
Abstract
The subtype C accounts for >50% of HIV type 1 (HIV-1) infections worldwide and it is currently the predominant viral form in South Brazil. Subtype C has been reported in all Brazilian regions; however, the phylogenetic relationship among strains circulating in those regions still remains unclear. This study aimed to investigate the origin and dynamic dispersion of HIV-1 subtype C toward Northeast Brazil. Our phylogenetic analysis suggests that most subtype C strains circulating in Brazil (99%) are descendant from the main lineage whose entrance in the country was previously described in the 1970s. According to the literature, additional introductions of subtype C were reported in the country through the Southeast region and in this study we identified another entry event that occurred most likely through the North region. Furthermore, our analysis suggests that the spread of subtype C to Brazilian Northeastern states occurred through multiple independent introductions of the main lineage that originated in South Brazil between mid-1980s and late 1990s. Despite the observation of eventual new HIV-1 subtype C introductions, our results highlight the predominance of a single lineage of this subtype in Brazil and the importance of South region in its dissemination throughout the country.
Introduction
Infection with HIV type 1 (HIV-1) usually results in a progressive destruction of the immune system, establishing a state of immunodepression that allows the appearance of opportunistic infections. 1 Although the advent of antiretroviral therapy has increased the expectancy and quality of life of HIV-1-infected individuals, there is still no cure or vaccine capable of preventing infection. 2 The intense genetic variability and capacity to establish reservoirs in vivo are the main obstacles in the fight against HIV/AIDS. 3 The HIV-1 genetic diversity is represented by the existence of two viruses (HIV-1 and HIV-2), groups, subtypes, and recombinant forms, which have different dispersion patterns around the world. 4
The HIV-1 epidemic in Brazil is predominantly characterized by the cocirculation of B, C, and F1 subtypes and the BF1 and BC recombinant forms. 5 Owing to the continental extent of the country, the Brazilian geographic regions present unique patterns of subtype distribution. Subtypes B and F1 predominate in the North region, whereas B, F1, and BF recombinants are the most frequent forms in the Northeast. 6,7 In the Southeast and Central West regions, B, C, F1, and BF1 cocirculate, 8,9 whereas in the South region, HIV-1 infections are caused mainly by subtypes B, C, and BC recombinants. 9
The subtype C is the most prevalent form of HIV-1 worldwide and it was first recognized in South Brazil by early 1990s, in the Rio Grande do Sul state. Initial studies in that region showed that 22% of the HIV infections were caused by subtype C between 1994 and 1997, 10 whereas most recent reports described a prevalence of 44%–66%, 11 pointing to an increase of subtype C prevalence in South Brazil over the last two decades, as previously reviewed. 12 Furthermore, the subtype C prevalence has also been reported in Southeast (16%), 13 Central West (11.5%), 8 Northeast (5%), 14 and North (2.9%) 15 Brazilian regions. Previous studies have reconstructed the history and the dynamics of the subtype C epidemic in Brazil. A close phylogenetic relationship has been demonstrated between strains circulating in Brazil and sequences from countries located in the eastern region of the African continent, such as Burundi and Kenya. 16 –18
The Northeast Brazil consists of nine states and is the third region with the highest number of HIV-1 infections in the country. 19 The increase of HIV-1 cases in this region seems to be related to factors such as low economic and educational levels, sex tourism, and prostitution, which might have facilitated the introduction and the spread of new viral forms. Despite previous studies already reporting the circulation of subtype C in Northeast Brazil, 7,14,20,21 the origin of these viruses remains unknown.
This study uses nucleotide sequences from all five Brazilian regions and aims to reconstruct the virus migration movement to the Northeast region, which has the largest number of federative units and coastal extension, attracting tourists from the other four regions. The generated data highlight the importance of molecular surveillance studies for the development of measures to control HIV-1 spread and for providing important information for the development of prophylactic agents.
