
Abstract
Multiple HIV-1 genotypes were found circulating in Guangdong Province, China, as this province is located in South China and has a high frequency of international trade. In this study, we report the near full-length genome (NFLG) of CRF12_BF that was identified from a male patient in Guangzhou city, Guangdong Province; this is the first time CRF12_BF has been reported in mainland China. The NFLG was amplified, and then PCR products were sequenced by Sanger sequencing. The CRF12_BF strain was confirmed by the Basic Local Alignment Search Tool and a neighbor-joining phylogenetic tree. In addition, this CRF12_BF strain was confirmed to contain the non-nucleoside reverse transcriptase inhibitor mutation E138A associated with potential low-level resistance against efavirenz and low-level resistance against rilpivirine by the Stanford University HIV Drug Resistance Database program. The analyzed sequence data in this study will provide more information on the HIV epidemic in China.
HIV-1
Guangdong Province is one of the commercially developed and populous areas located on the southern coast of China, with frequent foreign exchanges and a large transient population. HIV-1 spread quickly in Guangdong Province after the first HIV/AIDS case was identified in 1986. 6,7 There are several HIV-1 genotypes circulating in Guangdong Province. The predominant genotype in Guangdong Province was CRF01_AE, and subtypes A1, B, C, CRF07_BC, CRF55_01B, CRF08_BC, and CRF59_01B have also been reported. 8,9
Previous genomic analysis of the partial pol gene indicated that an HIV strain in Guangzhou city, Guangdong Province, might be classified as CRF12_BF (GenBank accession number: MT589616). In this study, we obtained the near full-length genome (NFLG) sequence for this CRF12_BF strain and present the genetic characteristics of this strain.
Serum was collected from a 48-year-old male patient (Patient ID: 10-097) who is a resident of Guangzhou and was diagnosed with HIV-1 on June 22, 2018. This study was approved by the institutional review board of the Guangzhou Eighth People's Hospital, and written informed consent of the patient was obtained before collecting serum samples. After serum collection, the patient was started on first-line antiretroviral drug therapy, comprising tenofovir (TDF), lamivudine (3TC), and efavirenz (EFV).
The RNA template was extracted using the QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany) and reverse transcribed to cDNA using SuperScript IV VILO Master Mix (Invitrogen, CA) according to the manufacturer's instructions. The NFLG of HIV-1 was amplified with Platinum SuperFi II PCR Master Mix (Invitrogen) using the same conditions and primers as previously described. 10,11 PCR products were purified and then sequenced by Tianyi Huiyuan Company (Guangzhou, China) with 28 specific primers and the Sanger sequencing method. Sequencing fragments were assembled by ContigExpress (Vector NTI software) with a minimum overlap of 100 bp. The assembled NFLG sequence was deposited in GenBank (accession number: MT712270).
The NFLG sequence of the HIV-1 isolate 10-097 was 8,835 bp (from 780 to 9,588 bp according to the HXB2 calibrator) in length. To check for potential contamination and to confirm the sequence quality, the sequence was submitted to the HIV-1 Sequence Quality Control Tool (
The sequence was then submitted to the HIV Basic Local Alignment Search Tool (BLAST) available at the Los Alamos HIV Database website (
This was further confirmed by a neighbor-joining (NJ) phylogenetic tree in Mega 6.06 software.
