
Abstract
Unique recombinant forms (URFs) are more likely developed among HIV-1 infections through men who have sex with men (MSM) because of cocirculation of multiple subtypes. In this study, two novel URFs deriving from two HIV-positive subjects (HB010014, HB010063) were identified in Shijiazhuang, Hebei province, China, and two sequences formed a distinct monophyletic cluster. Further recombination analysis showed that of two new URFs were consisted of circulating recombinant form (CRF)01_AE and CRF07_BC. The subregion phylogenetic analysis indicated that CRF01_AE segments were traced back to cluster 4 of CRF01_AE strains, which were prevalent among HIV-1 infections through MSM in China. New URFs being developing gradually and spreading released that more and more novel recombinant strains of HIV-1 could be developed, which means that the past prevention strategies need to be adjusted.
Introduction
Since the first HIV-1/AIDS case was reported in 1981, HIV-1 epidemic gives rise to a severe public health problem globally. 1,2 Up to date, total of 75.7 million infections with HIV-1 are reported and >32.7 million people have died of AIDS. 1 There are four groups of HIV-1, M, N, O and P, respectively. M group, consisting of nine subtypes (A, B, C, D, F, G, H, J, and K), 3 was more popular than others and resulted in the global HIV-1 pandemic.
Because of the coinfection or infected two HIV-1 subtypes concurrently, it was possible to develop the circulating recombinant forms (CRFs) in one's body. More than 110 CRFs and many unique recombinant forms (URFs) of HIV-1 have been reported (
Hebei is an economically underdeveloped province with >75 million population in north China. Meanwhile, Hebei is the gateway that people get in and out of Beijing and Tianjin. With the comprehensive implementation of Beijing–Tianjin–Hebei integration formulated by the Chinese government, the flow of people between Beijing and Hebei is more and more frequent, which increased the opportunities for HIV-1 transmission. The main genotypes of HIV-1 in Hebei were CRF01_AE (49.6%) and CRF07_BC (29.7%). And 98.9% of patients in Hebei are infected with HIV-1 through sexual contact, especially among MSM (77.5%). 9 The cocirculation of CRF01_AE and CRF07_BC among MSM provides an ideal condition for the generation of the second-generation recombinant forms composed of CRF01_AE and CRF07_BC among MSM in this region. 10
In this study, we identified two novel recombinant strains from two HIV-1–positive MSM individuals named HB010014 and HB010063. Both patients signed informed consent before being investigated, then, their plasma samples were collected. The demographic information of the patients is summarized in Table 1. For the near full-length genome (NFLG) amplification, viral RNA was extracted from plasma samples using a high pure viral RNA kit (Roche; Basel, Switzerland) and reverse transcribed into cDNA by using Superscript IV first-strand synthesis system (Invitrogen; CA). And then, the NFLG was amplified in two halves with High Fidelity Taq (Invitrogen) as described previously. PCR positive products were purified and sequenced by SinoGenoMax (Beijing, China) with several specific primers. All the sequence fragments were edited and assembled into contiguous sequences by using Contig Express software. Finally, the HB010014's NFLG of 8,804 bp (from 772 to 9,601 nt according to HXB2 calibrator) and the HB010063's NFLG of 8,903 bp (from 609 to 9,601 nt according to HXB2 calibrator) were obtained.
Demographic Characteristics of HIV-1–Infected Participants
The NFLG sequences were submitted to a Basic Local Alignment Search Tool to search for more similar sequences, but no sequence with high similarity (>95%) was found. The NFLG sequences collected from this study were further aligned with reference sequences using HIV align tool.
This alignment was then manually edited using BioEdit software. The phylogenetic tree was constructed by the neighbor-joining method based on the Kimura 2-parameter model with 1,000 bootstrap replicates in MEGA6 software. To analyze the recombination structures of the two strains, the sequences were submitted to Recombinant Identification Program (RIP) (
The phylogenetic tree showed that each of the two sequences formed a distinct and well-supported (bootstrap value = 100%) monophyletic cluster, distantly related to HIV-1 reference sequences (Fig. 1). As shown in the recombination analysis result by RIP, both NFLG sequences of HB010014 and HB010063 were composed of subtypes CRF01_AE and CRF07_BC (Fig. 2). The mosaic structure of the HB010014 is composed of nine segments as follows (Fig. 3A): ICRF01_AE (772–1,196 nt), IICRF07_BC (1,197–3,032 nt), IIICRF01_AE (3,033–5,286 nt), IVCRF07_BC (5,287–5,378 nt), VCRF01_AE (5,379–5,735 nt), VICRF07_BC (5,736–6,194 nt), VIICRF01_AE (6,195–6,953 nt), VIIICRF07_BC (6,954–7,564 nt), and IXCRF01_AE (7,565–9,601 nt), using HXB2 as a reference. Phylogenetic analyses of each subregion indicated that both CRF01_AE segments are mainly originated from CRF01_AE cluster 4 and cluster 5 strains prevalent in China (Fig. 4A), which are mainly circulating among the MSM population in China. 6 The same method is used for HB010063, the mosaic structure of the recombinant genome is composed of eight segments as follows (Fig. 3B): ICRF01_AE (633–1,171 nt), II07_BC (1,172–1,840 nt), IIICRF01_AE (1,841–5,089 nt), IV07_BC (5,090–5,666 nt), VCRF01_AE (5,667–6,317 nt), VI07_BC (6,318–8,586 nt), VIICRF01_AE (8,587–9,246 nt), and VIII07_BC (9,247–9,601 nt). Subregion phylogenetic analyses indicated that both CRF01_AE segments are mainly originated from CRF01_AE cluster 4 strains among the MSM population in China (Fig. 4B). Therefore, these recombinant strains are more likely to be formed in MSM. The cocirculation and dual infections of different HIV-1 subtypes are prone to result in the generation of URFs.
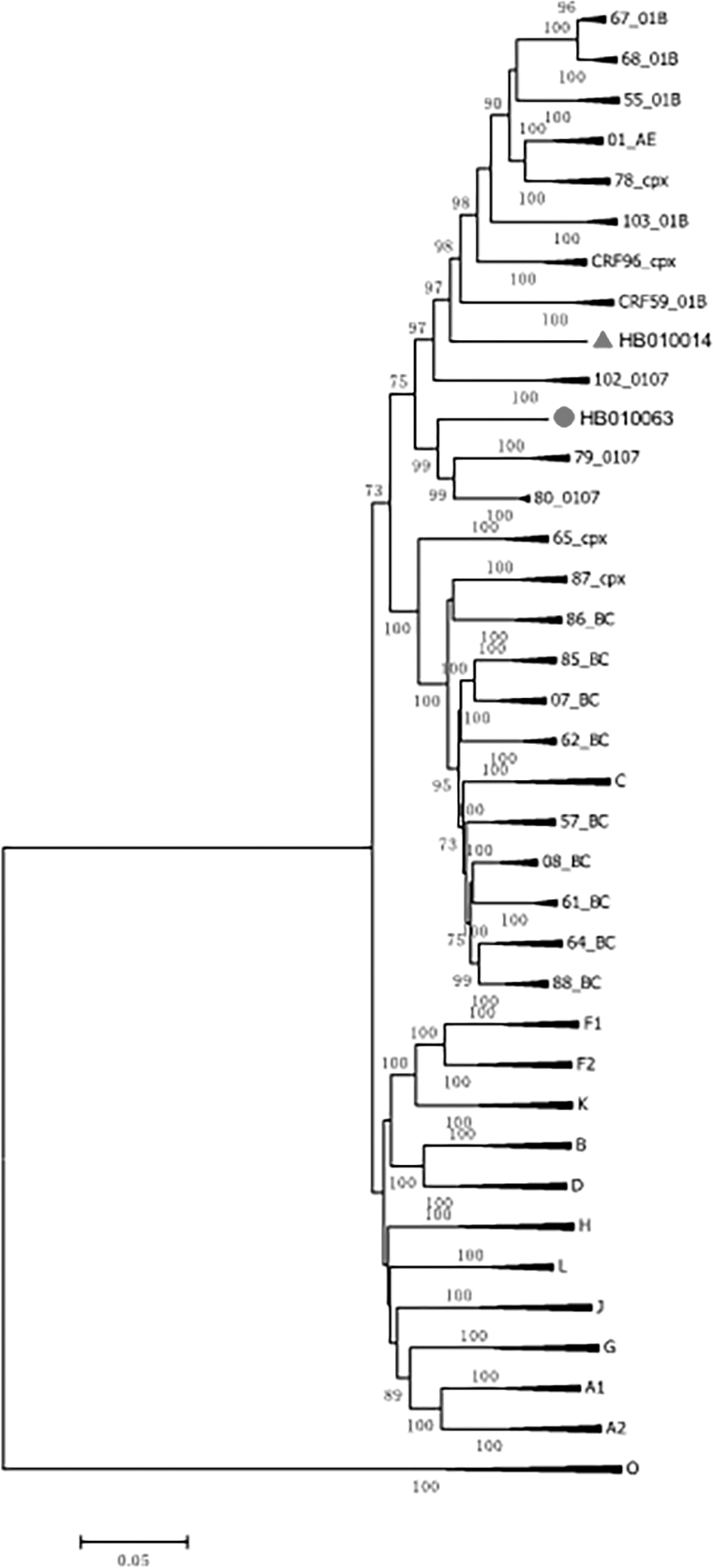
Phylogenetic tree analysis. A neighbor-joining tree was created with full-length genomes of our HB010014 and HB010063 strain sequences and the reference sequences of subtype