Material and Methods
Subtype C Brazilian dataset
To characterize the HIV-1 subtype C epidemic in Northeastern Brazil, a search for sequences from this region was carried out in the Los Alamos Database (
Twenty-two sequences from the Northeast region were identified covering the genomic fragment of pol gene corresponding to positions 2301–3200 relative to HXB2 reference strain. To compose the Brazilian dataset, a total of 948 subtype C sequences covering the genomic fragment aforementioned were initially collected. Subsequently, sequences were excluded according to the following criteria: absence of information regarding the year and location of sampling, duplicated entries, clones from the same sample, and sequences with high rate of gap, stop codons, and degenerate bases. All subtype C sequences were analyzed through the jpHMM tool 22 to exclude possible recombinant samples. The final Brazilian dataset had 460 subtype C sequences from five geographic regions (South n = 336, Southeast n = 63, Central West n = 32, North n = 7, and Northeast n = 22) isolated between 1998 and 2017 (Fig. 1).
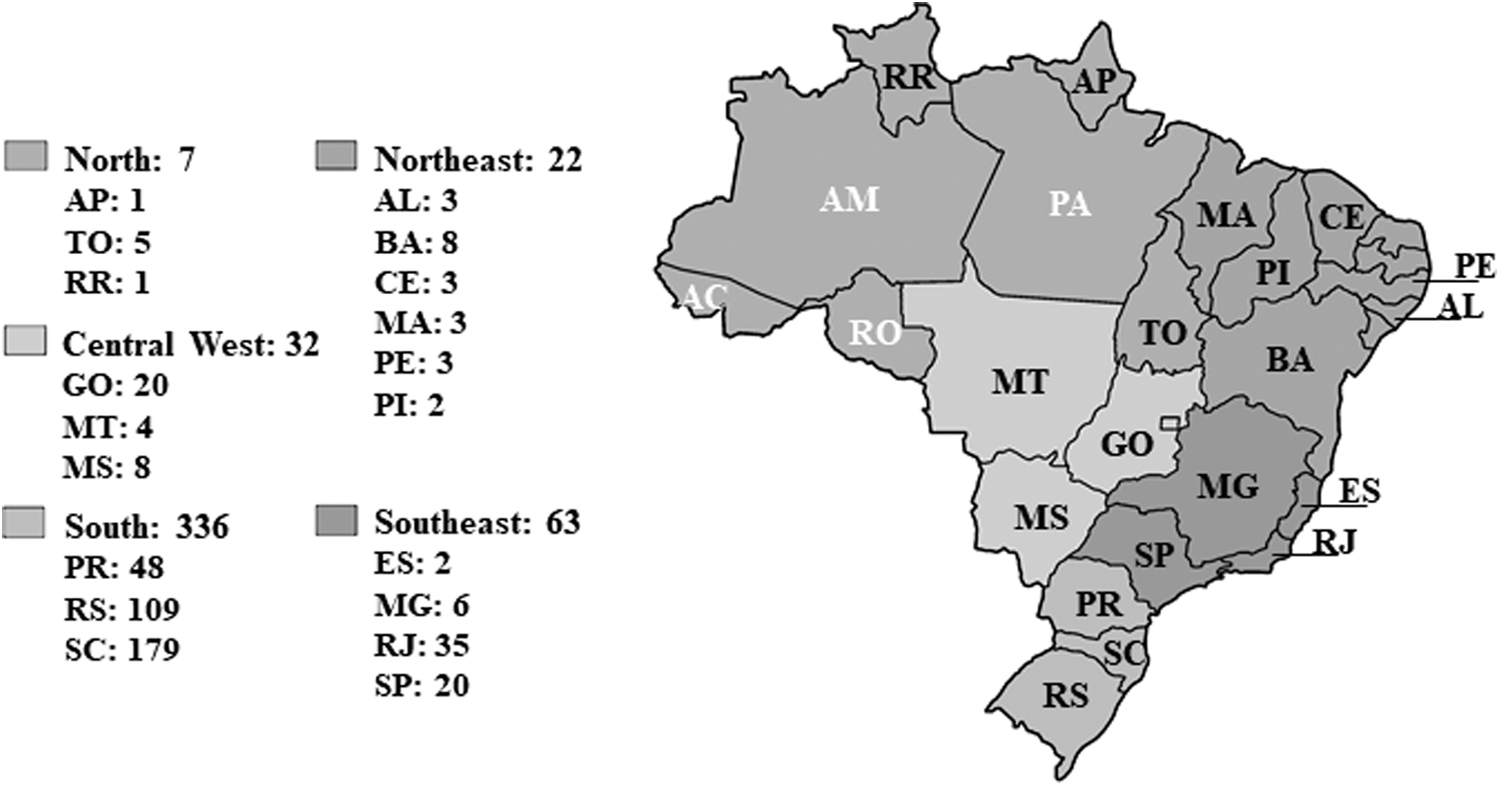
Distribution map of analyzed sequences by Brazilian regions and states (n = 460). The geographic regions are identified by different shades: North, Northeast, Central West, Southeast and South. The states are indicated by two letters code: AL (Alagoas), AP (Amapá), BA (Bahia), CE (Ceará), ES (Espírito Santo), GO (Goiás), MG (Minas Gerais), MS (Mato Grosso do Sul), MT (Mato Grosso), PE (Pernambuco), PI (Piauí), PR (Paraná), RJ (Rio de Janeiro), RS (Rio Grande do Sul), SC (Santa Catarina), SP (São Paulo), and TO (Tocantins). Unrepresented states are labeled in white: AC (Acre), AM (Amazonas), PA (Pará), and RO (Rondônia). The number of HIV-1 subtype C pol sequences from each location included in the present study are indicated. HIV-1, HIV type 1.
Subtype C reference dataset
To identify the different HIV-1 subtype C strains circulating in Brazil, a consensus sequence of Brazilian dataset (n = 460) was created in the GENEIOUS software
23
and submitted to HIV BLAST online tool against all subtype C sequences (
Sequence alignment and phylogenetic analyses
Alignment was performed using MAFFT online program 24 under the command: mafft —thread 8 —threadtb 5 —threadit 0 —reorder —auto input > output and manually edited using BioEdit software. 25 The dataset was assessed for presence of phylogenetic signal by applying the likelihood mapping analysis implemented in the TreePuzzle Program. 26
Maximum likelihood (ML) phylogenies were reconstructed using IQ-TREE 1.6.8 webserver. 27 The reconstruction was performed under the GTR+I+G nucleotide substitution model that was inferred in Modelfinder. 28 Subtree pruning–regrafting branch-swapping algorithm was used to perform Heuristic tree search. The reliability of each cluster was evaluated by analyzing 1,000 bootstrap (BS) replicates and with the approximate likelihood ratio test (aLRT) 29 based on the Shimodaira–Hasegawa-like procedure. The ML trees were visualized by FigTree program version 1.4.4.
