
Abstract
MicroRNAs play an important role in the interaction between viruses and hosts. In this study, we found that the expression level of miR-33b-5p was markedly increased in human immunodeficiency virus type 1 (HIV-1)–infected cell lines and the serum of person with HIV-1. Further investigation revealed that the level of ATP-binding cassette transporter (ABCA1), which transports cholesterol between intracellular and extracellular compartments to maintain cholesterol homeostasis, was reduced in HIV-1–infected target cells, as the target gene of miR-33b-5p. Furthermore, HIV-1 infection stimulated abnormal lipid transport in macrophages, resulting in lipid accumulation in cells. These changes can be reversed by an miR-33b-5p inhibitor. We discovered a mechanism through which HIV-1 infection caused miR-33b-5p to target ABCA1 and caused aberrant lipid transport, providing a novel method for diagnosing and treating poor lipid metabolism in person with HIV-1.
Introduction
Human immunodeficiency virus type 1 (HIV-1) causes AIDS by infecting immune cells such as CD4+ T cells and macrophages. 1,2 Antiretroviral therapy (ART) has transformed AIDS into a manageable disease and increased the life expectancy of people living with HIV-1. 3,4 Recently, growing clinical studies have shown that non-HIV/AIDS-related diseases, such as diabetes, atherosclerosis, and lipid metabolism disorders, increase in incidence and occur earlier in person with HIV-1, compared with HIV-1–negative patients. 5 –8
MicroRNAs (miRNAs) are noncoding, single-stranded, 20–24-nucleotide long RNAs. miRNAs are a class of post-transcriptional regulators, that is, they do not directly act on the DNA but on the mRNA transcribed by the DNA to downregulate the expression of the target gene. When miRNA and the mRNA 3′UTR of target gene are complete complementarity, miRNA directs the specific cleavage of mRNA and leads to degradation. And when they are not sufficiently complementary, the miRNA directs the repression of translation, that is, it only affects the level of protein expression and does not affect the stability of mRNA. 9 –11
miRNA expression has been linked to diseases, such as cancer, diabetes, and atherosclerosis, 12,13 hence it has emerged as a possible therapeutic target. 14 miRNAs have been implicated in the replication and pathogenicity of HIV-1 infection through direct or indirect interactions with viral or host genes. 15,16 For example, HIV-1 tat protein leads to microglial activation by upregulation of NLRC5 expression via miR-34a. 17 In addition, miR-1236 inhibits HIV-1 replication by suppressing the translation of cellular factor VprBP in monocytes. 18 These findings imply that miRNAs play a significant role in the interaction between HIV-1 and target cells. 19
We performed microarray analysis and quantitative polymerase chain reaction (qPCR) detection of HIV-1–infected human T cell leukemia cells (MT4 cells) and found that the expression level of several miRNAs was upregulated by >2.5-fold in HIV-1–infected cells. In our previous study, miR-210-5p, induced by Vpr, has been demonstrated to suppress the expression of TGIF2, which partly explains the mechanism of G2 arrest induced by HIV-1 infection. 20 After HIV-1 infection, miR-33b-5p was one of the highly induced miRNAs in our microarray analysis. One study reported that miR-33b-5p is one of the dominant mature forms of miR-33b, which targets SREBP, a gene involved in lipid metabolism by participating in cholesterol transport, cholesterol synthesis, and fatty acid oxidation. 21
Further analysis suggested that ATP-binding cassette transporter 1 (ABCA1) was a target of miR-33b-5p. 22 ABCA1 transports cholesterol between intracellular and extracellular compartments to maintain cholesterol homeostasis, and it is found in a variety of cells, including macrophages, T cells, liver cells, and mesenchymal cells. 23 Studies have reported that macrophages are the major factor affecting cholesterol level in the blood. 24,25
In this study, we focused on macrophages and found that HIV-1 infection downregulates ABCA1 expression by raising the expression of miR-33b-5p to affect cholesterol metabolism. This study provides a novel explanation and some suggestions for the lipid abnormalities of person with HIV-1.
Materials and Methods
Ethics statement
All human-participant research experiments were approved by the Ethics Committees of the Sixth People's Hospital of Xinjiang Uygur Autonomous Region and Jianghan University. Every participant provided informed consent.
