF treatment resulted in significantly higher levels of both proteins in HIV Tg macrophages but did not significantly change wild-type macrophage levels. Data presented as MFI ± SEM, n = 4 animals and 100–150 cells/group. *p < .05 versus HIV. Grp75, glucose-regulated protein 75; MFI, mean fluorescence intensity; VDAC, voltage-dependent anion channel.
Alveolar macrophages from human subjects with HIV exhibit impaired mitochondrial function
Our group has previously determined that HIV adversely affects alveolar macrophage phagocytosis, 7 and our data here indicated that mitochondrial bioenergetics might be responsible. Therefore, to determine the translational potential of the mitochondrial studies presented above, we conducted research bronchoscopies to obtain alveolar macrophages from subjects with and without HIV for OCR assessment of mitochondrial bioenergetics (Table 2). Although subjects with HIV were well-controlled on ART (regimens presented in Table 3), alveolar macrophages from these individuals demonstrated impaired OCR (Fig. 5A) and mitochondrial bioenergetics, including basal respiration (Fig. 5B) and ATP-linked respiration (Fig. 5C). However, maximal respiration was unchanged (Fig. 5D) and spare respiratory capacity was increased (Fig. 5E).

HIV impairs alveolar macrophage mitochondrial bioenergetics in human subjects. Alveolar macrophages from people living with and without HIV were obtained by bronchoalveolar lavage (n = 5/group). The rate of change in OCR in alveolar macrophages was then assessed (three wells/subject).
Characteristics of Study Participants
At the time of study enrollment.
BMI, body mass index; SD, standard deviation.
Antiretroviral Therapy Regimen of Subjects with HIV
NRTI, nucleoside reverse transcriptase inhibitor.
Discussion
This study provides evidence that loss of GM-CSF and subsequent mitochondrial dysfunction are novel mechanisms by which HIV may suppress lung immunity and increase susceptibility to both infectious and noninfectious pulmonary diseases. Treatment with GM-CSF improves alveolar macrophage GM-CSF receptor expression (Fig. 1) and phagocytosis (Fig. 2) by improving mitochondrial bioenergetics (Fig. 3). Given the profound innate immune impairments in people living with HIV and their substantial contribution to global morbidity and mortality, these findings have the potential to open new avenues of research into host-directed immune therapeutics.
As the resident innate immune effector of the lung, the alveolar macrophage identifies and eliminates pathogens in the alveolar space and activates the adaptive immune system. Although multiple studies have demonstrated impaired macrophage function in the setting of HIV infection, the mechanism by which HIV drives these impairments remains unknown. Given that few macrophages are primarily infected with the virus, 5 we suspect that HIV proteins produced by those infected cells and present in the alveolar space are likely involved. This hypothesis is supported by the fact that HIV proteins have deleterious effects on a variety of organ systems, including the brain, 20 vasculature, 21,22 and alveolar epithelium. 23,24 Further, in the lungs specifically, the HIV proteins Tat and gp120 not only cause macrophage dysfunction, 7 but also mimic the effects of direct infection on antioxidant defenses. 6 It is within this context that we have found in the HIV Tg rat model, which produces HIV proteins in the alveolar space, 24 a compelling way to study the effects of HIV on macrophage function.
In this study, we build on known impairments in phagocytosis and GM-CSF signaling in HIV 10 by linking macrophage dysfunction in HIV to impairments in mitochondrial bioenergetics. The bioenergetic profiles seen in our studies (Figs. 3 and 5) recapitulate the decreases in ATP-linked respiration seen in in vitro differentiated human macrophages inoculated with HIV for 24 h. 25 Our studies extend these findings by demonstrating profound mitochondrial bioenergetic derangements in primary alveolar macrophages isolated from both HIV Tg rats and people living with HIV on ART. In a novel finding, we have also linked GM-CSF-mediated mitochondrial function to phagocytic capacity of these cells. Specifically, the profound impairments in OCR impede efficient cellular energy utilization and compromise the ability of alveolar macrophages to carry out energy-intensive immune processes such as phagocytosis and pathogen killing.
The dramatic response of the HIV Tg rat alveolar macrophage phagocytic function to exogenous GM-CSF administration highlights the importance of GM-CSF to the alveolar macrophage inflammatory cascade, 17 in which GM-CSF is necessary but not sufficient to drive macrophage innate immune function. 26 Once myeloid lineage cells have matured into alveolar macrophages, GM-CSF remains important for allowing cells to respond to pathogens and other threats in the alveolar space by increasing production of inflammatory cytokines, recruiting cells from the adaptive immune system, and phagocytosing and killing pathogens. Clinically, these effects have been harnessed to great effect in the treatment of the paradigmatic disease of impaired GM-CSF signaling, pulmonary alveolar proteinosis, 27 which causes immunodeficiency, and a build-up of uncleared surfactant in the lung. In animal models, GM-CSF treatment has also been successfully used to modulate immune responses in bacterial pneumonia. 28 In the context of the data we present here, we note that HIV infection reduces the size of macrophages, 29 suggesting that HIV may induce alveolar macrophage immaturity and immune dysfunction via decreases in GM-CSF. Our future studies will delve into the molecular mechanisms by which GM-CSF mediates these effects.