13
The NFLG sequence was aligned with reference sequences downloaded from the Los Alamos HIV database (
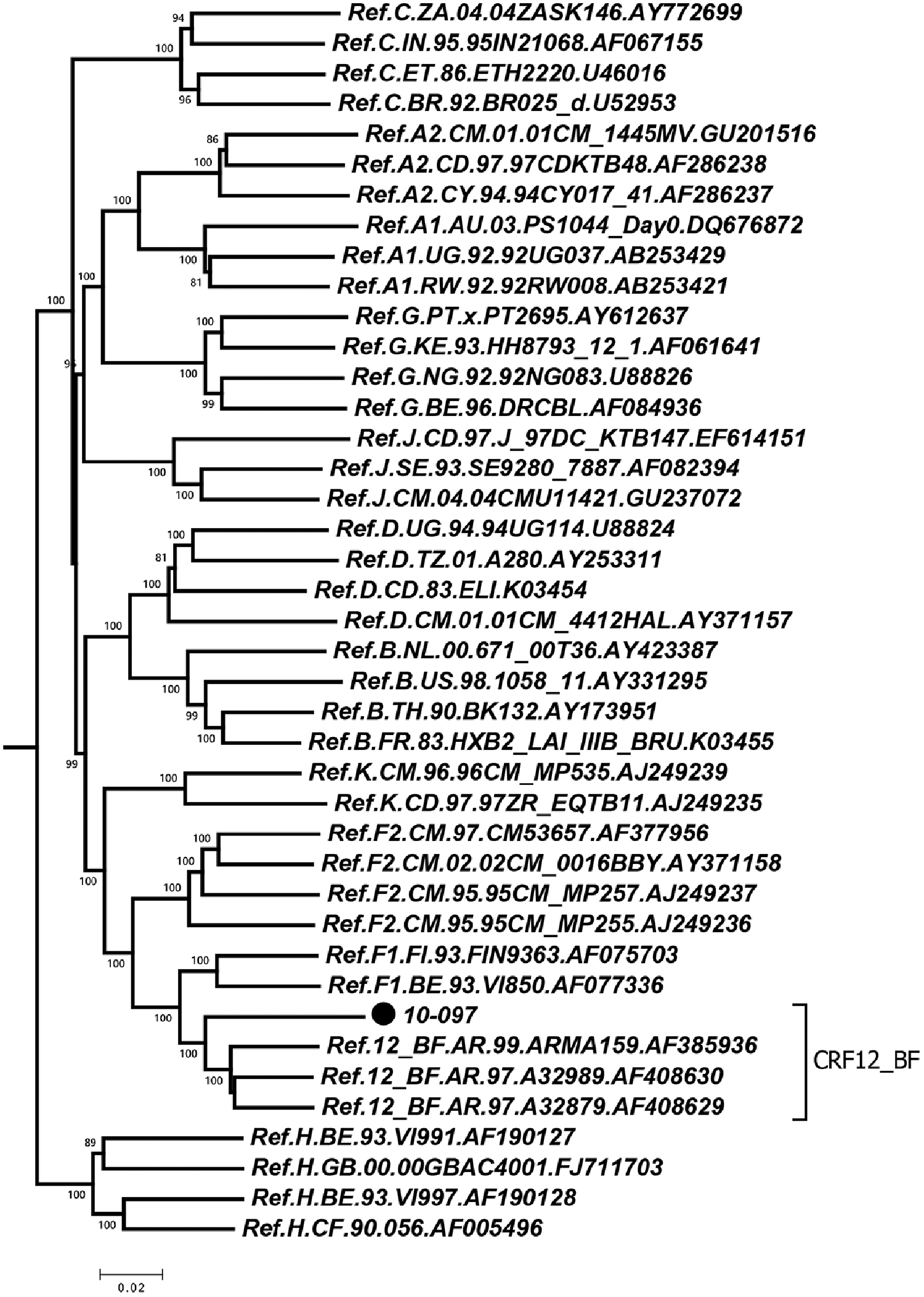
The NJ tree was constructed using the NFLG sequences of HIV-1 isolates by the program MEGA 6.0.6. The reference sequences of subtypes A–D, F–H, J, K, and CRF12_BF were downloaded from the Los Alamos HIV database (
The patient was divorced and claimed to have a history of obtaining dental fillings abroad. He denied that he had been infected through sexual behavior, and his ex-wife was HIV antibody negative. The patient was negative for syphilis, hepatitis B virus, hepatitis C virus, and tuberculosis. The baseline CD4+ and CD8+ T cell counts of the patient were 369 and 1,624 cells/μL, respectively, whereas the HIV-1 viral load was 3,500 copies/mL.