Analysis of structural motifs and amino acids
For the purpose of investigating and comparing amino acids patterns and signatures, the sequences representing different subtype C introductions were translated in GENEDOC
30
in association with the biological information package of the Prosite Database (
Phylodynamic and phylogeographic dispersion of HIV-1 subtype C toward Northeast region
To investigate the temporal signal of the dataset, we regressed root-to-tip genetic distances from the ML trees against sample collection dates using TempEst v 1.5.3. 31 The Bayesian analyses were conducted in BEAST 1.10.4 package. 32 –34 The ML phylogeny was used as a starting tree for Bayesian time-scaled phylogenetic analysis using BEAST 1.10.4 in CIPRES SCIENCE GATEWAY. 32,33,35 The tree reconstruction was inferred with the GTR + Γ4 nucleotide substitution model, an uncorrelated relaxed molecular clock, and the demographic model of logistic growth, according to previous findings investigating the fittest models for HIV-1 subtype C in Brazil. 36
The percentage of fully resolved probability mapping quartets totals 85.8% and because of the dataset small temporal signal (r = 0.38), we applied an informative normal prior (mean = 41, SD = 5.1) on the root height based on previous study that estimated the introduction HIV-1 subtype C in Brazil to 1976 (1966–1983, 95% high posterior density). 18 Four runs of Markov chain Monte Carlo (MCMC) with 100 million states each were computed, sampling every 10 million steps. The log and trees files were combined in LogCombiner discarding 40% as burn-in and the convergence of MCMC chains was checked using Tracer v.1.7.1. 37 The value of effective sample size of proper mixing was 282.2 (>200) and the maximum clade credibility (MCC) trees were summarized from the MCMC samples using TreeAnnotator by CIPRES. 35
To estimate the transmission dynamics of subtype C toward the Northeast region of Brazil, phylogeographic analysis was performed on the set of empirical trees obtained by the Bayesian phylogenetic analysis. A discrete diffusion model with Bayesian stochastic search variable selection as implemented in BEAST was applied to reconstruct the ancestral location states of the trees. Tip locations were defined as the Brazilian geographic region from which each sequence originated: North, Northeast, Central West, and Southeast regions. Owing to the large sample size, sequences from the South region were further classified according to the state of origin: Paraná (PR), Rio Grande do Sul (RS), and Santa Catarina (SC). The migratory events were summarized and visualized using the SPREAD3 tool. 38
Results
Phylogenetic inference of the HIV-1 subtype C epidemic in Brazil and introduction in Northeast region
