
Abstract
The prevalence of recombinant forms has greatly enhanced HIV-1 genetic diversity. Under co-circulation of major epidemic HIV-1 strains (CRF01_AE and CRF07_BC) in China, more CRF01_AE and CRF07_BC as the backbone of HIV-1 second-generation recombinants (SGRs) are also emerging. In this study, we identified three similar novel HIV-1 SGR strains composed of CRF01_AE and CRF07_BC from HIV-1 positive individuals in Shenzhen, China. Near full-length genome phylogenetic and recombinant analysis confirmed that these unique recombination forms were CRF01_AE and CRF07_BC strains recombined. Further subregion phylogenetic analysis indicated that all CRF01_AE fragments were from CRF01_AE cluster 4 prevalent among men who have sex with men, and all subtype B and C fragments derived from CRF07_BC. The emergence of novel recombinants of CRF01_AE/CRF07_BC indicates the increased genetic diversity of the HIV epidemic in Shenzhen. It is necessary to monitor HIV-1 SGR strains among high-risk populations for the epidemic dynamics of HIV-1 in Shenzhen, China.
The high incidence of dual infection leads to the frequent occurrence of HIV-1 recombination, resulting in increasing novel circulating recombinant forms (CRFs) and unique recombination forms (URFs), which promotes HIV-1 genetic polymorphism to a higher level.
1
Over time, the HIV-1 M group, as the most popular HIV-1 group in the world, has become increasingly complex. It is reported that 10 pure subtypes (A–D, F–H, and J–L) and 118 CRFs have been identified among the HIV-1 M group worldwide so far, all of which are included in the Los Alamos National Laboratory HIV Sequence Database (
Of these, 36 CRFs were identified in China, and nearly half of them (15/36) were reported from China since 2018. In China, the HIV epidemic is dominated by HIV recombination strains, and CRF01_AE and CRF07_BC strains have played a huge role in both the national and regional HIV epidemic in China. 2 At the same time, more and more URFs, especially HIV-1 second-generation recombinant (SGR) strains, have been constantly appearing, hinting that HIV-1 recombination is active in China. 3 –5
Shenzhen is a first-tier city located in Guangdong Province of southern China. As China's first special economic zone, Shenzhen is also one of the HIV epidemic hotspots in China. It has a population of 13.438 million, 63.18% of whom are nonlocal residents with registered residence in other areas. 6 Owing to rapid economic growth, large population movements, and a relatively tolerant social atmosphere, the city attracts men who have sex with men (MSM) from different regions of China. The MSM population plays an important role in the rapid changes of HIV-1 main epidemic strains in Shenzhen.
CRF01_AE (42.93%) and CRF07_BC (33.98%) were the main epidemic strains in Shenzhen. In 2018, CRF07_BC (43.50%) has exceeded the prevalence of CRF01_AE (33.24%). 7 Both two strains are undergoing adaptive evolution in the HIV epidemic in Shenzhen. In addition, the prevalence of other subtypes and other recombinant strains also provides more ideal conditions for the emergence of new recombinant strains. In this study, three HIV-1 novel recombinant strains composed of CRF01_AE and CRF07_BC with similar recombination patterns were identified from three epidemiologically unlinked HIV-1 positive individuals based on the HIV-1 near full-length genome (NFLG) sequences.
All three HIV-positive patients were from Shenzhen, China. The patient LS11585 was a 43-year-old married male who was infected through MSM. The patient LS12824 was an unmarried male aged 49 years who was also infected through MSM. The patient LS14734 was an unmarried male aged 39 years who was infected through injection drug use (IDU). Their peripheral blood sampling years were May 8, 2014, August 26, 2014, and February 27, 2015, respectively, and all the patients were newly diagnosed at that time. Additional information about the patients are shown in Table 1. All participants signed informed consent before sample collection.
Demographic Characteristics of Study Subjects Infected with HIV-1
IDU, injection drug use; MSM, men who have sex with men.
Viral RNAs were extracted from plasma samples using the High Pure Viral RNA Kit (Roche, Germany). Then, the cDNA was reverse transcribed utilizing the SuperScript™ IV First-Strand Synthesis System (Invitrogen). The NFLGs were amplified in two halves with 1 kb overlapping region by nested polymerase chain reaction (PCR) with Platinum™ Taq DNA Polymerase High Fidelity (Invitrogen) as described before. 8 After PCR positive products were purified and sequenced by SinoGenoMax Company (Beijing), all sequence fragments were assembled by using the ContigExpress software. Finally, NFLGs of LS11585 (8821 bp, HXB2:774nt-9601nt), LS12824 (8775 bp, HXB2:772nt-9601nt), and LS14734 (8838 bp, HXB2:772-9613nt) were obtained.
