
Abstract
In 2019, an outbreak of HIV infection predominantly affecting children occurred in Larkana district, Pakistan. This is the largest outbreak ever reported in this age group in Pakistan. In this study, we report two HIV-1 unique recombinant forms identified during the outbreak. Blood samples were collected from HIV-positive children as part of a case–control study to investigate the outbreak. The pol gene was sequenced and used to detect HIV subtype/recombinant forms using subtype, recombination, and phylogenetic analyses. Drug resistance mutation (DRM) analysis was performed to characterize the DRMs in each sequence. We observed the emergence of two unassigned unique recombinant forms related to CRF36_cpx in 15 individuals of the 344 samples collected. Genotype analysis revealed the presence of multiple DRMs associated with resistance to reverse transcriptase inhibitors. The discovery of these unassigned unique recombinant forms in our population highlights the need for comprehensive molecular epidemiological studies to fully understand the distribution and drug resistance patterns to aid control efforts.
Introduction
HIV-1
HIV infection with multiple strains can occur in an individual with repeated exposure, for example, due to high-risk sexual behavior, and can result in the emergence of new or complex recombinant forms. 4 Over 100 circulatory and unique recombinant forms (CRFs and URFs) of HIV-1 have been identified. Approximately 20% of HIV-1 infections worldwide are caused by URFs, while in some African countries, this proportion is as high as 40%. 5
In Pakistan, HIV-1 currently exists as a concentrated epidemic in certain high-risk groups, such as persons who inject drugs and men who have sex with men, and transgender sex workers (Hijra sex workers), with frequent spillovers into low-risk populations in the form of isolated outbreaks. 6 –9 Molecular epidemiological studies from Pakistan have shown the presence of diverse subtypes and URFs in the country, namely B, C, D, G, A1D, A1G, 01G, CG, 01_AE, CRF02A1, CRF02_AG, and CRF35_AD, while subtype A1 is the predominant subtype. 3,10 In addition to these known subtypes, several new and unique CRFs and URFs have been reported: URF_DG, 11 CRF56_cpx, and CRF02_A1. 8,12
In April 2019, an outbreak of HIV-1 infection predominantly affecting children was reported in Larkana district, Sindh province, Pakistan. This is the largest outbreak ever reported in this age group in Pakistan, where more than 1,000 children tested positive for HIV-1. 13 As part of the investigation of the outbreak in Larkana, epidemiological and molecular studies were carried out. 14
In this study, we report two unassigned URFs related to CRF36_cpx from individuals identified with HIV infection in the outbreak, which possibly emerged from recombination between CRF02_AG and CRF36_cpx and CRF02_AG, CRF36_cpx, and subtype H, respectively.
Methods
As part of a case–control study to investigate the outbreak, blood samples were collected from children aged below 15 years diagnosed with HIV-1 infection (cases) in Larkana and uninfected age- and sex-matched community controls. 15 The samples were collected after obtaining informed consent from the participants and/or the guardians of the participant. 16 A questionnaire was used to obtain demographic and relevant risk factor information from the study participants.
The study was approved by the Aga Khan University Ethics Review Committee (ERC# 2019-1536-4200), and all experiments were performed in accordance with relevant guidelines and regulations. In this study, we focus on two unassigned complex URFs. The findings of the complete molecular epidemiological analysis will be reported separately.
Following DNA extraction from 344 blood samples collected from cases, the pol (covering protease and reverse transcriptase regions) gene of HIV-1 was amplified using two sets of primers using a two-step nested polymerase chain reaction (PCR).
The following two sets of outer primers were used: forward (POLOF CAGCATGYCAGGGAGTRGGRGGACC, nt 1832–1856, HXB2, IBF1 5′-AAATGATGACAGCATGTCAGGGAGT-3′. nt 1823–1847, HXB2) and reverse (IBR1 5′-AACTTCTGTATATCATTGACAGTCCA-3′. nt 3303–3328, HXB2); the first-round product was used as a template in the second round with the primer set, forward (POLIF 5′-AGGCTAATTTTTTAGGGAARATYTGGCCTTCC-3′; nt 2078–2109, HXB2) and reverse (RTOUT3 5′-TATGTCATTGACAGTCCAGCT-3′; nt 3300–3320, HXB2).
PCR Mastermix (ABM) Bestaq (2 × ) and Hotstart (2 × ) were used to prepare a 25-μL reaction mixture with the primer at 0.8 and 0.6 pmol for the first and second rounds, respectively. Thermocycling conditions were as follows: 95°C for 5 min, followed by 40 cycles of denaturation at 95°C for 1 min, annealing at 50°C for IBF1/IBR1 and 55°C for POLOF/IBR1 sets (round 1), 60°C (round 2) for 20 s, and extension at 72°C for 1 min with a final extension at 72°C for 7 min. For confirmation/validation, each run included positive and negative controls. The amplicons were subsequently sequenced on a Sanger sequencing platform (Macrogen, Korea).