To investigate the evolutionary history of subtype C in Brazil, a preliminary ML tree was reconstructed using 460 Brazilian subtype C sequences plus 110 subtype C sequences worldwide (Fig. 2). Full details of the sequences analyzed in this study are provided in the Supplementary Material S1. No recombination events were found among the sequences. The phylogenetic reconstruction showed that 455 (98.9%) Brazilian subtype C sequences fall within a single well-supported major clade (BS = 88; aLRT = 99) comprising isolates from all five Brazilian regions sampled between 1998 and 2015. Moreover, five Brazilian viral sequences grouped separately from the main monophyletic clade, clustering with subtype C sequences from other countries (Kenia, Senegal, South Africa, and Uganda), were supported by both BS (70–89 interval) and aLRT (47–99 interval) values. Of note, all sequences from Northeast region branched within the larger Brazilian subtype C clade (Fig. 2).

Maximum likelihood analysis of HIV-1 subtype C pol sequences (876bp) from Brazil (n = 460) and worldwide (n = 110). The color of the branches represents the geographic origin of the subtype C sequences. A major monophyletic group formed by subtype C sequences from Brazil is shown as a collapsed cluster (455) while five Brazilian samples grouped outside this cluster. The clusters formed by sequences from Africa, Asia (India), and the North America are collapsed and presented in different colors. The tree was built under the evolutionary model GTR+I+G and visualized in the software Figtree v1.4.4. The statistical support is indicated only at key nodes as bootstrap/aLRT values. Horizontal branch lengths are drawn to scale with the bar at the bottom indicating 0.02 nucleotide substitutions per site. aLRT, approximate likelihood ratio test.
The presence of characteristic amino acid substitutions was investigated among the sequences representing the six putative subtype C introduction in Brazil (Supplementary Material S2). In the fragment of pol analyzed here, 14 motifs were conserved in all sequences. These predicted regions correspond to cyclic adenosine monophosphate or guanosine monophosphate kinase protein phosphorylation sites, Casein kinase II and tyrosine kinase phosphorylation sites, active sites of protease, N-myristoylation and amidation sites.
Considering only the substitutions with frequency ≥75%, the consensus sequence (CONS_C_BR) of the major Brazilian monophyletic cluster (n = 455) differed at five positions from the world consensus C [protease: I75L, N93K, K97N; reverse transcriptase (RT): E92D and G174D], five of which represent exclusive signatures of this group. The other five samples representing additional entries of HIV-1 subtype C into Brazil (KF255847, KF255848, KF255855, KF255861, and KX443060) have at least five unique amino acid signatures each, thereby showing different signature patterns (Supplementary Material S2).
Inference of HIV-1 subtype C spread toward Northeast region
To further investigate the introduction events and the dynamic dispersion of subtype C toward the Northeast region, phylogeographic analyses were conducted. For these analyses, the dataset composed of 455 sequences branched inside the Brazilian monophyletic clade of subtype C.
