
Abstract
In recent years, men who have sex with men (MSM) have been identified as the primary source of HIV-1 transmission in Hebei Province, China. Co-circulation of multiple subtypes in HIV-1 positive MSM populations may contribute to the emergence of the second generation of recombinant HIV-1 strains, indicating the occurrence of dual infections or superinfections in MSM populations. Thus, the discovery of new recombinant strains is important to indicate the appearance of multiple infected individuals and the prevalence caused by changes in the parent strains. In this study, we present two new unique recombinant forms (URFs) from two HIV-1-positive subjects (HB070052 and HB070056) infected through homosexual contact in Hebei Province, China. The near full-length genome of the two URFs revealed that HB070052 was divided into seven segments by six breakpoints in the gag, pol, vif, and vpr genes; HB070056 was separated into five fragments by four breakpoints, with two regions of CRF07_BC inserted into a CRF01_AE backbone's gag, pol regions. The subregion tree showed CRF01_AE segments were traced back to the cluster 4 and 6 of the CRF01_AE phylogenetic tree, which were prevalent among HIV-1 infections through MSM in China. The continued emergence of the novel CRF01_AE/CRF07_BC recombinant forms indicates the HIV-1 epidemic is complex and long-term surveillance of recombinant strains is necessary among MSM in this region.
Introduction
Since the first case of HIV/AIDS was identified in the 1980s, the rapid replication and high mutation rate of HIV type 1 (HIV-1) has led to an epidemic of strains of multiple subtypes and their recombinant forms. There are four groups of HIV-1: M, N, O, and P, respectively. M group, consisted of 10 subtypes (A, B, C, D, F, G, H, I, J, K, and L), was more popular than others and caused global HIV-1 pandemic. Meanwhile, the recombination of different subtypes formed a large number of circulating recombinant forms (CRFs) and unique recombinant forms (URFs). 1 Up to now, 132 CRFs and a large number of URFs have been reported. 2
Hebei Province is a northern Chinese province that surrounds Tianjin and Beijing. The HIV-1 transmission mechanism and the level of social development in the surrounding areas will affect the HIV-1 prevalence in Hebei due to its unique geographical location. The first case of HIV-1 infection in Hebei Province was reported in 1989, and it soon became an epidemic, mainly through sexual transmission. In Hebei, the first recombinant form (CRF07_BC) was found in 2002. 3 The first recombinant form of CRF01_AE and CRF07_BC was found in 2008, 4 and URFs were found in 2011. 5 CRF07_BC, CRF01_AE, and B subtypes were the most common genotypes. 6 HIV-1 was introduced into the men who have sex with men (MSM) population in Hebei from neighboring metropolises in recent years and quickly spread among the population.
The proportion of MSM among newly reported HIV-1 infections increased from 4.9% in 2005 to 62.6% in 2013. 7 MSM has obviously become the main population of HIV-1 infection in Hebei. Furthermore, high-risk sexual behaviors in MSM caused the AIDS epidemic and increased HIV-1 genetic diversity, which may lead to a high level of recombination. 8 A previous study published in 2019 found that 77.5% of HIV-1 infections in Hebei were MSM, with 49.6% infected with CRF01_AE strains and 29.7% infected with CRF07_BC strains. 9 As a result, the co-circulation and dual infections of CRF01_AE and CRF07_BC in MSM will certainly facilitate the emergence of the second-generation recombinant strain. Besides, new recombinants are not inferior to other HIV-1 M strains in pathogenicity and therapeutic efficacy. The appearance of new recombinants suggests that dual infections or superinfections occur in MSM.
In this study, two new URFs derived from CRF01_AE and CRF07_BC in MSM from Hebei Province were detected by near full-length genome (NFLG) sequence analysis. Plasma samples were collected from two HIV-1 positive individuals (HB070052 and HB070056) of 867 samples infected through homosexual contacts in Tangshan, Hebei. The patients HB070052 and HB070056, a 31-year-old male and a 62-year-old male, were both diagnosed as HIV-1 positive in October 2020 The NFLG sequences of HB070052 and HB070056 were amplified based on subtype inconsistencies in gag, pol, and env refer to Table 1.
Demographic Characteristics of HIV-1-Infected Participants
MSM, men who have sex with men.
