
Abstract
The HIV-1 epidemic was mainly driven by men who have sex with men (MSM) recently in Beijing, China, with high genetic diversity. Novel recombinant strains were frequently reported at 3.4%–9.9%. It is imperative to interpret the recombinant modes and the putative transmission sources by near full-length genome (NFLG). Four individuals from the MSM population were identified as novel recombinant strains during surveillance of pretreatment drug resistance. NFLG sequences were harvested by near end-point dilution and nested PCR with two overlapping half fragments. Phylogenetic inference was performed with subtyping reference sequences and major parental strain sequences, to explore the patterns of genetic recombinant and potential sources of parent strains. The breakpoints were determined using SimPlot 3.5 to draw genome mosaic map, and the potential parental strains were confirmed by Mega 6.0 using segmental neighbor-joining trees. BL19487-00 and BL1948-00 sequences were obtained from epidemiologically linked individuals and shared similar breakpoints (HXB2 nt 4,497 ± 8 to 4,722) with substitution of subtype B pol gene segment in the backbone of CRF55_01B. BL3104-00 and BL4307-00 carried seven and eight breakpoints, respectively, in the backbone of CRF65_cpx with g5 CRF01_AE substitutions. The recombinant fragments were located around gag, pol, and env genes, with vpr-tat and nef-3′-LTR genes only for BL4307-00. No transmitted drug resistance was observed with the four unique recombinant forms (URFs), except for some drug resistance associated mutations. The advent of URFs around CRF55_01B and CRF65_cpx identified in recent years implied that the sexual behaviors were active and the epidemic of HIV was complicated among MSM in Beijing. Molecular epidemiological surveillance and precise control should be reinforced for this population.
Introduction
Recently, HIV-1
Super-infections and unique recombinant forms (URFs) may be closely associated with disease progression. 15,16 CRF01_AE expressed rapid disease progress to AIDS and advanced immunodeficiency among MSM, with 39.5% coreceptor switch within 3 years after acute infections in China. 4,17 It is imperative to analyze the HIV-1 genetic characteristics of the near full-length genome (NFLG) for the novel recombinant strains.
In this study, we utilized the near end-point dilution method to amplify HIV-1 NFLG sequences by two-segment nested PCR from the candidate novel recombinant strains. We intend to understand the genetic recombination modes and infer the possible origins of transmission.
Methods
Case recruitment and sample collection
In the Fifth Medical Center of PLA General Hospital, HIV-1 individuals were recruited for the initiation of ART. Plasma samples were separated from intravenous anticoagulated whole blood and stored at −80°C for pretreatment drug resistance. Written informed consent was available for ART and blood testing. HIV-1 pol gene fragments from four cases were previously classified as undetermined subtypes or CRFs, suggesting novel recombinant strains.
Nucleic acid extraction, amplification, and sequencing
Viral RNA was extracted from 280 μL plasma samples using Qiagen QIAamp viral RNA Mini kit according to the instructions. Complementary DNA (cDNA) was synthesized with Invitrogen SuperScript III Reverse Transcriptase, and serial PCR amplifications were performed using the near end-point dilution of cDNA (1:1, 1:3, and 1:9) with four repeat wells for each dilution. Two overlapping half segments were amplified for the HIV-1 NFLG, using the primer pairs as follows. 18 –20 The 5′ half segment outer primer pairs: 1.U5.B1F: CCTTGAGTGCTTCAAGTAGTGTGTGCCCGTCTGT (HXB2 nt 538–571), 07Rev8: CCTARTGGGATGTGTACTTCTGAACTT (HXB2 nt 5,219–5,193); the 5′ half segment inner primer pairs: Upper1A: AGTGGCGCCCGAACAGG (HXB2 nt 634–650), Rev11: ATCATCACCTGCCATCTGTTTTCCAT (HXB2 nt 5,066–5,041). The 3′ half segment outer primer pairs: 07For7: CAAATTAYAAAAATTCAAAATTTTCGGGTTTATTACAG (HXB2 nt 4,875–4,912), 2.R3.B6R: TGAAGCACTCAAGGCAAGCTTTATTGAGGC (HXB2 nt 9,636–9,607); the 3′ half segment inner primer pairs: VIF1: GGGTTTATTACAGGGACAGCAGAG (HXB2 nt 4,900–4,923), Low2c: TGAGGCTTAAGCAGTGGGTTCC (HXB2 nt 9,612–9,591). The positive amplicons at the highest dilutions were chosen for amplification to 200 μL volume products and sent to Beijing SinoGenoMax Company for Sanger's sequencing.