ML analysis showed that the 22 subtype C sequences from Northeast region appeared to be distributed among 19 independent lineages that were intermixed between the sequences from other Brazilian regions (Supplementary Material S3). All 19 clusters involving Northeastern sequences were well supported with both BS and aLRT values, except one clade (BS = 47/aLRT = 46), which grouped a sequence from Alagoas state (Northeast) with a sequence from Mato Grosso do Sul (Central West). Most subtype C lineages from Northeast Brazil composed of only one sequence from this location and only three clades grouped two sequences from this region. Of note, most clades containing sequences from the Northeast also had sequences from two, three, or four other geographic regions (Supplementary Material S3).
To estimate the time of subtype C introductions in Northeast region, Bayesian analyzes were conducted (Fig. 3). The estimated evolutionary rate was 2.068 × 10–3 substitutions/sites/year (95% Bayesian Credibility Interval 1.0–2.6 × 10−3), consistent with other estimates for this subtype. 17,18 In this reconstruction, sequences from Northeast were distributed in the tree along with sequences from South, Southeast, North, and Central West regions (Fig. 3). The topologies in this MCC tree were remarkably similar to those in the ML analysis (Supplementary Material S3). The clades formed by sequences from the Northeast presenting PP >0.8 had the time to the most recent common ancestor (tMRCA) estimated (Fig. 3 and Table 1). The oldest node (PP = 0.83) showing evidence of subtype C transmission to the Northeast region (Piauí state) contained sequences from three other regions: South (Paraná state), Central West (Goiás state), and North (Tocantins state) with the tMRCA = 1985 (1977–1988). A second branch (PP = 0.85) grouped two sequences from Bahia state (Northeast region) and one from Rio de Janeiro state (Southeast region) [tMRCA = 1987 (1979–1988)]. The third branch (PP = 1) was formed by sequences from Northeast (Alagoas and Maranhão states), Central West (Goiás and Mato Grosso do Sul states), and North (Tocantins) with the tMRCA estimated in 1988 (1980–1992).