The RNA was extracted from 200 μL plasma samples, then complementary DNA was reverse transcribed from the extracted RNA using the Superscript IV First Strand Synthesis System (Invitrogen). After that, both halves of the NFLG were amplified by nested PCR using ExTaq reagent (TaKaRa). Two rounds of PCR were performed under the following conditions: 94°C for 5 min, 94°C for 30 s, 60°C for 30 s, 72°C for 6 min for a total of 30 cycles, and 72°C for a 10 min extension. PCR products were detected by 1% agarose gel electrophoresis, then purified and sequenced by SinoGenoMax (China) with a series of special primers.
In addition, chromatographic data were assembled by ContigExpress software (a component of Vector NTI version 11.5.1; Invitrogen). Then, the two NFLG sequences were aligned with subtype reference sequences and the CRFs' sequences in China (
Furthermore, the phylogenetic tree and subregion tree were constructed using the neighbor-joining method based on the Kimura two-parameter model with 1,000 bootstrap replications by Mega6. Recombination form and breakpoints were identified by the Recombination Identification Program (RIP) (

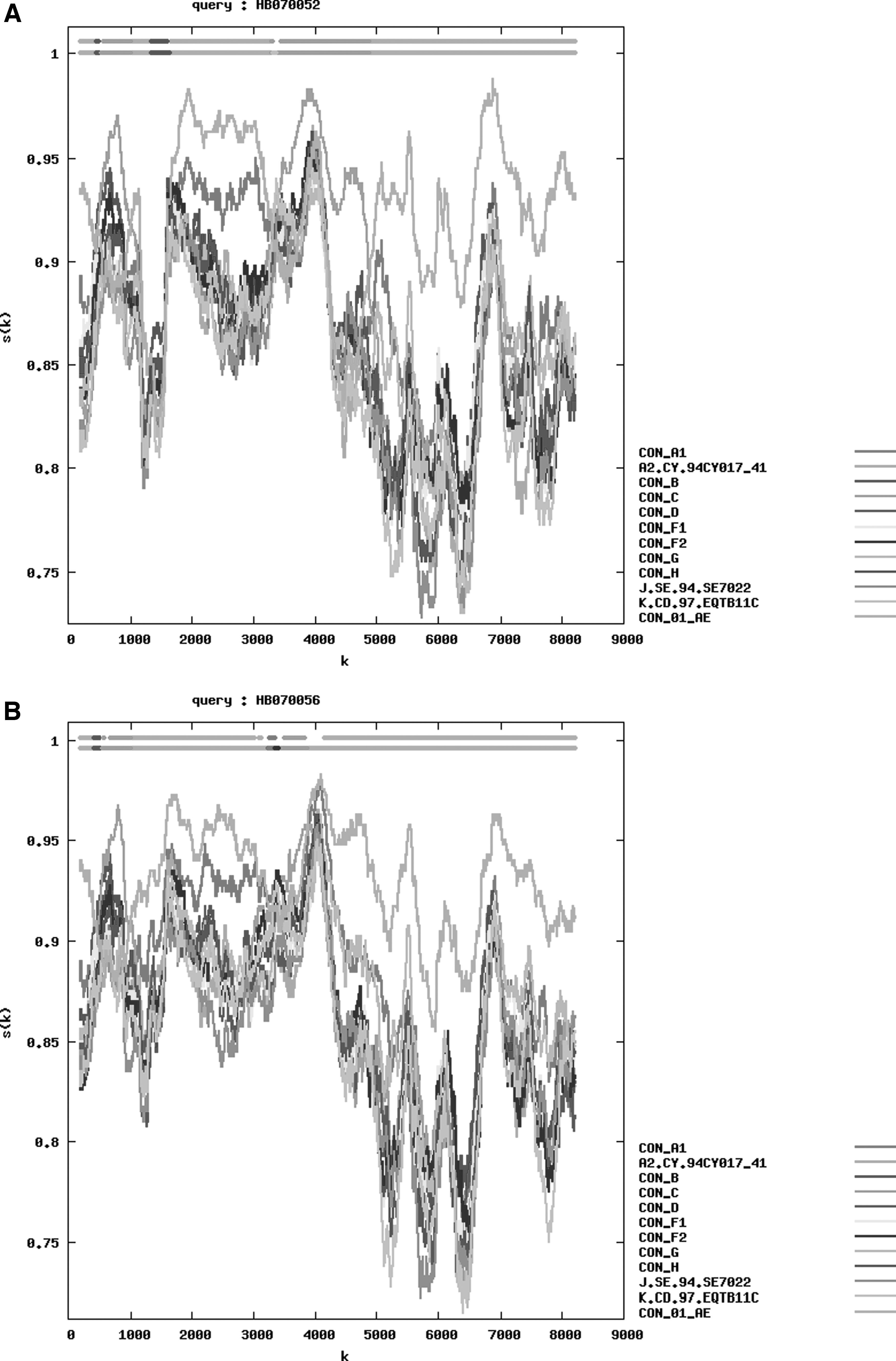
Phylogenetic tree analysis. A neighbor-joining phylogenetic tree of HB070052 (8,771 bp, ●) and HB070056 (8,937 bp, ▲) was constructed based on the NFLG sequences using Mega6.0 with 1,000 bootstrap replications. Bootstrap values ≥90% were shown at the corresponding nodes. The scale bar represents 5% genetic distance. CRF, circulating recombinant form; NFLG, near full-length genome.