Participant recruitment
This study recruited 76 patients who were diagnosed with HIV-1 infection in the Sixth People's Hospital in Xinjiang Uygur Autonomous Region between 2018 and 2019 without any other autoimmune disease. In addition, 30 healthy volunteers of similar age and gender, who were confirmed to be HIV-1 negative, were recruited as a control group. Serum (1 mL) was collected from each participant, before they accepted ART, and stored at −80°C for further analysis.
Cell culture and antibodies
MT-4 (human T cell leukemia cell line), THP-1 (human monocytic cell line), and HEK-293T (embryonic kidney cell line) cells were obtained from the Chinese Academy of Science's Cell Bank (Shanghai, China). MT-4 and THP-1 cells were cultured in the Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco). HEK-293T cells were cultured with Dulbecco's modified Eagle's medium (DMEM; Gibco). Cells were cultured at 37°C with 5% CO2, and the medium was supplemented with 10% heat-inactivated fetal calf serum (Gibco). Cells were transfected with the corresponding plasmid using lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions or infected with pseudotyped HIV-1 according to the experiment objective.
The antibody against HIV-1 p24 (sc-69728) was provided by Santa Cruz Biotechnology (Santa Cruz). The antibody against ABCA1 (ab18180) was obtained from Abcam (Abcam). The antibody against β-actin (20536-1-AP) was obtained from Proteintech (Proteintech, China).
Plasmid construction
The 3′UTR of ABCA1 was amplified from cDNA of THP-1 cells using matching primers (forward: CCGAGCTCAGAATCCTGTTCATACGGGGT, reverse: GCGTCGACCTCACATAGGTACATTCCACAGG), and the PCR fragments were cloned into pmirGLO vectors to form the ABCA1 3′UTR luciferase reporter construct (3′UTR-Luc). The HIV-1 proviral vector
Pseudotyped HIV-1 preparation and single-round infection
HEK-293T cells (2 × 106/well) were seeded into a six-well plate before transfection, and cotransfected with 15 μg of
THP-1 cells (1 × 106/well) were seeded into a six-well plate with 100 mM phorbol-12-myristate-13-acetate (PMA; ThermoFisher) stimulation for 36 h to differentiate into macrophages (THP-1/PMA). 26 Infection was performed with 100 ng p24/mL of HIV-1-EGFP, followed by 2 h of incubation in serum-free medium. The culture medium was changed to fresh RPMI-1640 containing 10% fetal bovine serum at 6 h postinfection and cells were collected for the next step after 30 h.
Microarray assay of miRNA
MT-4 cells were transfected with HIV-1-EGFP (100 ng of p24/mL) or incubated with fresh medium as a control for 36 h, and total cellular RNA was purified with TRIzol (Invitrogen). The microarray assay was performed by Shanghai Kangcheng Biological Company. The Gene Expression Omnibus (GEO) accession number for these miRNA-seq datasets is GSE161444, as described previously. 20
RNA extract and qPCR
Total cellular RNA was purified using TRIzol reagent according to the manufacturer's instructions; serum RNA of person with HIV-1 was extracted using serum RNA extraction kits (Roche, Switzerland) according to the manufacturer's instructions. M-MLV reserve transcriptase (Takara, Japan) was used to reverse transcribe the mRNA into cDNA using the miR-33b-5p RT primer (RiboBio, China) at 42°C for 60 min, followed by 72°C extension for 15 min. The cDNA was used to determine the expression levels of the required RNA via qPCR using SYBR Kits (Bio-Rad). The primers for the miRNAs were synthesized by RiboBio. The relative level of the miRNAs was standardized to that of U6 small nuclear RNA (internal control) using the 2−ΔΔCt method.
Bioinformatics analysis
Two target prediction tools TargetScan (
miRNA mimics and inhibitors
miR-33b-5p mimics (double-stranded RNA oligonucleotides) and inhibitors (single-stranded chemically modified oligonucleotides) were synthesized by RiboBio and transfected into cells with Lipofectamine 2000. Nonspecific RNA mimics or inhibitors from RiboBio were used as negative controls (NCs). The sequences are as follows: miR-33b-5p-mimic, GUGCAUUGCUGUUGCAUUGC; miR-33b-5p-inhibitor, GCAAUGCAACAGCAAUGCACUU; NC-mimic, UUCUCCGAACGUGUCACGUTT; NC-inhibitor, CAGUACUUUUGUGUAGUACAA.