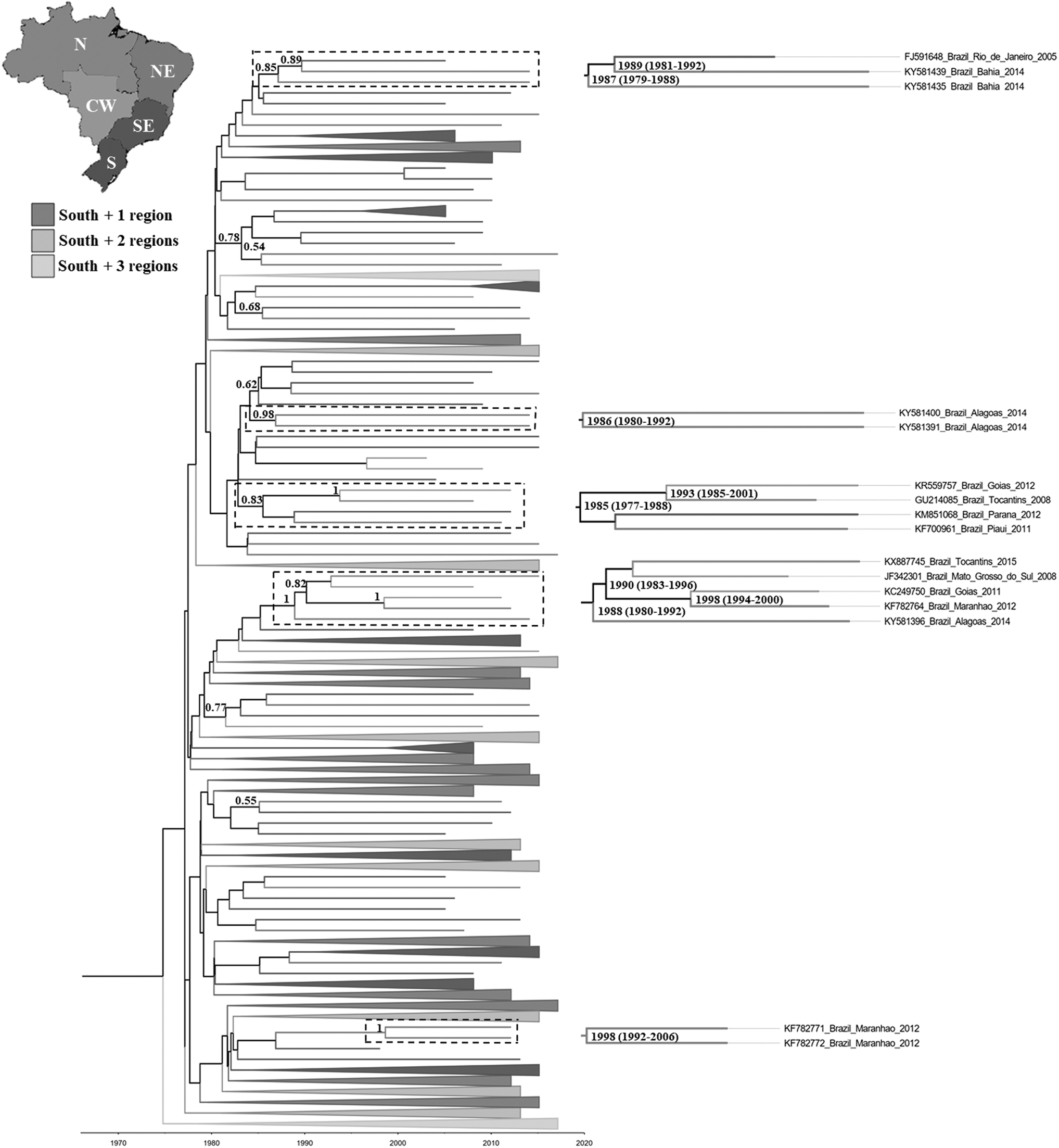
Time scale Bayesian maximum clade credibility tree showing phylogenetic relationships among subtype C viruses circulating in Brazil (n = 455). North (6), Northeast (22), Central West (32), Southeast (59), and South (336). Branch colors represent the geographic region from where the subtype C strain originated, according to the legend given in the figure. The collapsed clusters represent groups formed by sequences from the South region plus one, two or three other regions. The posterior probability (PP) is indicated only at key nodes and the dotted boxes highlight those with a high (>0.80) PP support. The median age (with 95% high posterior density interval in parentheses) for subtype C migration events into Northeast region are shown. Horizontal branch lengths are drawn to scale with the bar at the bottom indicating years.
Subtype C Data in the Northeast Region Based on Phylodynamic Tree
Only two nodes contained two sequences from the same state of Northeast region. The first node contained two sequences from the state of Alagoas, both isolated in 2014 [tMRCA = 1986 (1980–1992); PP = 0.97] and the second grouped two samples from Maranhão state collected in 2012 [tMRCA = 1998 (1992–2006); PP = 1] (Fig. 3). The result of phylogeography under SPREAD3 revealed an apparent migratory flow of HIV-1 subtype C mainly between the states of the southern region (Paraná, Santa Catarina, and Rio Grande do Sul). Subsequently, the dissemination occurred toward the states in the north of the country, reaching the Central West, Southeast, North, and Northeast regions (Fig. 4 and Supplementary Materials S4 and S5). Considering the value of PP > 0.80 as a cutoff in the phylogeographic tree, the node information points to the South region as the probable source of the subtype C strains found in the Northeast region.