To analyze whether mutations associated with drug resistance existed, the pol gene of this strain was submitted to the Stanford University HIV Drug Resistance Database (
Out of 362 sequences, 9 sequences (2.49%) also had the NNRTI mutation E138A. As the patient had no prior exposure to antiretroviral drugs, we speculate that the resistance site of this sequence is derived from transmitted drug resistance. On June 2019, 1 year after highly active antiretroviral therapy (HAART), the CD4+ count of the patient had increased to 595 cells/μL, and the HIV-1 RNA was below the low limit of detection (COBAS AmpliPrep/COBAS TaqMan HIV-1 Test v2.0, the quantitative range is 20 to 10,000,000 copies/mL; Roche, Switzerland). Currently, 2 years after HAART, the CD4+ count of the patient is 639 cells/μL, and the HIV-1 RNA is still below the low limit of detection. This suggested that the pre-existing mutation E138A may have a rare effect on the TDF/3TC/EFV combination treatment.
To explore the possible origins of the 10-097 strain, further analysis was performed to investigate the phylogenetic relationship of the 10-097 strain with strains from other countries. We downloaded all CRF12_BF sequences from the Los Alamos HIV database. There were 481 CRF12_BF sequences in total, 14 of which were complete genomes, and the other sequences were from the gag, pol, and env genes. The complete genome, pol gene, gag gene, and env gene were selected to maximize the length and the number of segments for further analysis. Segments spanning 8,690 bp of the NFLG (nucleotides 796-9485 according to the HXB2 calibrator) from 14 CRF12_BF patients, 567 bp of the pol gene (nucleotides 2661-3227 according to the HXB2 calibrator) from 378 patients, 422 bp of the gag gene (nucleotides 1616-2038 according to the HXB2 calibrator) from 26 patients, and 329 bp of the env gene (nucleotides 7064-7392 according to the HXB2 calibrator) from 24 patients were included in the final data set.
A total of 378 sequences of the pol gene were aligned with the 10-097 strain and reference F2 sequences by BioEdit Software. The alignment was submitted to the ATGC website to build a maximum likelihood (ML) phylogenetic tree by using the online program PhyML 3.0. 14 The ML tree was constructed using the general time reversible substitution model with branch support estimated using the approximate likelihood ratio test (aLRT). 15 The final trees were visualized using the program Figtree 1.4.2 (Fig. 2). The sequences of the complete genome, gag gene and env gene, were analyzed in the same way. As shown in the phylogenetic tree of the pol gene, the sequence from the 10-097 strain clustered with three CRF12_BF sequences from Argentina and formed a better-supported (aLRT branch support value 0.918) cluster with an Argentina sequence (accession number AY365719, Fig. 2). There were no clusters formed with the sequences of the complete genome, gag gene and env gene (data not shown). Therefore, we speculated that the 10-097 strain in this study may have originated from Argentina.