Sequence cleanup, phylogenetic analysis, and subtyping
The sequenced Applied Biosystems, Inc (ABI) files were cleaned up and assembled using Sequencher 5.0. The obtained NFLG sequences were codon-based aligned with online tool of Gene Cutter from the Los Alamos National Library (LANL) HIV sequence database, together with the subtyping reference sequences and the possible parental sequences of g5 CRF01_AE, CRF65_cpx and CRF55_01B sequences from LANL database. 21 –23 The neighbor-joining (NJ) tree was constructed using software MEGA 6.0, with the Kimura 2-parameter mode and the Bootstrap method of 1,000 replications to check the reliability of the nodes and determine the subtyping with reference to COMET HIV tool.
Recombinant breakpoint analysis
The aligned sequences were submitted to software Simplot 3.5 for the closest parental strains and recombinant modes, using similarity plotting and Bootscan analysis. We set the default parameters, including window size of 200 bp and step by 20 bp with Kimura (2-parameter) and NJ model. The recombination breakpoints were determined using the FindSite function. Meantime, the segmental NJ trees were constructed according to the breakpoints. We use the Map–Draw tool of the LANL HIV sequence database to draw the genome mosaic map according to the corresponding position of HXB2.
Genotype resistance analysis
The CPR program and HIVdb program of Stanford University HIV resistance database were used to analyze the transmitted drug resistance (TDR) and surveillance drug resistance mutation (SDRM), and drug resistance associated mutations (DRMs), including protease (PR), reverse transcriptase (RT), and integrase (IN) gene regions. This research was approved by Ethics Review Committee of Beijing Center Disease Prevention and Control (2013[2]).
Results
General information of the cases
The four cases (patient ID BL1947, BL1948, BL3104, and BL4307) were all infected through MSM, living in Beijing, yet without Beijing household registration. They were of ages between 27 and 57 years old, and began receiving ART soon after diagnosis. Epidemiological investigations showed cases BL1947 and BL1948 were sexual partners. The general information, including the HIV-1 viral load and CD4 cell counts at baseline of treatment, is listed in Table 1.