The analyses of RIP, jpHMM, and Boostcan demonstrated that both NFLG sequences were composed of CRF01_AE and CRF07_BC. Three CRF07_BC fragments inserted into the CRF01_AE backbone in the NFLG of HB070052. HB070056 was made up of three CRF01 AE fragments inserted with CRF07 BC (Figs. 2 and 3). Subsequently, the mosaic recombinant structure of two sequences was described as follows: ICRF01_AE (HXB2, 776–1,197 nt); IICRF07_BC (HXB2, 1,197–1,858 nt); IIICRF01_AE (HXB2, 1,858–2,065 nt); IVCRF07_BC (HXB2, 2,065–2,462 nt); VCRF01_AE (HXB2, 2,462–4,138 nt); VICRF07_BC (HXB2, 4,138–5,750 nt); VIICRF01_AE (HXB2, 5,750–9,511 nt), HB070052; ICRF01_AE (HXB2, 776–1,177 nt); IICRF07_BC (HXB2, 1,177–1,854 nt); IIICRF01_AE (HXB2, 1,854–4,044 nt); IVCRF07_BC (HXB2, 4,044–4,583 nt); VCRF01_AE (HXB2, 4,583–9,513 nt); HB070056 (Fig. 4).
RIP analysis of the NFLG sequence of HB070052
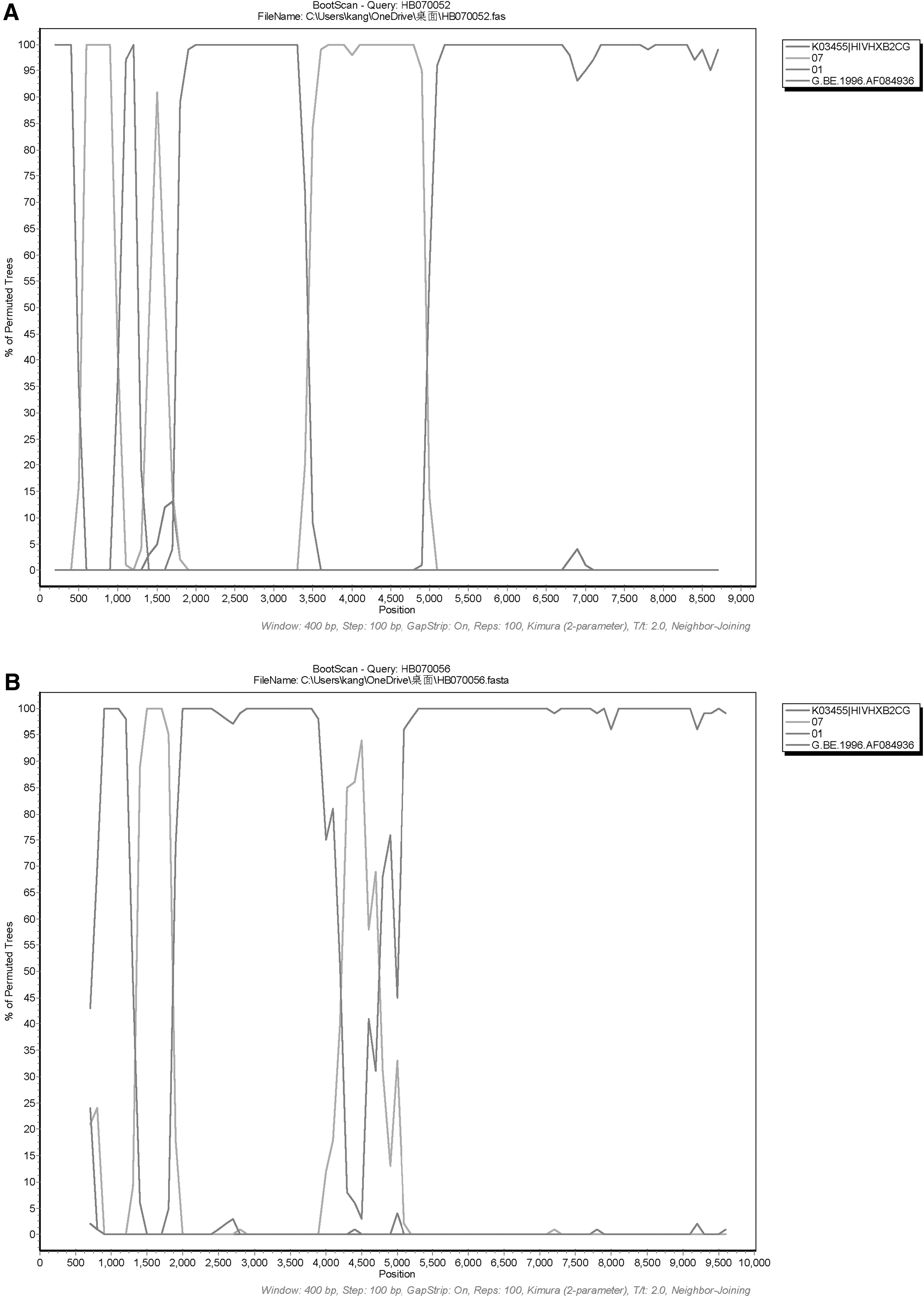
Bootscanning analysis of the NFLG sequence of HB0700052

Genetic maps of HB070052
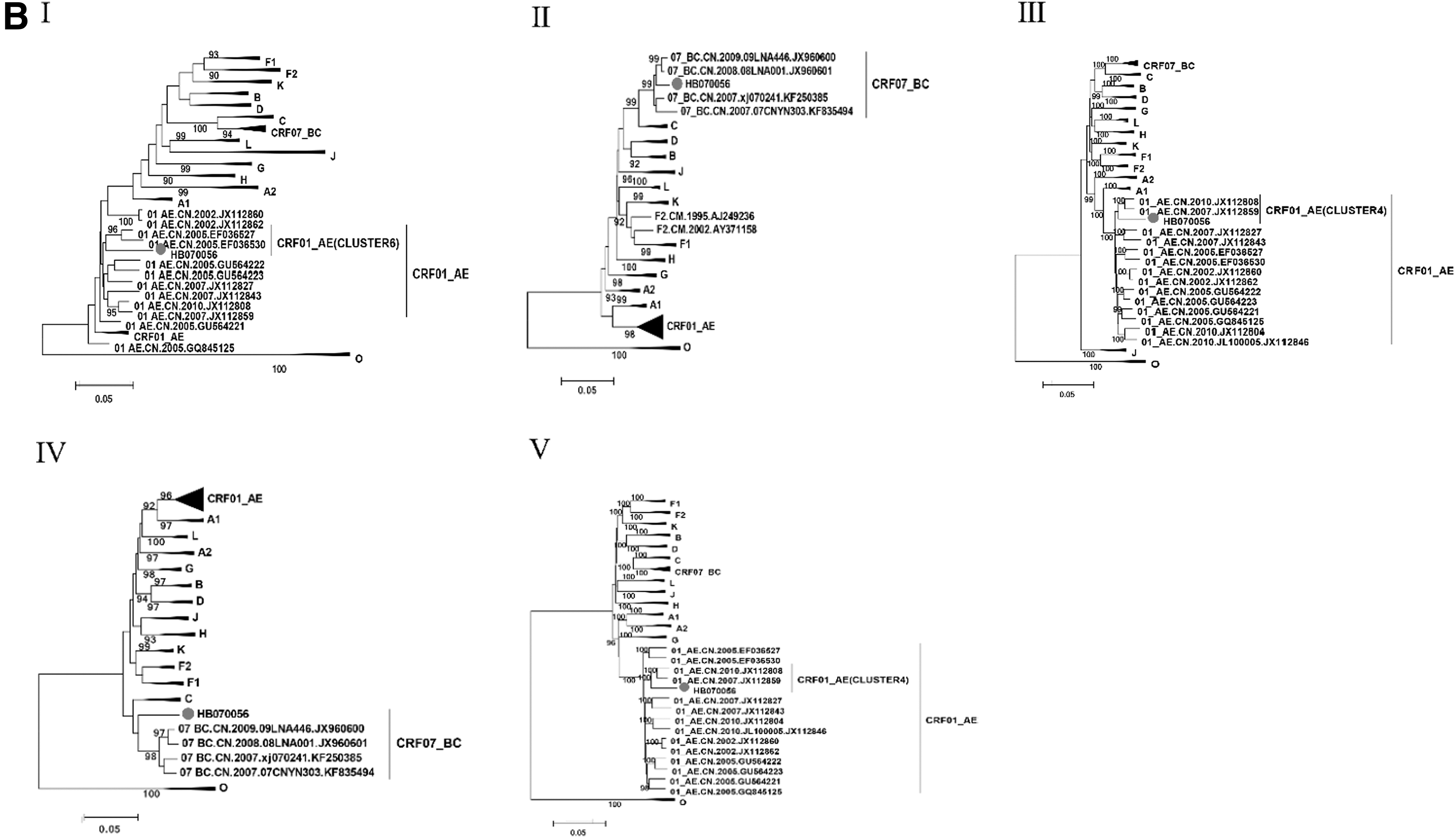