Viral dispersion dynamics of HIV-1 C in Brazil. Phylogeographic analysis showing subtype C transmission route from South and it to all other Brazilian geographic regions. Viral strains from Northeast originated from South. The lines represent the supported transitions with a PP = 1.0
Discussion
Among 10 different subtypes and 102 circulating recombinant forms of HIV-1, C subtype alone accounts for nearly one half of worldwide infections. 39 The high prevalence rates of this subtype are detected in countries from Asia and Africa, mainly in the East and South of the African continent. 39 In some countries, subtype C became the prevalent HIV-1 form after its introduction, surpassing another previously dominant genotype, such as in South Africa 40,41 and India. 42,43 In South Brazil, subtype C overcame, over the last two decades, the epidemic initially characterized by subtype B predominance and, in addition, it spread to other regions, being reported especially in the neighboring regions, Southeast and Central West. 11,18
This study investigated the phylogenetic relationships among subtype C strains isolated from all Brazilian geographic regions, representing 19 of the 26 state units in the country. Our analysis showed that majority (98.9%) of the subtype C viruses circulating in Brazil are descendants of a main lineage that, according to previous studies, has been introduced in the country through the South region between 1976 and 1983. 17,18
Of the 460 sequences, five Brazilian viral isolates grouped separately from the main major clade, providing evidence that multiple subtype C introductions have occurred in Brazil. Apart from these, four lineages have been previously identified, indicating that additional entry events of subtype C have occurred through Southeast region of Brazil. 18 Here, we bring evidence of an additional introduction of subtype C in Brazil represented by a strain isolated in Roraima 44 (North region), which grouped with a sequence from South Africa (BS = 79; aLRT = 96).
The comparative molecular characterization between the consensus sequence of Brazilian main subtype C clade (CONS_BR_C; n = 455) and the five sequences outside this group (KF255847, KF255848, KF255855, KF255861, and KX443060) revealed structural motifs associated with phosphorylation sites on pol gene. Previous reports have demonstrated that HIV-1 RT is a substrate for in vitro phosphorylation by several protein kinases, although its influence on RT activity is not completely understood. 45,46 According to previous studies, the Brazilian subtype C indeed reveals specific amino acid patterns. 47,48
Within the protease and the RT regions of the pol gene, the Brazilian subtype C consensus sequence presents four substitutions that were not shared with the consensus sequences from Botswana, India, South Africa, Tanzania, and Zambia. 47 In addition, 12 specific positions in gag and env genes were also not found in other consensus sequences from Botswana, Ethiopia, India, South Africa, and Tanzania. 48 Our results demonstrate that Brazilian consensus (CONS_C_BR) and the five sequences representing additional entries of HIV-1 subtype C in Brazil have five unique amino acid signatures each. Therefore, they exhibit different signature patterns among them. These substitutions in amino acid signatures found in CONS_C_BR and in each sample representing additional subtype C introductions in Brazil reinforces the independent origin of these strains (Supplementary Material S2).
Despite its current predominance in the southern region of Brazil, subtype C has been reported in Northeast with a 5% prevalence. 14 However, the origin and epidemiological relationships of the subtype C variants circulating in that region are unknown. In this study, we collected all the subtype C sequences from the Northeast available in public databases to assess its dynamic dispersion.
The ML reconstructions indicated that all subtype C viruses from Brazilian Northeast region resulted from the dissemination of the main subtype C lineage (C_BR) (Fig. 2), which was introduced multiple times in this part of Brazil (Supplementary Material S3). Although most samples from Northeast are isolated from each other in the phylogenetic trees, they grouped with sequences from the other four geographic regions (South, Southeast, Central West, and North) (Fig. 3 and Supplementary Material S3), pointing to a high level of genetic mixing of Brazilian subtype C viruses from different geographic locations.
Previous Bayesian analyses indicated the contribution of the South region to the subtype C epidemic toward the northernmost states of Brazil. 18,49 These studies indicated the South region as the main point of initial dispersion of the subtype C in Brazil, especially to Central West region, 17,18,49 which presents 7.8% of HIV-1 subtype C prevalence. 9 The factors that would have contributed to the spread of the virus were human mobility owing to geographical proximity and the job opportunities in the fields of agriculture and livestock. 50,51 In fact, it has been shown that most of the subtype C strains found in Central West region present close phylogenetic relationships with those isolated in the South. 8,50 Other studies reported that the Southeast region may also represent an additional strategic point for subtype C dispersion to other regions. 8,51 Delatorre and colleagues 18 have proposed that the Southeast could represent a secondary point for subtype C dissemination to Central West and back to South.