These NFLG sequences were submitted to the Basic Local Alignment Search Tool (BLAST) analysis for searching the highest similarity (>95%) sequences, but no sequence was found. Then, they were further aligned with HIV-1 reference sequences (HIV-1 M Group: pure subtypes and main CRFs in China, HIV-1 O Group) obtained from the Los Alamos National Laboratory HIV Sequence Database through MAFFT version 7 online and then edited by BioEdit7.2.5. The NFLGs maximum-likelihood (ML) phylogenetic tree was constructed under the General Time Reversible and Gamma distributed with Invariant sites (GTR+G+I) model with SH-like aLRT values by phyML 3.0. 9
To determine the recombination structures and recombination breakpoints, three NFLG sequences were submitted to Recombinant Identification Program (RIP,
The ML phylogenetic tree showed that these HIV-1 URF strains formed a well-supported (bootstrap value = 1) monophyletic cluster, distantly related to all HIV-1 reference sequences (Fig. 1). Preliminary recombination analysis by jpHMM showed each of the three NFLG sequences probably composed of CRF01_AE, Subtype B, and C (Fig. 2A). To verify whether the fragments of subtype B and C are derived from CRF07_BC or the pure subtypes (subtype B and C), we conducted further recombination analysis with RIP, and the results confirmed that all the B/C fragments were derived from CRF07_BC (Fig. 2B).

ML phylogenetic analysis based on three HIV-1 URFs NFLGs. Three HIV-1 URFs NFLGs were marked with a solid circle, and the corresponding sequence label was also marked with a light grey background. The ML tree was constructed by phyML3.0 based on the General Time Reversible and Gamma distributed with Invariant sites (GTR+G+I) model with SH-like aLRT values. HIV-1 reference sequences were HIV-1 M Group (pure subtypes and CRFs in China) and HIV-1 O Group obtained from the Los Alamos National Laboratory HIV Sequence Database. Only bootstrap values ≥0.9 are presented at the nodes of the tree. CRFs, circulating recombinant forms; ML, maximum-likelihood; NFLGs, near full-length genomes; URFs, unique recombination forms.
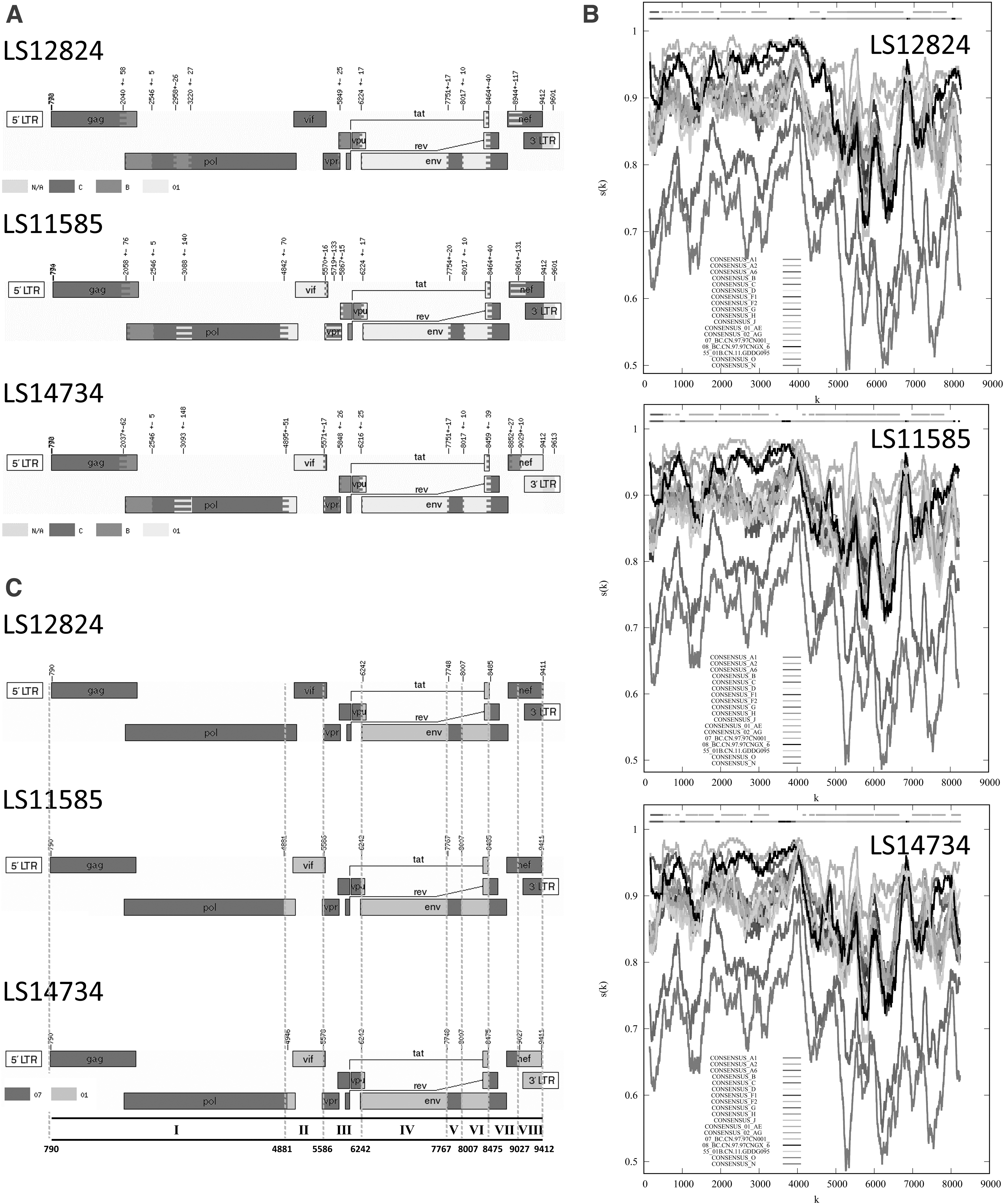
Recombinant analysis based on three HIV-1 URFs near full-length genomes.