The sequences (n = 15) were submitted to GenBank and were assigned the following accession numbers: MN698251, MN698252, MN698253, MN698255, MN698256, MN698257, MN698258, MN698259, MN698260, MN698261, MN698262, MN698263, MN698264, MN752136, and MN752137.
The obtained pol sequences were subtyped using the REGA HIV-1 subtyping tool, while the jpHMM tool was used to detect recombinations and recombination breakpoints within the pol gene of the HIV-1 genome. 12,13,15,17 The strain identity was investigated using phylogenetic analysis. For this, a multiple sequence alignment (MSA), containing outbreak sequences (ID: AKULO) as well as the HIV-1 subtype reference obtained from the Los Alamos HIV Sequence Database, was generated using the MAFFT program. 18,19
For one sequence (AKULO_387), for which two recombination breakpoints were predicted by the jpHMM tool, two separate MSAs were generated. The first and second MSAs of the AKULO_387 sequence comprised split nucleotides spanning positions 2236–3192 and 3193–3313 (with reference to the HXB2 genome), respectively. These two MSAs were aligned separately with the HIV-1 subtype reference obtained from the Los Alamos HIV Sequence Database using the MAFFT online MSA tool.
Subsequently, all three MSAs were used to generate individual maximum likelihood phylogenetic trees using PhyML software 3.0 (
Results
Of the 344 samples from cases, unassigned URFs were observed in 15. The median age of the 15 participants was 2.8 years (range: 0.8–9 years) and 33% were female. At the time of sample collection, two participants were ART-naïve, and the remainder had only recently started ART (Table 1). Subtyping analysis identified the 15 sequences as CRF02_AG with undefined recombination related to CRF36_cpx.
Data and Drug Resistance Mutation Profiles of 15 Individuals
Antiretroviral therapy drugs affected by mutations are shown as superscripts.
LNZ: lamivudine, nevirapine, and zidovudine. Nucleoside reverse transcriptase inhibitors (NRTIs): emtricitabine (FTC), lamivudine (3TC), abacavir (ABC), tenofovir disoproxil fumarate (TDF), and zidovudine (AZT). Non-nucleoside reverse transcriptase inhibitors (NNRTIs): efavirenz (efv), nevirapine (nvp), etravirine, 26 rilpivirine (rpv), and doravirine (dor).
Key: Bold font, high-level resistance; *intermediate-level resistance; ^low-level resistance; and ◊potential low-level resistance. Different drug resistance levels are separated by the “/” sign. The “—” sign indicates the absence of any DRM.
On analysis, 14 sequences clustered with CRF36_cpx sequences submitted from Cameroon (Accession Nos.: GU366128, EF087994, and EF087995) and CRF02_AG sequences submitted from Liberia and Nigeria (Accession Nos.: AB485636 and L39106, respectively) (Fig. 1A). These 14 sequences exhibited a strong node value of 0.87 (Fig. 1A), indicating that these sequences are an unassigned recombinant form related to CRF36_cpx, possibly emerging from recombination between CRF02_AG and CRF36_cpx.