Demographic Information of the Four Individuals with Baseline Blood Testing
Phylogenetic tree analysis
Finally, four NFLG sequences were obtained from baseline sampling (sample ID named after patient ID-00) with 8,913 (BL1947-00, HXB2 635-9590), 8,970 (BL1948-00, HXB2 636-9589), 8,982 (BL3104-00, HXB2 637-9600), and 8,981 (BL4307-00, HXB2 645-9606) nucleotides (nt), respectively. NJ trees were constructed with the four sequences aligned with the subtyping reference strains and CRF65_cpx, CRF55_01B, and g5 CRF01_AE (Fig. 1).
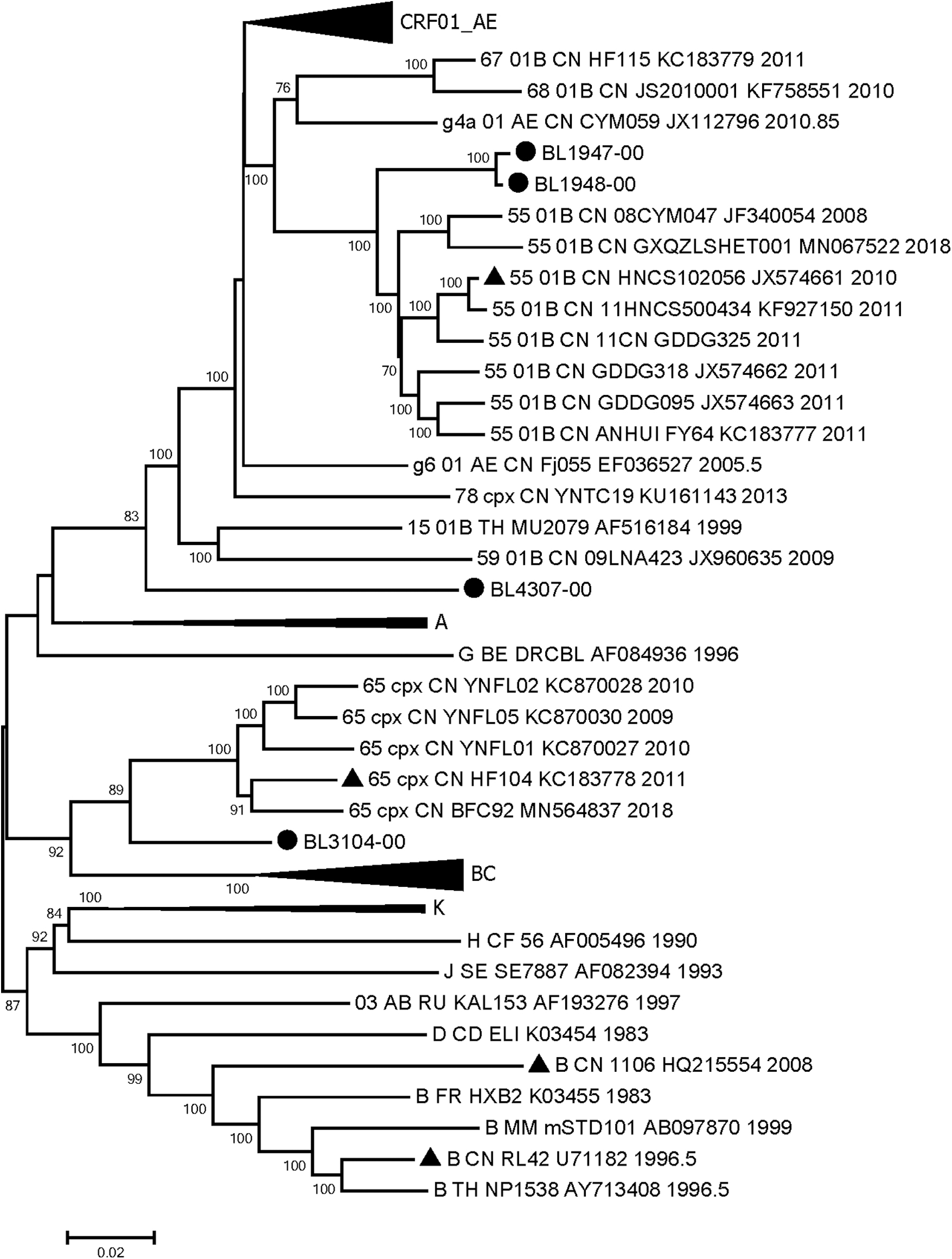
Neighbor-joining tree of HIV-1 NFLG sequences from four individuals. • Indicates NFLG sequences from the four individuals. ▴ Indicates sequences of potential parent reference strains. Bootstrap values >70% are shown at the nodes. NFLG, near full-length genome.
BL1947-00 and BL1948-00 formed a large monophylogenetic cluster with CRF55_01B sequences (bootstrap value 100%), indicating close genetic relative to CRF55_01B. However, the two sequences, forming a small subcluster (bootstrap value 100%), were located in the outer layer of CRF55_01B, suggesting genetic disparity with CRF55_01B, and the two individuals with a direct or indirect transmission relationship, as proven by epidemiology investigation.
BL3104-00 and CRF65_cpx reference sequences clustered together (bootstrap value 89%), indicating a close genetic relationship. Otherwise, the long branch of BL3104-00 to CRF65_cpx suggested its genetic heterogeneity with CRF65_cpx. BL4307-00 fell into the periphery of the CRF01_AE and serial 01B clusters (e.g., CRF67_01B, CRF68_01B, CRF55_01B, and CRF59_01B), suggesting certain genetic similarity. COMET HIV-1 and BLAST analyses showed that the HIV-1 pol gene fragments and NFLG sequences of these four samples were novel recombinant forms.
Recombinant breakpoint analysis
Simplot 3.5 software was used to analyze the recombination modes and putative parental sources of the NFLG sequences of the four strains. Cases BL1947 and BL1948 shared the same recombination modes and breakpoints, based on backbone of CRF55_01B, with subtype B fragment substitution in the pol gene (Fig. 2A; Supplementary Fig. S1). BL1947-00 and BL1948-00 genomes were composed of three fragments, and in the backbone of CRF55_01B, fragment II (HXB2 nt 4,497 ± 8 to 4,722) was derived from subtype B.