Luciferase reporter assay
The luciferase reporter plasmid (ABCA1-3′UTR-luc) and the corresponding miRNAs were cotransfected into HEK-293T cells, cells were lysed with 100 μL of passive lysis buffer (Promega). The luciferase activity of 20 μL of cell lysate was measured using a dual luciferase reporter assay system (Promega). The activity of firefly luciferase was normalized based on the activity of Renilla luciferase, and the quantitative value in each sample was normalized with the mock transfected control.
Western blot analysis
Cells were washed twice with phosphate-buffered saline (PBS) and lysed with RIPA lysis buffer (Beyotime, China). Protease inhibitor cocktail (10%; Roche) and phosphatase inhibitor (Beyotime) were added to the lysis buffer right before use. ABCA1 protein was separated from each sample using 8% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and p24 and β-actin proteins were separated using 10% SDS-PAGE. Protein was transferred to polyvinylidene fluoride membranes (Millipore) after performing SDS-PAGE. After blocking with 5% nonfat milk in TBS buffer containing 0.05% Tween 20, the blot was probed with target-specific primary antibodies, followed by a horseradish peroxidase–conjugated secondary antibody. Protein bands were imaged using ECL Substrate (Millipore) with a Gel Doc™ system (Bio-Rad).
Oil Red O staining
THP-1/PMA were cultivated in RPMI-1640 medium containing ox-LDL before 6 h of incubation with HIV-1-EGFP at 100 ng (p24)/mL and harvested for Oil Red O (ORO) staining using a staining kit (Solarbio, China) according to the manufacturer's instructions. Cells were washed with PBS and fixed with ORO fixation solution for 30 min. After washing with 60% isopropanol for 5 min, cells were stained with ORO staining solution for 30 min. Excess ORO staining solution was completely removed by washing with distilled water. To visualize the nuclei, cells were counterstained with Mayer hematoxylin for 2 min. Then, the cells were incubated with ORO buffer for 1 min before being covered with ultrapure water. The stained cells were visualized using a light microscope.
Enzyme-linked immunosorbent assay
THP-1/PMA cells were infected with HIV-1-EGFP at 100 ng (p24)/mL for 36 h. Cells and culture supernatant were harvested for total cholesterol (TC) and triglyceride (TG) quantification using enzyme-linked immunosorbent assay (ELISA). The TG Quantification Kit (Bio-Swamp, China) and Cholesterol Assay Kit (MyBioSource) were used to quantify TG and TC levels, respectively, according to the manufacturer's instructions.
Statistical analysis
Each experiment was carried out at least three times. Data are presented as mean ± standard deviation and were analyzed using GraphPad Prism 6. Significance of differences between two groups was estimated using a t-test; statistically significance was set at p ≤ .05.
Results
HIV-1 infection induces miR-33b-5p expression
To evaluate the miRNA expression profile of HIV-1–infected T cells, MT-4 cells were infected with HIV-1-EGFP for 48 h; then, the total cellular RNA was subjected to array-based miRNA profiles. The original data of the miRNA array have been submitted to the database, and our prior study about miR-210 has been published. 20 The expression level of miR-33b-5p was shown to be 3.6-fold increase in HIV-1-EGFP–infected cells, compared with that in uninfected cells (Fig. 1A). To confirm miRNA array analysis results, we performed time-dependent and dose-dependent infection in MT-4 cells using HIV-1-EGFP and determined the expression level of miR-33b-5p via qPCR. Compared with uninfected cells, the expression level of miR-33b-5p was significantly elevated in time-dependent (Fig. 1B, C) and dose-dependent manners (Fig. 1D, E).