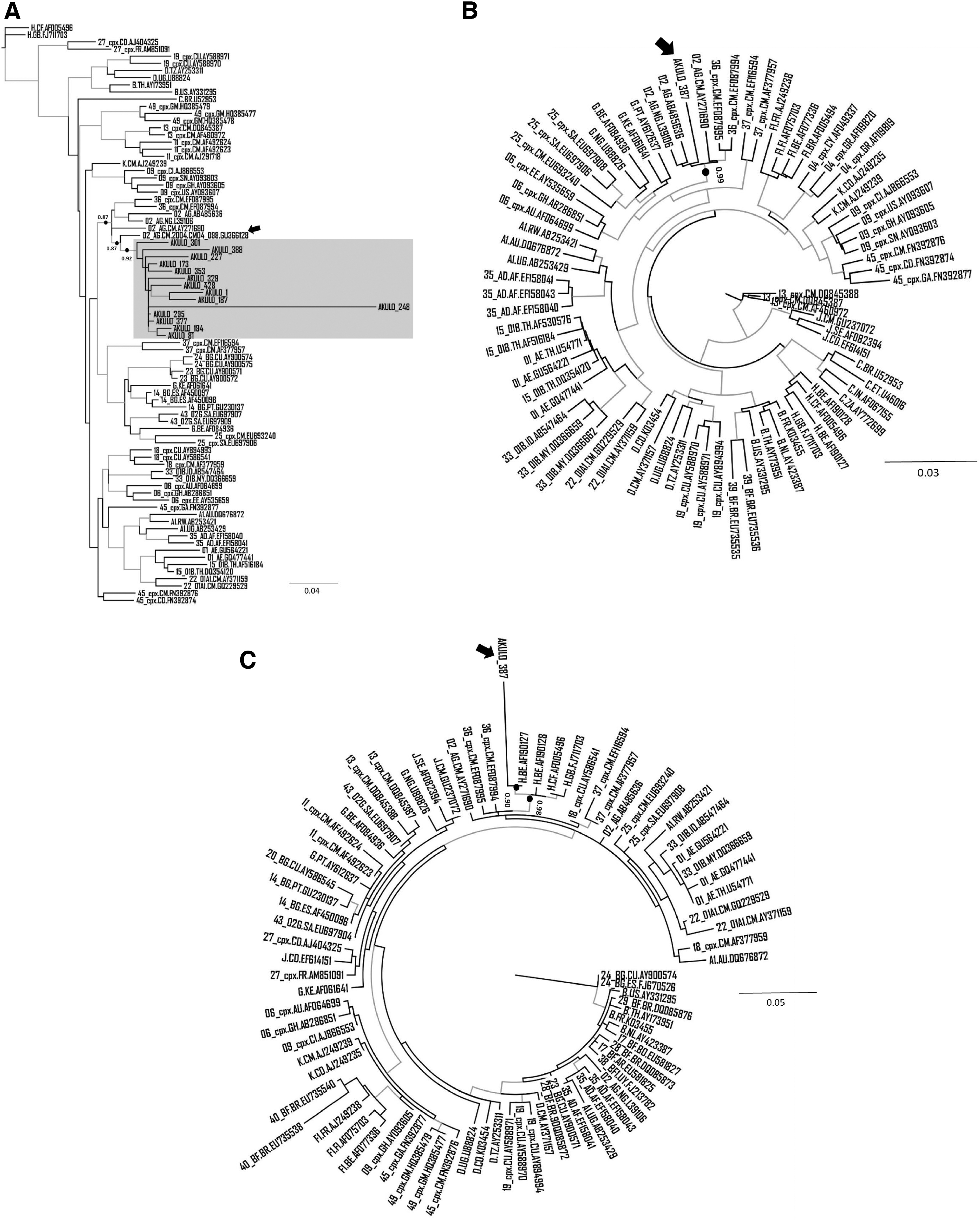
Phylogenetic analysis of unassigned URFs:
The Cameroonian URF (Accession No.: GU366128) most closely clustering with the 14 sequences generated in our study (Fig. 1A, black arrow) was previously reported in a 2004 cohort study where the authors identified this strain as CRF02_AG containing the 36cpx recombination at the 5′end of the pol region. 20 Interestingly, when we included this sequence in our phylogenetic tree, it became part of the larger cluster (node support value 0.87) that contained outbreak sequences as well as CRF02_AG and CRF36_cpx sequences (Fig. 1A), further supporting that the 14 outbreak sequences are unassigned URFs related to CRF36_cpx.
For the remaining sequence (ID: AKULO_387), the phylogenetic analysis was performed using the two MSAs developed on the breakpoints predicted by jpHMM. On the phylogenetic tree, based on the first MSA, this sequence clustered with CRF02_AG submitted from Cameroon, Nigeria, and Liberia (Accession Nos.: AY271690, L39106, and AB485636, respectively) and CRF36_cpx submitted from Cameroon (Accession Nos.: EF087994–EF087995) with a node support value of 0.99 (Fig. 1B).
Similarly, on the phylogenetic tree, based on the second MSA, the sequence clustered with subtype H submitted from Belgium (Accession No.: AF190127) with a node support value of 0.90 (Fig. 1C), indicating that the strain is a subtype H-like unassigned complex URF related to CRF36_cpx.
The branch length of the sequence in one of the 14 samples (ID: AKULO_248) in the cluster (Fig. 1A) suggested the presence of additional recombination(s) that was not predicted by any tool. The phylogenetic analysis of this sequence showed clustering with CRF18_cpx in addition to CRF02_AG and CRF36_cpx (Fig. 2), suggesting additional recombination; however, this warrants further investigation.

Phylogenetic analysis of unassigned URF in AKULO 248: ML phylogenetic tree showing the AKULO_248 sequence (red color). The blue-colored branches represent the aLRT SH-like support values ≥0.80. The AKULO_248 sequence is shown by the black arrow.