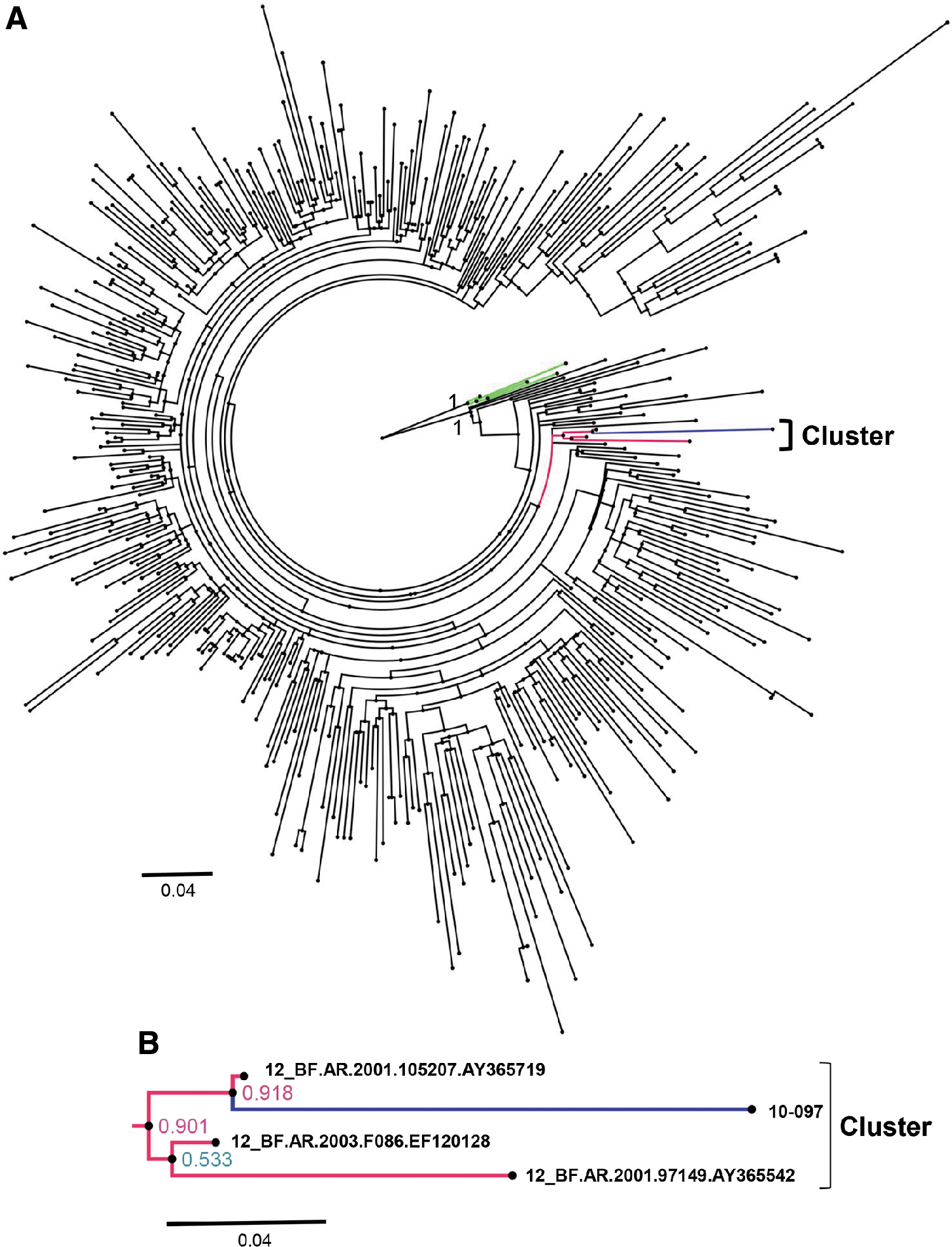
Phylogenetic tree analysis.
In this research, we obtained the sequence of one NFLG of CRF12_BF isolated in Guangdong Province. This is the first report on the CRF12_BF of the full-length genome in mainland China, suggesting that a more complex HIV genotype is circulating in Guangdong Province. Although this pre-existing NNRTI-mutated virus was fortunately suppressed after TDF/3TC/EFV combination treatment, surveillance of newly diagnosed patients for evidence of transmitted resistance is still recommended so that the appropriate treatment can be tailored to the patient to prevent the spread of drug-resistant viruses.
Footnotes
Acknowledgments
We thank the staff at the Infectious Disease Center of Guangzhou Eighth People's Hospital for recruiting this patient, collecting blood samples and epidemiology information, and conducting regular follow-ups.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was funded by Guangzhou Science and Technology Plan project (grant no. 202002030028) and the National Grand Program on Key Infectious Disease Control (grant nos. 2018ZX10302103-002, 2017ZX10202101-003, and 2018ZX10715004).