RIP was performed for HB010014 and HB010063 to identify parental subtypes. Similarity distance analysis was performed using RIP from the Los Alamos National Laboratory HIV Database with default setting except for the window size of 400. RIP, Recombinant Identification Program.

Genetic maps of HB010014

Subregion phylogenetic tree. Subregion phylogenetic analyses of different segments of HB010014 ) marks HB010014 and HB010063. Bootstrap values ≥70% are shown at the corresponding nodes. The scale bar represents 5% genetic distance. The segment IV of HB010014 and segment VIII of HB010063 were too short to construct neighbor-joining phylogenetic trees.

In summary, we identified two new HIV-1 URFs, the emergence of new complex recombinant forms increases the diversity of HIV-1 epidemic in this region of Hebei China, which indicated the very complexity of the local HIV epidemic.
Sequences Data
The gene sequences of HB010014 and HB010063 were deposited in the GenBank with the accession numbers MW728360 and MW728361, respectively.
Footnotes
Authors' Contributions
L.H. was in charge of article writing, experimental operation, and data analysis. H.L. was responsible for article correction and laboratory procedure. L.W. carried out plasma samples collection. L.J. carried out article correction. J.H. took care of sequence assembly. T.L. carried out RNA extraction. X.W. carried out RNA extraction. Y.L. provided HIV reference sequences. J.L. carried out article correction. J.L. was in charge of sample storage and transport. X.Q. took charge of sample information collection and sorting. X.H. and H.L. performed sample information collection. L.L. provided experimental conditions. E.D. was responsible for experimental design, article correction, and provided experimental samples.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the NSFC (31900157), the state key laboratory of Pathogen and Biosecurity (AMMS), School of Public Health and Affiliated Shijiazhuang Fifth Hospital, North China University of Science and Technology.