On genotypic analysis, 6 of the 15 (40%) sequences contained mutations associated with resistance to antiretroviral drugs (Table 1): the T215N, M184V, and Y115F mutations are associated with resistance to nucleoside reverse transcriptase inhibitors (NRTIs); and the K103N, Y181C, A98G, and V179L mutations are associated with resistance to non-nucleoside reverse transcriptase inhibitors (NNRTIs). No protease inhibitor resistance mutations were observed. 19
A similar analysis of the reference sequences that clustered with our outbreak sequences (Fig. 1) showed that none of the reference sequences harbored any drug resistance mutation (DRM), except the subtype H sequence (Accession No.: AF190127; Fig. 1C) that had DRMs (D67N, K70R, and K219Q) associated with resistance to all NRTIs.
Discussion
The current study reports the detection of novel unassigned URFs related to CRF36_cpx in children identified with HIV-1 during the outbreak in Larkana. CRF 02_AG is one of the main circulating strains in Pakistan; however, the current analysis suggests that it is recombining with other subtypes/CRFs, leading to the emergence of subtype H-like or CRF36_cpx-related URFs. CRF36_cpx is limited to Cameroon and has been found to recombine with CRF02_AG. 20
Subtype H has only been reported in Central Africa and the United Kingdom. 19,21,22 Recombination of subtype H with multiple other subtypes and CRFs has been observed, including recombination with CRF04_cpx and CRF27_cpx and complex recombination with U/CRF02_AG. 19,23
One limitation of this study is the use of the pol sequence only for assignment of the recombinant form. Analysis based on longer regions of the HIV-1 genome or whole genome, which could not be done due to funding constraints, would have been useful to accurately determine the recombinant form and region(s) of recombination. Nonetheless, our analysis supports the presence of subtype H-like or CRF36_cpx-related URFs, which suggest that the HIV epidemic in Pakistan is much more dynamic and complex than previously thought. Some of these strains may become more adaptive and emerge as major forms in the future.
Genotypic analysis of sequences suggested the presence of DRMs that cause resistance against multiple reverse transcriptase inhibitors. Three of 13 ART-experienced individuals harbored mutations associated with high-level resistance to NRTI and/or NNRTI DRMs (M184V, K103N, Y115F, and Y181C). Two of the 13 individuals also had mutations, T215N and Y179L, associated with resistance against zidovudine, and abacavir and tenofovir, respectively, which are used as the first-line regimen in Pakistan. The mutation, T215N, is a revertant mutation; the presence of this mutation suggests that the individual was previously infected with or had acquired HIV-1 in an area where the majority population had T215Y/F (a highly resistant NRTI mutation). 9
Similarly, the mutation, A98G (observed in two individuals), is associated with resistance to multiple NNRTIs. No DRMs were observed in ART-naïve individuals. While all but two individuals were ART-experienced, the remainder had started ART recently (mean ART duration was 1 month; range 8 days to 3 months). Therefore, it is unlikely that these mutations emerged as a result of suboptimal adherence and may have emerged due to transmitted drug resistance. 24,25 This may have possible implications for the treatment and control of HIV in Pakistan.
The presence of multiple DRMs in these strains, especially to first-line ART drugs, is alarming as it limits treatment options. Large-scale transmission of resistant strains can hamper efforts to control the spread of the HIV epidemic in Pakistan, where second-line drugs are not easily available.
The discovery of multiple URFs/CRFs in this outbreak highlights the need for comprehensive molecular epidemiological studies and molecular surveillance to understand the distribution of different genotypes as well as their origin, transmission, and drug resistance patterns. This will inform the appropriate treatment of individuals with HIV and strategies for preventing further outbreaks and controlling the spread of the HIV epidemic in the country.
Footnotes
Authors' Contributions
S.H.A., D.S., and R.A.F. performed the experiments, analyzed the data, and wrote the article. S.F.M., R.S., S.A.S., S.H.A., R.A.F., P.K., and F.M. were involved in the outbreak investigation. D.S. and A.A.N. cleaned the data and prepared the final datasheets. S.H.A., R.A.F., and F.M. conceived the study. S.H.A. and D.S. made equal contributions to all experiments and data analysis, hence they are cofirst authors of the article. R.A.F. and F.M. made an equal contribution to project supervision, coordination of sample collection, and conception of the study, hence they share senior authorship of the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The study was funded by the Higher Education Commission, Grant No. 5217/Sindh/NRPU/R&D/HEC/2016; Pakistan Science Foundation, Grant No. PSF/Res/S-AKU/Med (488); and World Health Organization, Grant No. 2019/969219-0. R.A.F. is funded by the Wellcome Trust through a Senior Fellowship in Clinical Science (206316/Z/17/Z).