These observations together with the phylogenetic relations shown in the present analysis (Fig. 3) indicate that Southeast and Central West regions may also constitute HIV-1 subtype C transmission routes toward Northeast, in addition to the main route represented by the South region. Of note, there is one clade composed of sequences from Northeast (Piauí), South (Paraná), North (Tocantins), and Central West (Goiás). This cluster had tMRCA estimated around 1985 (PP = 0.83 95% BCI, 1977–1988) and may represent the oldest introduction of subtype C in Northeast. In combination, these data provide additional evidence that subtype C has expanded from South to northern regions of the country, as previously suggested. 49 The two nodes containing sequences from Northeast sequences may represent intraregional transmission networks within the same region. In both cases the estimated year is dated after the oldest entry event in this region.
The phylogeographic analysis using SPREAD3 revealed that in the beginning of the 1980s, subtype C was mainly concentrated in the South region, within an intraregional transmission trajectory, whereas an apparent migratory flow of the virus from South to Southeast and Central west regions occurred around the mid-1980s (Fig. 4 A, B and Supplementary Materials S4 and S5). Similarly, Delatorre and collaborators 18 have suggested that viral strains from southern states (Santa Catarina and Paraná) were transmitted to Southeast and Central West regions between 1983 and 1988. Another report indicated that HIV-1 subtype C had already reached the Central West region in 1981 (from Rio Grande do Sul), the Southeast region (São Paulo) during the early 1980s, and the North region (Amazonas and Tocantins states) between late 1980s and early 1990s. 49
In this sense, there was a gap with respect to the origin and time of subtype C introduction in the Northeast region. The present phylogeographic analysis (Supplementary Materials S4 and S5) suggests that all viral sequences involved in the dispersion of subtype C to Northeast region most likely originated from South region between mid-1980s and late 1990s and that Santa Catarina was the main hub of subtype C dissemination to other locations, corroborating previous studies. 17,18 However, in the view that Santa Catarina state presents the highest number of sequences in our dataset (Fig. 1), we cannot rule out the impact of sampling heterogeneity in the ancestral states reconstruction of our phylogeographic analysis, as shown in the study by Graf and colleagues. 49 Therefore, future studies with a more balanced sample distribution should be performed to investigate the HIV-1 dissemination patterns in a finer scale (i.e., states or cities).
Conclusion
The Northeast is the Brazilian region with the highest number of federative units (n = 9) and the largest coastal extension in the country. According to the Brazilian Ministry of Tourism, the Northeast is the preferred tourist destination for most Brazilians, 52 attracting millions of visitors every year, 20% of which are from South and Southeast regions. 53 Most subtype C strains circulating in Brazil are descendant of a main lineage; however, this study confirmed the occurrence of additional independent introductions represented by punctual cases. One of these lineages represents evidence of an independent entry of subtype C in Brazil throughout the North region.
The HIV-1 subtype C epidemic in Northeast region resulted from multiple independent introductions of a main Brazilian subtype C lineage that originated in South region between mid-1980s and late 1990s. These Northeastern strains clustered together with viruses isolated from all other four geographic regions, pointing to a high level of genetic mixing of Brazilian subtype C viruses. Taken together, these results provide additional evidence that subtype C has been expanding from the south to the north of the Brazilian territory. These data contribute to the continuous monitoring of the molecular epidemiology of HIV-1, which is of great importance for the surveillance and control of HIV/AIDS epidemic.
Footnotes
Authors' Contributions
R.C.O.: conceptualization, investigation, writing—original draft preparation; T.G.: methodology, formal analysis, writing—review and editing; F.F.A.R.: methodology, validation, writing—review and editing; G.P.S.A.S.: investigation; M.G.: conceptualization, methodology, validation, writing—review and editing; J.P.M.C.: conceptualization, validation, investigation, writing—original draft preparation, supervision.
Acknowledgment
R.C.O. thanks Fundação de Amparo à Pesquisa do Estado da Bahia (FAPESB) for the masters scholarship. M.G. thanks Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ). The authors thanks Allan Botura Brennecke Leite for the English proofreading.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
R.C.O. and M.G., were supported by scholarships. This research was not supported by any funding.
Supplementary Material
Supplementary Material S1
Supplementary Material S2
Supplementary Material S3
Supplementary Material S4
Supplementary Material S5
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.