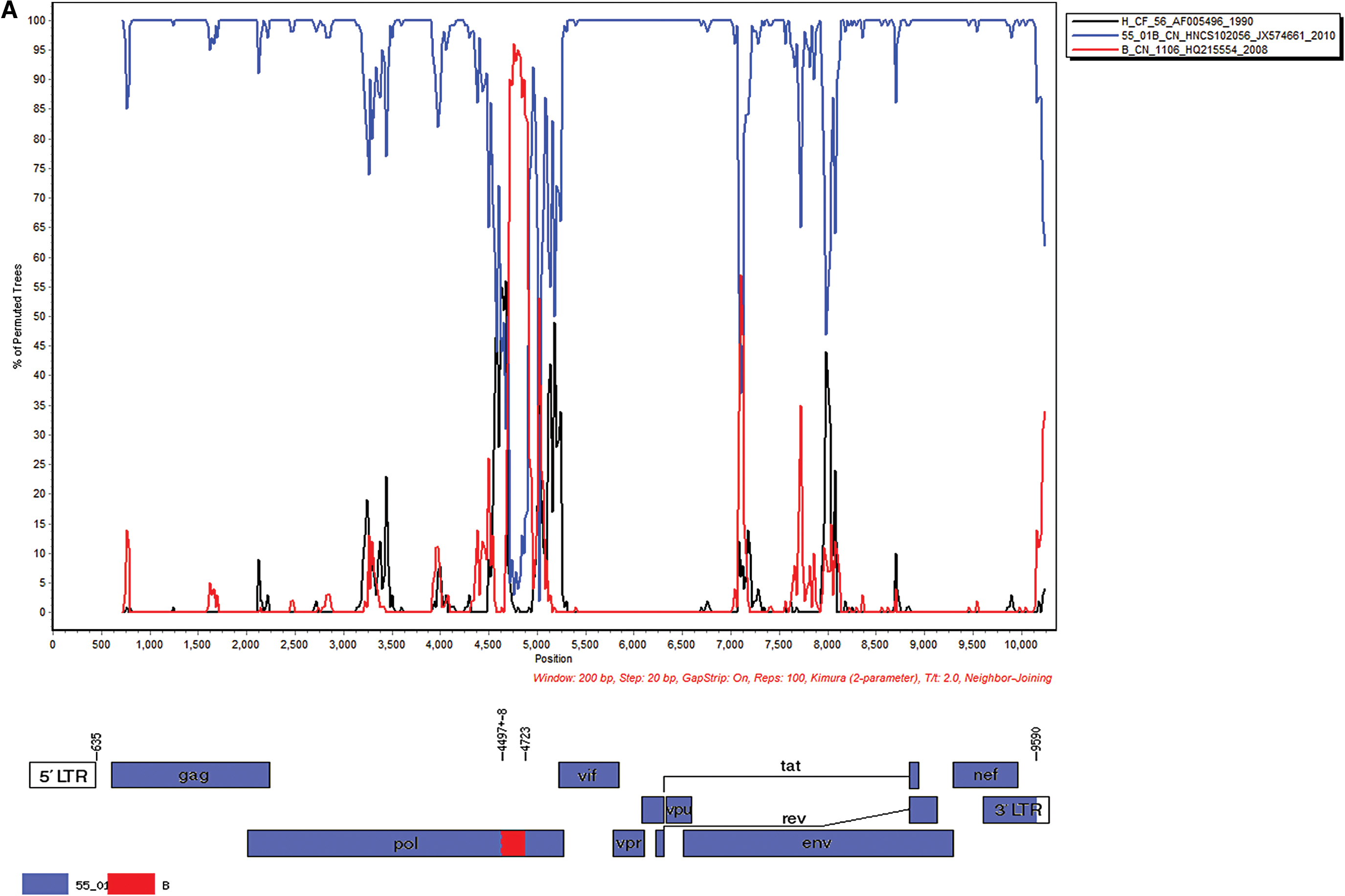
Bootscan analyses and genome mosaic map of the four URF NFLG sequences.
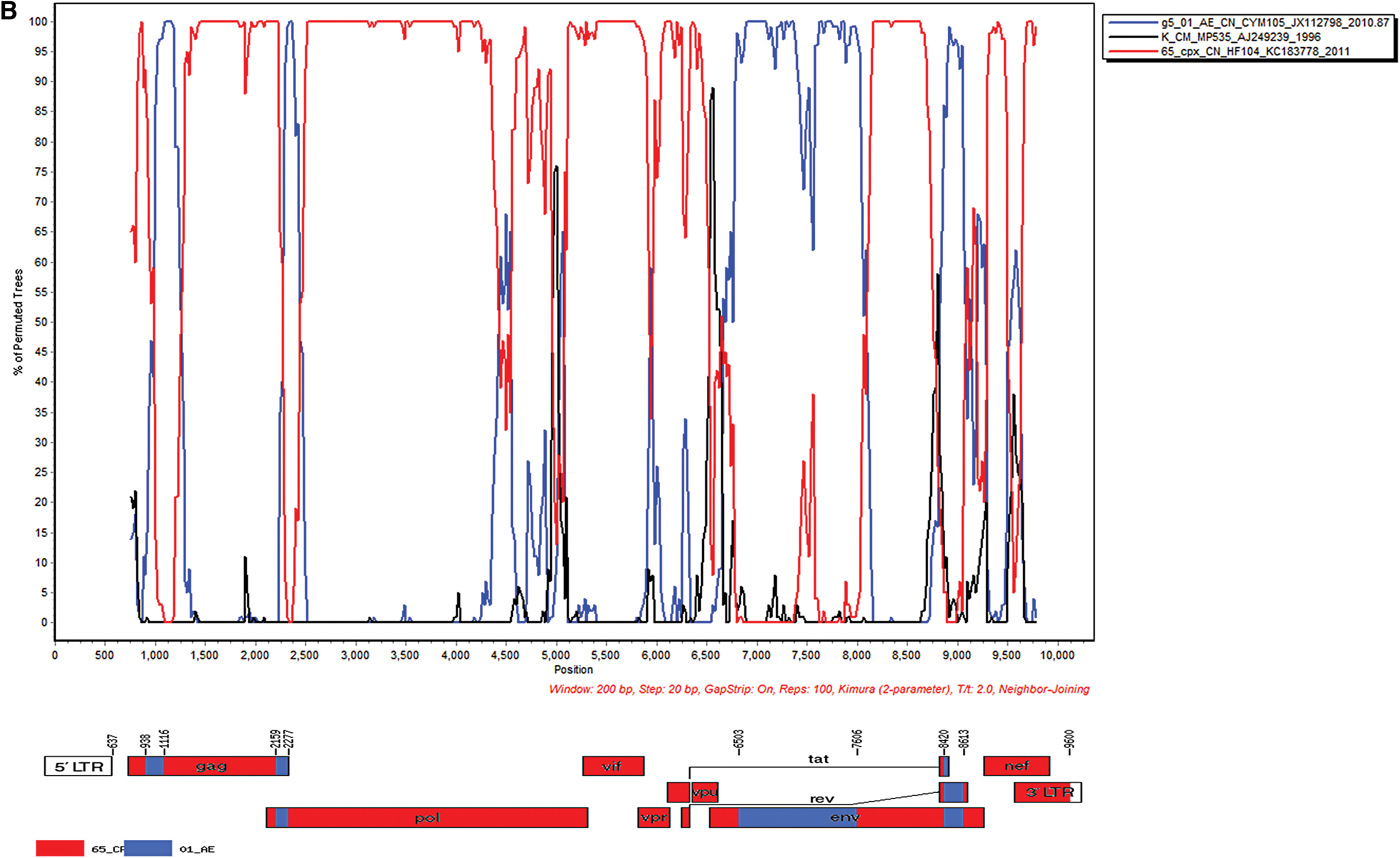
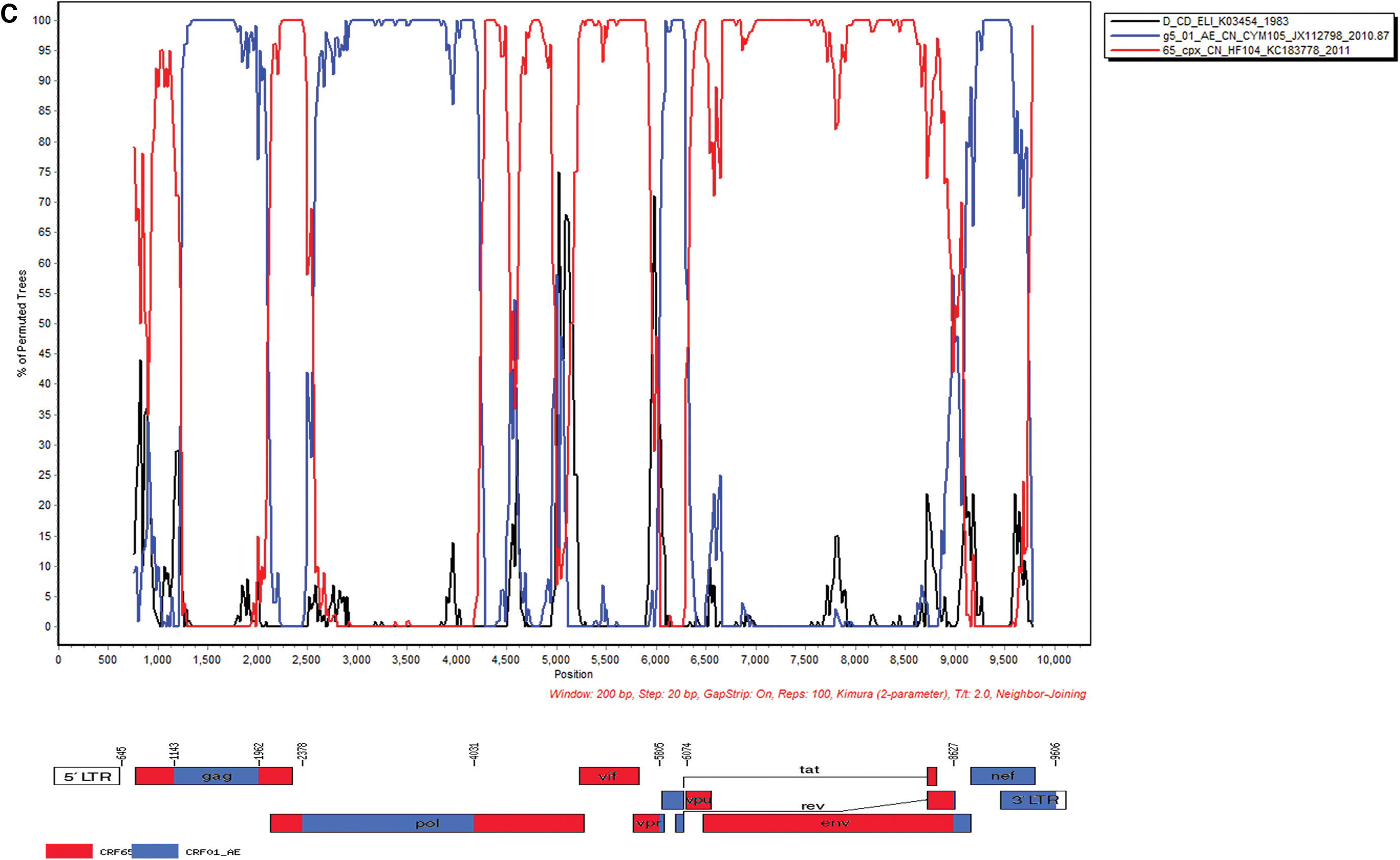
BL3104-00 and BL4307-00 sequences shared similar parental strains of CRF65_cpx and g5 CRF01_AE, yet with different recombinant modes and breakpoint positions. The BL3104-00 genome was divided into nine fragments, with eight breakpoints, HXB2 nt 938, 1,116, 2,159, 2,277, 6,503, 7,606, 8,420, and 8,613. Fragments I, III, V, VII, and IX of the backbone were originated from CRF65_cpx, and fragments II, IV, VI, and VII were substituted by g5 CRF01_AE (Fig. 2B; Supplementary Fig. S1). BL4307-00 genome was divided into eight fragments, with seven breakpoints of HXB2 1143, 1962, 2378, 4031, 5805, 6074, and 8627. Fragments I, III, V, and VII were originated from CRF65_cpx strain as the backbone, and fragments II, IV, VI, and VII were substituted by g5 CRF01_AE strain (Fig. 2C; Supplementary Fig. S1).