As shown in the recombination structure pattern (Fig. 2C), all the three NFLG sequences (Patient ID: LS11585, LS12824, and LS14734) are composed of CRF01_AE and CRF07_BC, and contain 7, 5, and 8 recombination fragments, respectively. Their recombination breakpoints at HXB2 positions were as follows: 790nt, 4881nt, 5586nt, 6242nt, 7767nt, 8007nt, 8485nt, and 9412n for LS11585; 790nt, 6242nt, 7748nt, 8007nt, 8485nt, and 9412nt for LS12824; 790nt, 4946nt, 5578nt, 6242nt, 7748nt, 8007nt, 8475nt, 9027nt, and 9412nt for LS14734.
Interestingly, there are very similar recombination breakpoints between each of the three NFLGs in the region from 6242nt to 8485nt. To further analyze the recombinant subregion, we divided the URF gene maps into eight recombinant regions (Fig. 2C) as follows (according to HXB2 positions): I: 790-4880nt, II: 4881-5585nt, III: 5586-6241nt, IV: 6242-7766nt, V: 7767-8006nt, VI: 8007-8474nt, VII: 8475-9026nt, VIII: 9027-9412nt. Subregion phylogenetic analysis showed that all the CRF01_AE segments were from the CRF01_AE cluster 4 strains mainly prevalent in the MSM population in China (Fig. 3). 10 Therefore, all the CRF01_AE recombinant segments are most likely originated from the MSM population. All the CRF07_BC regions are mainly clustered with the CRF07_BC reference sequences (Fig. 3).

Subregion tree analysis of three HIV-1 URFs recombinant gene segments. The subregion neighbor-joining tree was constructed by the Kimura 2-parameter model of nucleotide substitution with 1,000 bootstrap replicates in MEGA v6 and visualized by iTOL. CRF01_AE segments are mainly distributed in segment IV (6242-7766nt) and VI (8007-8474nt). CRF07_BC segments are mainly distributed in segment I (790-4880nt), III (5586-6241nt), V (7767-8006nt), and VII (8475-9026nt).
Combining the background information of these patients in Table 1, all three patients are males with high-risk factors (MSM or IDU). Therefore, with the co-circulation of CRF01_AE and CRF07_BC, and high-risk factors acting as bridges, more and more second-generation recombination strains emerge and even may lead to new trends of the HIV epidemic in China in the future.
In short, our research discovered three URFs with very similar NFLG recombinant forms. All of them were SGR strains composed of CRF01_AE and CRF07_BC recombinant strains. The emergence of these recombinant strains suggests that the predominant HIV strains in China such as CRF07_BC and CRF01_AE are undergoing adaptive changes and evolution. At the same time, the genetic diversity of HIV-1 has been continuously improved. This will interfere with the prevention and treatment of HIV infection. The monitoring for HIV prevalence in Shenzhen should be strengthened, especially for the high-risk populations such as MSM and IDUs with specific recombinant strains, and this will make local AIDS prevention and control more directly and effectively.
Sequences Data
The gene sequences of LS11585, LS12824, and LS14734 were deposited in the GenBank with the accession numbers OL314397, OL314398, and OL314396, respectively.
Footnotes
Authors' Contributions
B.Z., J.H., and L.L. designed the study; B.Z. and J.Z. performed the experiments; J.Z, C.Z., and L.C. collected samples; H.L., Y.L., and B.H.Z. participated in sample storage, sequences assembly, and provided HIV reference sequences; L.J., T.L., X.L.W. collected the demographic data; B.Z., X.R.W., J.L., J.H., and L.L. participated in the article correction.
Acknowledgments
The authors thank all of the participants and peer workers.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the National Key Research and Development Program of China (2020YFA0907000), the National Nature Science Foundation of China (NSFC: 81773493, 31800149, and 31900157), the State Key Laboratory of Pathogen and Biosecurity (AMMS), the National 13th Five-Year Grand Program on Key Infectious Disease Control (2018ZX10721102 and 2018ZX10732101-001-003).