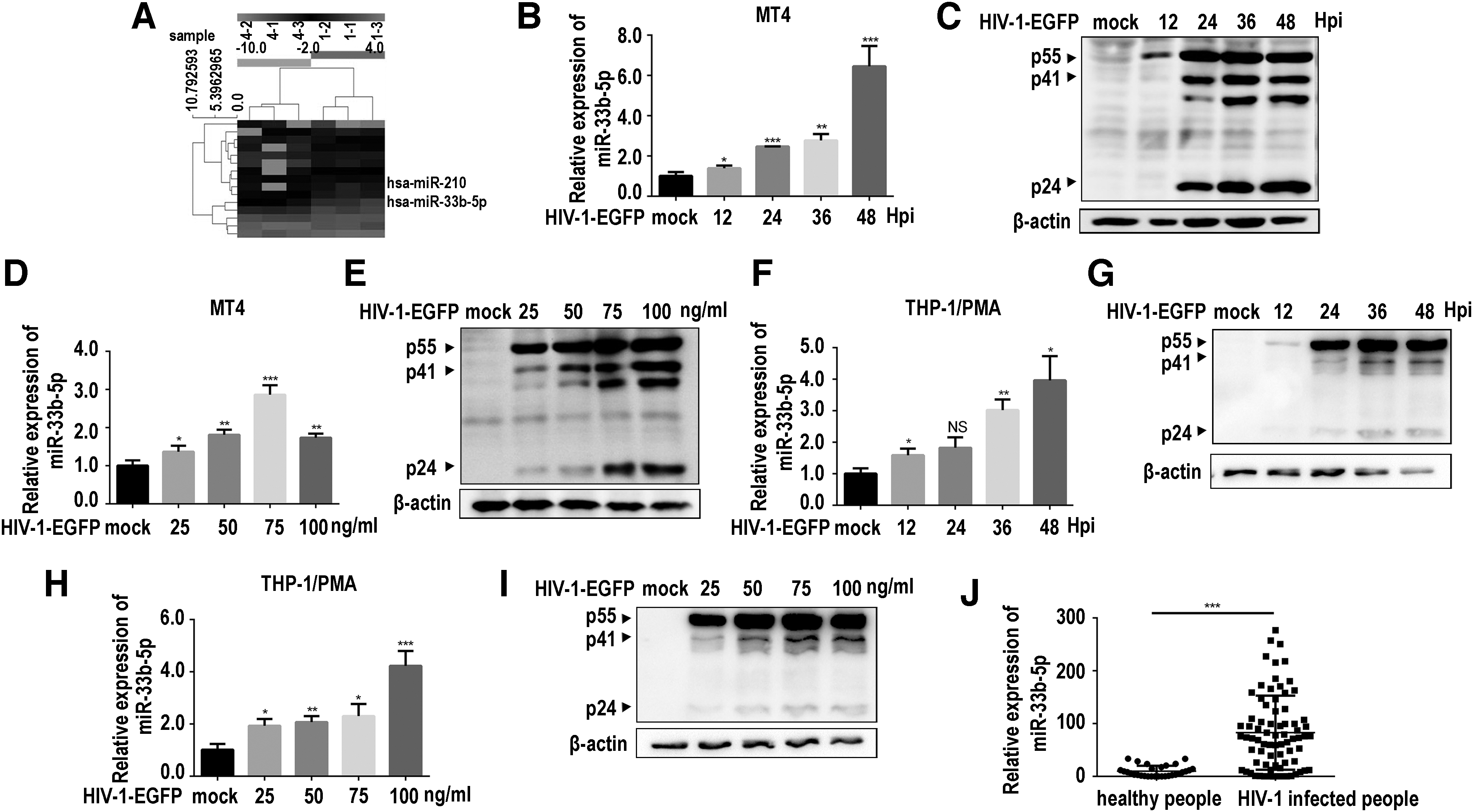
HIV-1-EGFP infection enhanced miR-33b-5p expression.
To corroborate these results, we performed similar experiments in THP-1/PMA cells. Our results also showed that miR-33b-5p increased in time-dependent and dose-dependent manners in THP-1/PMA cells (Fig. 1F–I). Furthermore, we detected miR-33b-5p in clinical samples with qPCR and found that serum levels of miR-33b-5p of person with HIV-1 were significantly higher than those of HIV-1–negative patients (Fig. 1J). These in vivo and in vitro results suggested that HIV-1 infection enhanced miR-33b-5p expression.