Using the Map–Draw tool to draw the URFs genome mosaic map, the results showed that the BL1947-00 and BL1948-00 recombination sites were located in the pol gene. The BL3104-00 recombination sites were located in the gag, pol, and env gene regions, whereas the breakpoints of BL4307-00 were located in the gag, pol, env, vpr-tat, and nef-3′-LTR gene regions (Fig. 2; Supplementary Fig. S1).
Segmental phylogenetic analysis
Segmental or chimeric NJ phylogenetic trees were constructed according to the breakpoints (Fig. 3; Supplementary Fig. S2).
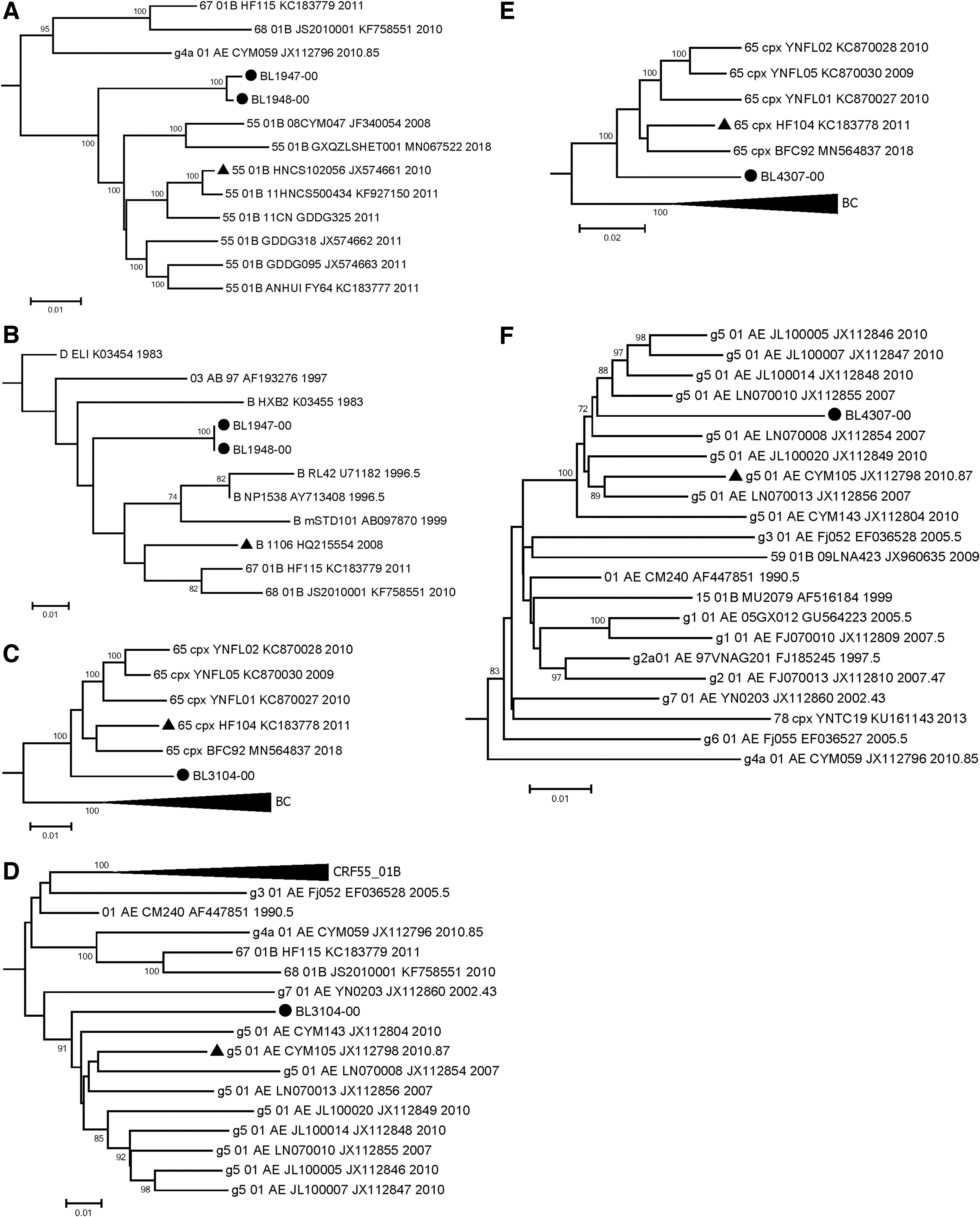
The segmental chimeric phylogenetic analyses of four URF NFLG sequences. •: NFLG sequences from four individuals. ▴: Sequences of potential parent reference strains.