Subregion tree analysis of two NFLGs revealed that the parental origin of all CRF01_AE fragments was from the MSM-related CRF01_AE cluster 4 linkage and cluster 6 linkage (Fig. 5). CRF07_BC reference sequences clustered with CRF07_BC regions in HB070052 (II, IV, and VI) and HB070056 (II and IV). In conclusion, the parental origins of the two novel second-generation URFs were CRF01_AE and CRF07_BC.

Subregion phylogenetic tree. Subregion phylogenetic analysis of different segments of HB070052
In this study, we reported two novel CRF01_AE/CRF07_BC recombinant forms from MSM in Hebei Province, China. MSM has become the main source of HIV-1 transmission in Hebei, and the diversity of HIV-1 among MSM is more complex than in other provinces. 10 A previous study found that the URF, composed of CRF01_AE and CRF07_BC, is the most common second-generation recombination modes of HIV-1 in MSM. 11
Therefore, the two novel URFs in Hebei Province provided the complexity of high-risk behaviors and HIV-1 genetic diversity in MSM. It is critical to continuously survey the prevalence of recombinant HIV-1 subtypes among MSM to control and prevent the HIV-1 epidemic in this region. Although the collected information indicates that the patients were infected through homosexual contact, the actual cause of the infection may still be open to question. Currently, we are developing a sequence analysis-based method to identify the real source of the infected strains, distinguish whether they are from the MSM populations or not, and this may be helpful for finding the most appropriate strategy to interrupt the local transmission of the strains.
Sequences Data
The gene sequences of HB070052 and HB070056 were deposited in the GenBank with the accession numbers ON529529 and ON529530, respectively.
Footnotes
Authors' Contributions
Article writing, experimental operation, and data analysis by Z.L. Experimental operation and data analysis by Q.K. RNA extraction by X.W. Y.L. provided HIV reference sequence. Article correction by L.J. Sample information collection and sorting by T.L. Sample storage and transport by J.L. B.Z. conducted HIV confirmatory tests. Article correction and laboratory procedure by J.H. Experimental design, article correction, and providing experimental samples by H.L. L.L. provided experimental conditions.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the NSFC (Grant Nos. 81773493 and 31800149), the State Key Laboratory of Pathogen and Biosecurity (AMMS), National Grand Program on Key Infectious Disease Control (Grant Nos. 2017ZX10201101, 2018ZX10721102, and 2018ZX10732101-001-003).