HIV-1 infection downregulates ABCA1 expression by increasing miR-33b-5p expression
To explore the potential target gene of miR-33b-5p, we used Target Scan and miRanda for bioinformatic prediction. ABCA1 received the highest score among the anticipated target genes. Sequence analysis indicated complementary sequences between ABCA1 and miR-33b-5p (Fig. 2A). To explore the regulation of ABCA1 by miR-33b-5p, we cotransfected ABCA1-3′UTR-Luc into HEK-293T cells with miR-33b-5p mimics. After 24 h, the cells were lysed and used to perform the luciferase reporter assay. The expression of ABCA1 was suppressed in miR-33b-5p mimic-transfected cells, in a dose-dependent manner, implying ABCA1 mRNA to be a potential target of miR-33b-5p (Fig. 2B). Next, we infected THP-1/PMA cells with a serial concentration of HIV-1-EGFP or for varied durations of infection time with a single concentration of HIV-1-EGFP; then, we quantified the protein level of ABCA1. The amount of ABCA1 protein decreased in both dose-dependent (Fig. 2C, D) and time-dependent manners (Fig. 2E, F).
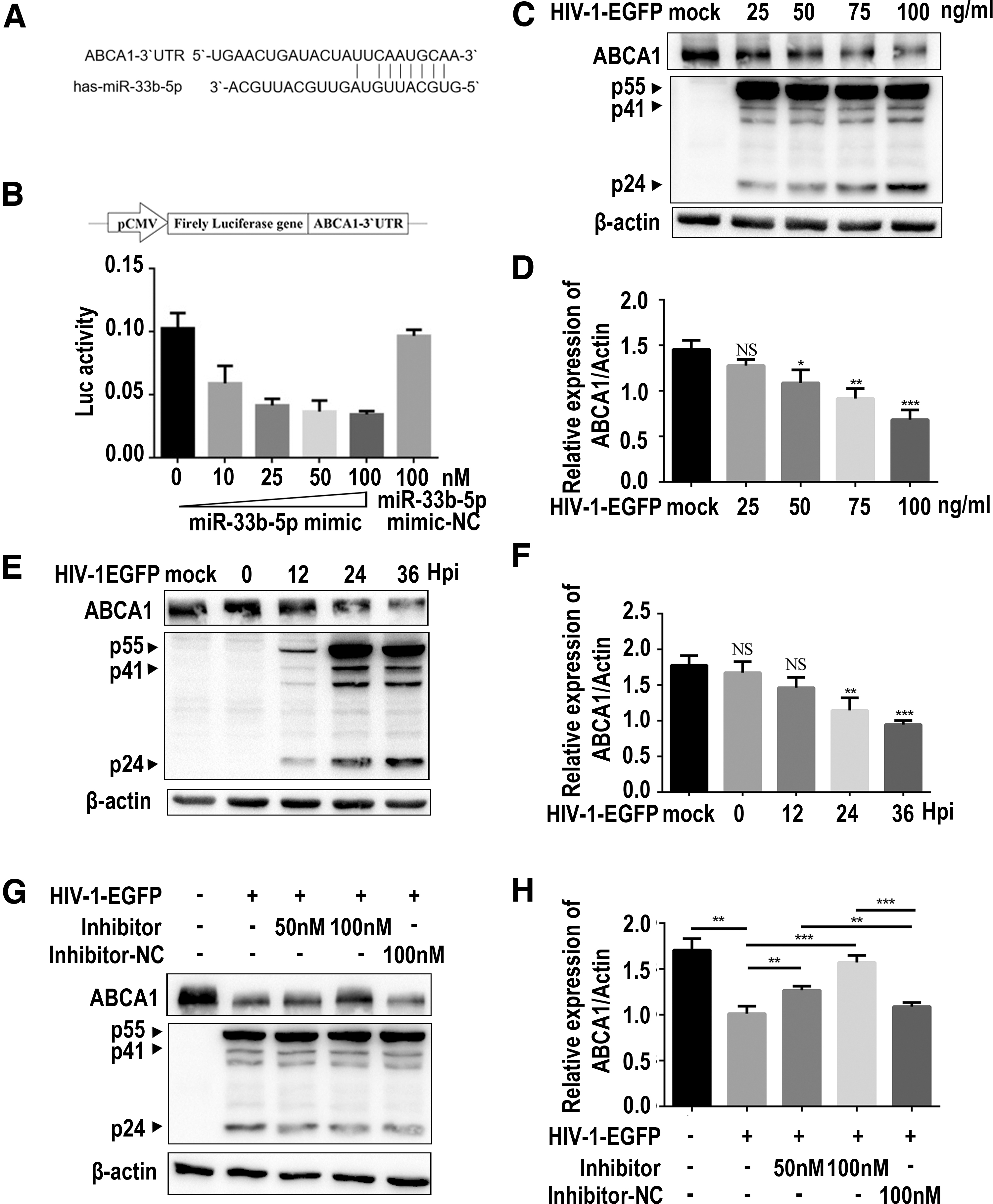
HIV-1-EGFP infection downregulates ABCA1 expression by enhancing miR-33b-5p.