The chimeric sequences comprising fragments I and III of BL1947-00 and BL1948-00 were gathered together in the outer layer of the phylogenetic cluster of CRF55_01B (bootstrap value 100%), similar to the whole genome (Fig. 1). Fragment II, containing less genetic information (only 225 nt in length relative to HXB2), clustered with subtype B reference sequences in the phylogenetic tree, with its Bootstrap value <70% (Fig. 3A, B; Supplementary Fig. S2 ).
The chimeric sequence composed of fragments I, III, V, VII, and IX of BL3104-00 was clustered with CRF65_cpx (bootstrap value 100%), genetically closer than the whole genome (Fig. 1) and that of fragments II, IV, VI, and VII clustered with g5
The chimeric sequence composed of the fragments I, III, V, and VII from BL4307-00 was clustered with CRF65_cpx in the NJ tree (bootstrap value 100%), obviously deviating from the location in the whole genome NJ tree (Fig. 1). Otherwise, the fragments II, IV, VI, and VII were merged into the cluster of g5 01_AE. CN (bootstrap value 100%) (Fig. 3E, F; Supplementary Fig. S2).
Genotypic drug resistance interpretation
No TDR was observed in the four NFLG sequences for PR/RT and IN as interpreted by Stanford University HIV drug resistance database. However, both BL1947-00 and BL1948-00 shared the consistent mutations E138G and V179E in the RT region, which may express low-level resistance to NNRTIs drugs, for example, efavirenz (EFV), etravirine (ETR), and nevirapine (NVP), ripavirin (RPV). BL3104-00 carried a mutation of L33F in the PR gene region, which was potentially low resistant to fusanavir, nelfinavir, and tipranavir, and V179D mutation in the RT region that may lead to intermediate resistance to EFV and NVP, low resistance to RPV, and potential low resistance to ETR.