Closer examination of two key pathways involved in mitochondrial bioenergetics yielded interesting results that point toward a possible mechanism for GM-CSF in this setting. Our findings that exogenous GM-CSF upregulates the mitochondrial proteins Grp75 and VDAC in HIV Tg macrophages (Fig. 4) offers compelling evidence of the mechanism by which GM-CSF may improve mitochondrial bioenergetics, thereby providing alveolar macrophages with the energy stores required to phagocytose and kill pathogens. Grp75 is a known regulator of mitochondrial calcium homeostasis and mitochondrial integrity, and its increased expression in GM-CSF treated macrophages suggests improvements in redox homeostasis as well. 18 Although Grp75 levels were unchanged by HIV transgene expression, alveolar macrophages from the transgenic animals responded to exogenous GM-CSF treatment with a robust increase in protein level. This marked improvement in Grp75 levels with GM-CSF may help explain the GM-CSF-mediated improvements in mitochondrial function we observed by promoting calcium homeostasis and mitochondrial integrity (that were perhaps compromised by deficiencies in other unidentified pathways). However, assuming the normal protein levels in HIV Tg macrophages corresponds to normal activity levels, we believe it is unlikely that Grp75 contributes to the impaired mitochondrial bioenergetics we observed at baseline in HIV.
VDAC, also known as mitochondrial porin, is an essential facilitator of crosstalk between mitochondria and the wider cellular environment and is also a key player in cellular apoptosis. 19 VDAC was impaired at baseline in HIV Tg macrophages and may have contributed to the baseline defect in mitochondrial bioenergetics. Increased VDAC levels in response to GM-CSF treatment in these macrophages may therefore explain the improvement in mitochondrial bioenergetics, as enhanced VDAC expression would improve mitochondrial communications and provide better regulation of cellular death signaling in HIV. 30 Understanding the mechanism by which HIV transgene expression alters the response of VDAC and Grp75 to GM-CSF treatment will be an important next step in our work on these pathways, as will a recapitulation of the effects of GM-CSF on mitochondrial bioenergetics, VDAC, and Grp75 in human subjects with and without HIV.
Our ability to provide further mechanistic evidence is somewhat limited by the fact that inhibiting expression of these essential genes (i.e., with silencing RNA) leads quickly to cell death. Further, there are as yet no neutralizing antibodies commercially available. However, we believe the evidence we provide here will stimulate further inquiry into the mechanisms by which HIV impairs mitochondrial function (potentially through dysregulation of mitochondrial dynamics and mass, which will be a key focus of our future studies) and will eventually lead to the development of novel therapeutic strategies to improve lung function in people with HIV.
The animal model data presented here are both compelling and novel and they are strengthened by our finding that humans with chronic HIV infection have comparable defects in mitochondrial function. Despite the expected heterogeneity among human subjects and the limited number of subjects in our cohort, these human data effectively provide confirmation that the effects of viral proteins in the animal model approximate the effects of chronic HIV infection in human subjects in many important ways. Our trust in the model is further enhanced by recent data suggesting a decrease in the mitochondrial bioenergetics of myoblasts from simian immunodeficiency virus-infected rhesus macaques. 31
The etiology of the marked increase in spare respiratory capacity in subjects with HIV is unclear based on our current data but is intriguing and requires further investigation. It may be related to direct effects of viral infection (which does not occur in the transgenic model) or to the stage of infection, as all of the subjects were well-controlled on antiretroviral therapy. Interestingly, prior studies of the effects of ART on mitochondrial function have found that non-nucleoside reverse transcriptase inhibitors (NNRTIs), nucleoside reverse transcriptase inhibitors (NRTIs), and protease inhibitors (PIs) tend to reduce spare capacity and maximal mitochondrial respiration. 32 Because we did not study subjects off ART, we are unable to determine whether ART had an effect on OCR, but we do note that those studies were not performed in alveolar macrophages and may not reflect the situation in the lung compartment specifically. Regardless, the fact that the spare capacity is higher while the baseline level of oxygen consumption is lower suggests that the alveolar macrophages are able to be activated but are relatively quiescent at baseline, a fact that would render them relatively slow to respond to the need for energy-intensive activities. These data will be better contextualized through the use of cell-energy phenotyping and the assessment of extracellular acidification rate, both of which will be important corollaries to the experiments presented here.
Conclusion
In summary, we have determined that chronic exposure to HIV impairs alveolar macrophage mitochondrial bioenergetics and innate immune function. The persistence of these deficits despite effective viral suppression in the human subjects underscores the urgent need to further investigate pathways mediating macrophage dysfunction, as these deficits will not be reversed with ART alone. In the animal model, these deleterious effects are mediated, at least in part, by downregulation of GM-CSFRβ and mitochondrial proteins. Treatment with GM-CSF restores receptor expression, innate immune function, and mitochondrial bioenergetics. In light of recent data on the importance of the mitochondrial fusion-fission balance, our future investigations will focus on those pathways and pathways regulating mitophagy. 33,34 Based on our current evidence, though, we are hopeful that the GM-CSF pathway can be harnessed for therapeutic benefit to mitigate pulmonary immune complications in people living with HIV.
Footnotes
Authors' Contributions
Experiments were performed by B.S.S., M.A., X.F., N.S., and S.M.Y. Data were interpreted and analyzed by B.S.S., S.C.A., M.A., X.F., N.S., and S.M.Y. Article was drafted by B.S.S., S.C.A., and S.M.Y.
Acknowledgments
We thank Robert Raynor and S. Todd Mills for their invaluable technical support.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by grants from the National Institutes of Health: K08 AA 024512 (Bashar S. Staitieh), K23 AI134182 (Sara C. Auld), R00 AA 021803 (Samantha M. Yeligar), and R01 AA 026086 (Samantha M. Yeligar).