To further verify that HIV-1-EGFP infection downregulated ABCA1 expression through miR-33b-5p, we added miR-33b-5p inhibitor in HIV-1-EGFP–infected THP-1/PMA cells (Fig. 2G, H). Similarly, HIV-1-EGFP–infected cells downregulated ABCA1 expression compared with its expression in uninfected cells (Fig. 2G line 1, and line 2). Under miR-33b-5p inhibitor treatment, ABCA1 expression was progressively restored (Fig. 2G lines 3 and line 4), but there was no change in the cells treated with the NC (Fig. 2G line 5). These findings indicated that ABCA1 expression was downregulated in HIV-1–infected THP-1/PMA cells upon enhancing miR-33b-5p expression.
HIV-1 infection causes lipid accumulation
Because ABCA1 is essential for cholesterol transport and HIV-1-EGFP infection decreased the expression level of ABCA1, we questioned whether HIV-1 infection affected lipid levels in macrophages. We analyzed lipid levels in the HIV-1-GFP–infected THP-1/PMA cells by ORO staining assay. Lipid accumulated more in HIV-1-EGFP–infected cells than in uninfected cells (Fig. 3A). Using ELISA, we determined the levels of TG and TC in the culture supernatant and in HIV-1-EGFP–infected THP-1/PMA cells. Compared with uninfected cells, TG and TC (Fig. 3C, D) levels were significantly decreased in the culture supernatant and increased in HIV-1-EGFP–infected cells. These results indicated that HIV-1 infection causes intracellular lipid aggregation, which might be because of ABCA1 levels decreasing after HIV-1 infection.

HIV-1-EGFP infection leads to abnormal lipid metabolism.
HIV-1 infection induces lipid metabolic disorder by increasing miR-33b-5p expression
To determine whether intracellular lipid aggregation induced by HIV-1 infection was owing to enhancement of miR-33b-5p expression, we analyzed lipid levels in HIV-1-EGFP–infected THP-1/PMA cells in the presence of miR-33b-5p inhibitor (Fig. 4A, B). HIV-1-EGFP–infected cells showed intracellular lipid aggregation (Fig. 4B, lane 1 and lane 2). Using additional miR-33b-5p inhibitors remarkably reduced intracellular lipid aggregation, compared with the addition of inhibitor to NC and uninfected cells (Fig. 4B, lanes 2–5). Finally, we used ELISA to determine TC and TG levels in the culture supernatant and in THP-1/PMA cells (Fig. 4D, F).

HIV-1-EGFP infection affects lipid metabolism through increasing miR-33b-5p.
The results revealed that following HIV-1-EGFP infection, intracellular TG levels increased, whereas TG levels in cell culture supernatant decreased, as given in Figure 3C. Whereas intracellular TG concentration reduced in infected cells with additional miR-33b-5p inhibitor to a similar level of uninfected cells, culture supernatant TG concentration was stably maintained in infected cells with or without miR-33b-5p inhibitor (Fig. 4D). Similar changes were found in cell culture supernatant and intracellular TC after addition of miR-33b-5p inhibitor (Fig. 4F). These findings suggested that HIV-1 infection affects the outflow of cholesterol and other lipid compounds by upregulating miR-33b-5p expression.