Discussion
The epidemic of HIV is still expanding currently in Beijing, even after the administration of rigorous strategies of “treat all” and surveillance of drug resistance at baseline of ART from 2014, the uprising incline of the epidemic tends to slow down. 1,2,24 Numerous researches revealed the top three subtypes or CRFs spreading in Beijing were CRF01_AE (44.4%–56.4%), CRF07_BC (20.0%–32.5%), and B (5.4%–20.6%), 2,3,10,11,24 making the epidemic complicated with high genetic diversity. Meanwhile, MSM are the most affected population accounting for 70% infections. Hence, novel recombinant strains sprung up frequently, with the prevalence of novel recombinant strains at 3.4%–9.9%. 3,9,10,24 Several NFLG sequences were harvested, to reveal the possible parental strains, mainly around the main top three strains. 12 –14,25
In this study, we obtained four URF NFLG sequences from Beijing MSM HIV-1 infections sampling during 2015–2019. The recombinant modes were determined, confirming the second-generation recombination based on the newly reported CRF55_01B and CRF65_cpx in recent years. 22,23
Sequences BL1947-00 and BL1948-00 shared genetically high similarity with the consistent recombination modes. The short substitute fragment (HXB2 nt 4,497 ± 8 to 4,722) offered less information about the exact putative parental strain (B or Thai B), meanwhile, the breakpoint of HXB2 nt 4,497 ± 8 was just located next to the breakpoint of HXB2 4453 (3,767–4,452 from Subtype B) in CRF55_01B. Liang B and colleagues reported a case from Guangdong Province was heterosexually infected by a URF with CRF55_01B and subtype B recombinant in nef-3′ LTR region. 26 CRF55_01B is first identified in MSM originated from CRF01_AE and subtype B around 2013, 22 and in Shenzhen, CRF55_01B was mainly circulating from MSM. 6 CRF55_01B accounted for ∼0.8%–2.4% infections in Beijing. 9,24
Both BL3104-00 and BL4307-00 shared CRF65_cpx as the backbone, carried eight and seven breakpoints, respectively. Genome mosaic maps and Simplot results indicated that the partial gag, pol, and env gene regions from the two strains, and tat and the nef-3′-LTR gene regions in BL3104-00 were substituted by g5 CRF01_AE (Fig. 2B, C). The length and modes of recombination gene regions from the two strains were genetically different. A novel recombinant form comprising CRF65_cpx was also reported in MSM in Jilin, China. 27
CRF65_cpx, a complex CRF formed by multiple recombination of CRF01_AE, subtypes C and B, containing a total of 13 recombination breakpoints, 22 was spreading among the MSM population at 1.5%–2.3% in Beijing. 9,28 CRF01_AE was first introduced to Yunnan and Guangxi from Thailand, currently serving top one strain prevalent in Beijing. 2,9 At present, it can be further divided into seven epidemic clusters (clusters 1–7) in China. 21 Cluster 4 and cluster 5 were mainly MSM populations prevalent in northern China including Beijing and Tianjin. 21 The backbones of BL3104-00 and BL4307-00 were originated from cluster 5 (g5 CRF01_AE, CYM105).
Although the parental strains of BL3104-00 and BL4307-00 were both CRF65_cpx and CRF01_AE, the topological structure positions of the two whole sequences were not and genetically close in the phylogenetic tree (Fig. 1). The main reasons for the disparity were first, although the parental strains were homologous, the positions of the recombination breakpoints and the proportions in the sequence of the parents in the two NFGL sequences were not the same; second, NJ phylogenetic tree is based on the average pairwise distance among the NFLG sequences. The proportion of CRF01_AE counterpart in the BL4307-00 strain was relatively higher, thus close to the CRF01_AE cluster in the NJ tree of the whole genome (Fig. 1). The proportion of CRF01_AE in the BL3104-00 strain was comparatively lower than that of BL4307-00, correspondingly, with higher proportion of CRF65_cpx, genetically close to CRF65_cpx shown in Figure 1. This difference was derived from different genetic recombination patterns, which may lead to deviations or false appearances in the topological position of the phylogenetic tree. Of course, this kind of deviation or illusion might remind researchers to strengthen the phylogenetic research and in-depth analyses of the sequences, especially for long length of gene fragments.
No SDRMs for pol gene or integrase gene were observed in the four URFs, and three individuals were detected with the DRMs by HIVdb algorithm, which may lead to different degrees or levels of resistance to first-line antiretroviral drugs. However, these mutations have little impact on the current first-line regimen. 9 To avoid the occurrence of acquired drug resistance, continuous surveillance of drug resistance pre- or post-treatment is still suggested to help patients (especially for cases with CD4 cell counts <200 cells/μL) obtain better virus suppression effects.
The emergence of novel recombinant strains and genetic diversity among MSM indicates that the epidemic is active and the circulating strains were complicated. 9,29 It is urgent to reinforce the surveillance of molecular epidemiological investigation, and provide an insight into the recombinant mode and putative parental sources, to implement precise prevention and control strategies, 30,31 finally to end the epidemic of HIV-1.
Sequence Data
The NFLG nucleotide sequences of four URFs (BL1947-00, BL1948-00, BL3104-00, and BL4307-00) of HIV-1 are available from the GenBank database accession numbers MW110767–MW110770, respectively.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by Beijing Natural Science Foundation (Grant No. 7202074), Beijing Municipal Science and Technology Project (Grant No. D161100000416002), and Beijing Talents foundation (Grant No. 2018000021223ZK38).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.