Discussion
Various studies have demonstrated that miRNAs are involved in the interaction between virus and host, and these miRNAs might play a role in pathology, virus replication, and virus clearance. 18,27,28 We investigated the impact of miRNA in HIV-1–infected cells and found that HIV-1 infection upregulated miR-33b-5p expression, which led to suppression of ABCA1 expression, ultimately leading to lipid metabolic abnormalities in macrophages.
Increased expression of miR-33b-5p can diminish the expression of HMGA2 to reduce the development of gastric cancer cells, 29 which can also inhibit GLUT4, and thus, affect glucose transport. 30 ABCA1, as the target of miR-33b-5p, had also been reported. 22,31 Our dual fluorescence reporting system and overexpression experiments proved that miR-33b-5p could reduce the expression of ABCA1 by targeting its 3′UTR. Furthermore, we found that ABCA1 expression was suppressed in THP-1/PMA cells infected with HIV-1 in a viral dose-dependent and infection time-dependent manner. Addition of miR-33b-5p inhibitor interfered with ABCA1 suppression by HIV-1 infection. Our findings provide a new understanding of the interaction between HIV-1 and miRNA of host cells.
Critical for regulating cholesterol homeostasis, ABCA1 is involved in the early stages of high-density lipoprotein biogenesis, which enhances intracellular excess cholesterol outflow after binding to apolipoprotein AI and is a critical rate-limiting component in cholesterol reverse transport. 32,33 Our study showed that HIV-1 infection causes intracellular miR-33b-5p expression. Previous clinical research indicated that HIV-1 patients had metabolic dysfunction. 34,35 Hence, our results shed light on the molecular mechanism of cholesterol aggregation in person with HIV-1.
Despite the fact that ART efficiently suppresses HIV-1 replication, several antiviral agents have been reported to be capable of inducing lipid metabolism disorders. 36 –39 Protease inhibitors impair the hydrolysis of TG-rich lipoproteins through lipase, which reduces free fatty acid storage and interferes with normal postprandial metabolism of free fatty acids. 38,40 Our clinical samples were from person with HIV-1 without ART. Previous studies reported that HIV-1 Nef protein inhibits ABCA1-impaired cell cholesterol export. However, the virus used in this study lacks the Nef gene. It is suggested that HIV-1 infection suppresses ABCA1 expression by disturbing lipid metabolism in multiple ways. All these results suggest that before ART, an assessment of lipid metabolism is important for an HIV-1–infected patient, and anti-HIV-1 agents should be chosen to avoid ritonavir and other protease inhibitors for patients with a lipid disorder.
miRNA are abundant, stable, and detectable in the bloodstream, whether in plasma or serum, and have been identified as biomarkers. 41 The ABCA1 involved in lipid abnormality had been reported. 42 The reported and our data demonstrated that miR-33b-5p can target the 3′UTR of ABCA1 to regulate the lipid metabolism, and miR-33b-5p is significantly upregulated in the serum of HIV-1–infected individuals. Because the miRNA is easy to determine and the serum is easy to get, miR-33b-5p could be used as the lipid metabolism-related markers in HIV-1–infected individuals. We are building a patient cohort to investigate the relationship between the level of miR-33b-5p and the lipid metabolism of HIV-1–positive patients.
In summary, we discovered that HIV-1 infection interfered with lipid metabolism by inducing the expression of miR-33b-5p to suppress ABCA1. This implicates miR-33b-5p as one of the molecular mechanisms of HIV-1–induced lipid metabolism disease and offers fresh insights into the virus–host interaction. miR-33b-5p might be a useful biomarker for lipid metabolism prediction and reasonable ART selection.
Footnotes
Authors' Contributions
J.Q., Z.S., and B.S. conceived and designed the experiments; J.L. and Z.S. collected the samples; Q.Y., Q.P., and W.L. performed the experiments; Q.Y. analyzed the data; Q.Y., J.Q., and B.S. wrote and edited the article. All authors have read and approved the final version of the article.
Research Involving Human Participants
The authors declare that no human and/or animal material, data, or cell lines were involved in this study.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the National Natural Science Foundation of China [Grant No. 3167016], Wuhan Science and Technology Bureau [Grant No. 2020020601012318], and Jianghan University [Grant Nos: 8190006, 6210035, 2021yb138, and 2019037].