
Abstract
It was confirmed that the sterile alpha motif and HD domain 1 (SAMHD1) limits human immunodeficiency virus type 1 (HIV-1) replication. In contrast, viral protein x (Vpx) in HIV-2 and some simian immunodeficiency viruses can counteract this effect. The possible interaction between SAMHD1 and Vpx was suggested by previous studies; however, there are no data to confirm this interaction. Therefore, this study aimed to study the interaction between two proteins and the properties of Vpx protein for the first time using bioinformatic tools. Vpx and SAMHD1 sequences were obtained from the National Center for Biotechnology Information GenBank. Several software were used to define Vpx properties and the interaction between Vpx and different SAMHD1 isoforms. Our findings indicated the difference in interaction sites among different Vpx. However, in all Vpx proteins, this region is from amino acids 4 to 90. In addition, two regions (26–31 and 134–139) and two amino acids 425 and 429 in SAMHD1 are vital in the possible interaction. In addition, our analysis determined the physicochemical and immunological properties of the Vpx. Considering all factors, this study could confirm that Vpx interacts with SAMHD1, which could inhibit SAMHD1. Moreover, our findings can pave the way for future studies to express and purify Vpx in the laboratory and study this protein in vitro.
Introduction
Human immunodeficiency virus type 1 (HIV-1)
Different proteins in human cells that restrict lentiviral replication include apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3 (APOBEC3), tripartite motif protein 5 alpha (TRIM5α), tetherin, and most recently sterile alpha motif and HD domain 1 (SAMHD1), which is expressed in myeloid cells and blocks viral replication at the level of reverse transcription. 5 In myeloid cells, SAMHD1 restricts HIV-1 replication and does not restrict HIV-2 replication. This is indicated in the role of Vpx to antagonize restriction imposed by SAMHD1 by proteasomal degradation of SAMHD1 in the myeloid lineage (dendritic cells, monocytes, and macrophages) and quiescent CD4+ T cells. In addition, it was suggested that the SAMHD1-dependent and SAMHD1-independent functions are controlled by different regions of the Vpx. 1,6,7
Bioinformatic tools are an effective means to study viral proteins and natural inhibitors. These tools could offer valuable data to understand the function of different inhibitors by analysis of structures, modification sites, and possible interactions. 8,9
While some studies suggested different Vpx regions to interact with the SAMHD1 protein, the interaction between two proteins, Vpx and SAMHD1, has not been investigated and this study aimed to examine this interaction in Vpx of different SIV strains as well as provide information about physicochemical properties, structure, and postmodification site of Vpx.
Materials and Methods
Ethical approval
The study was approved by the Ethics Committee of Shiraz University of Medical Sciences (IR.SUMS.REC.1398.1127).
Sequence alignment and phylogenetic tree
Table 1 displays all the accession numbers of 118 SIV VPX sequences, and 3 sequences of SAMHD1 isoforms (isoforms 1, 2, and 3) were retrieved from the National Center for Biotechnology Information (NCBI) GenBank (www.ncbi.nlm.nih.gov/).
The Accession Numbers of All 118 Sequences That Were Used in This Study
With the following parameters: gap open cost, 10, gap extension cost, 1.0, and very accurate progressive alignment algorithm, homology was examined among sequences using CLC-sequence viewer software (version CLC Genomics Workbench 20). In addition, phylogenetic trees were constructed via CLC-sequence viewer software, using the neighbor-joining method (Bootstrapping: 1,000) to confirm phylogenetic trees' reliability. 10
Physicochemical analysis
General physicochemical properties of VPX were determined by employing “Expasy's ProtParam” (http://expasy.org/tools/protparam.html). 11
Immunoinformatic prediction
All Immune Epitope Database (IEDB) epitope prediction methods (http://tools.immuneepitope.org/tools/bcell/iedb_input), including Chou and Fasman, Karplus and Schulz, Kolaskar and Tongaonkar, Emini, Parker, and BepiPred methods were run for prediction of B cell epitope positions. 12 In addition, all features provided by BcePred (www.imtech.res.in/raghava/bcepred) such as hydrophilicity, flexibility/mobility, accessibility, polarity, exposed surface, and turns were considered to predict B cell epitopes. Finally, ABCpred software (www.imtech.res.in/raghava/abcpred/) predicted 16-meric B cell epitopes. Furthermore, the probability of antigenicity and IgE epitopes and allergic properties were assessed by VaxiJen software (www.ddg-pharmfac.net) and AlgPred (www.imtech.res.in/raghava/algpred/index.html), respectively. 13
Postmodification translation
Serine, threonine, and tyrosine phosphorylation site prediction was done using DISPHOS (www.dabi.temple.edu/disphos/pred.html) and NetPhos (www.cbs.dtu.dk/services/NetPhos/). NetNGlyc (www.cbs.dtu.dk/services/NetNGlyc/) and GlycoEP (www.imtech.res.in/raghava/glycoep/submit.html) were run for N-glycosylation site prediction.
Secondary and tertiary structure prediction
To predict secondary and tertiary structures of VPX and SAMHD1, SOPMA (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_sopma.html) and I-TASSER (http://zhanglab.ccmb.med.umich.edu/I-TASSER) were used. The suggested model by I-TASSER was refined by GalaxyRefine and 3D refine, and finally, the refined models were evaluated by “Qmean” (http://swissmodel.expasy.org/qmean/cgi/index.cgi), “Rampage” (http://mordred.bioc.cam.ac.uk/∼rapper/rampage.php), ERRAT (https://servicesn.mbi.ucla.edu/ERRAT/), and ProSA-web (https://prosa.services.came.sbg.ac.at/prosa.php). 14
Docking analysis
Docking analysis for Vpx and all SAMHD1 isoforms was done using Hex and PatchDock servers. 15 The parameters used for the Hex docking process were as follows: Correction type—Shape only, FFT Mode—3D fast lite, Grid Dimension—0.6, Receptor range—180, Ligand Range—180, Twist range—360, and Distance Range—40. The parameters used for the PatchDock docking process were as follows: Clustering RMSD: 1.5, Complex Type: Protein small ligand.
Results
Sequence alignment and phylogenetic tree results
The alignment results showed that the most frequent substitutions in the Vpx were located in the C-terminal. Figure 1 shows the phylogenetic tree of 118 Vpx sequences categorized into 3 main clusters, the middle cluster includes 3 major clades, which include the majority of sequences. In addition, the middle cluster contains several clades, which include the majority of selected sequences. In terms of phylogenetic properties, almost all sequences appeared to be relatively close and originated from similar ancestors. SAMHD1 sequences were aligned and results are illustrated in Supplementary Data 2, some deletions were defined in C-terminal.
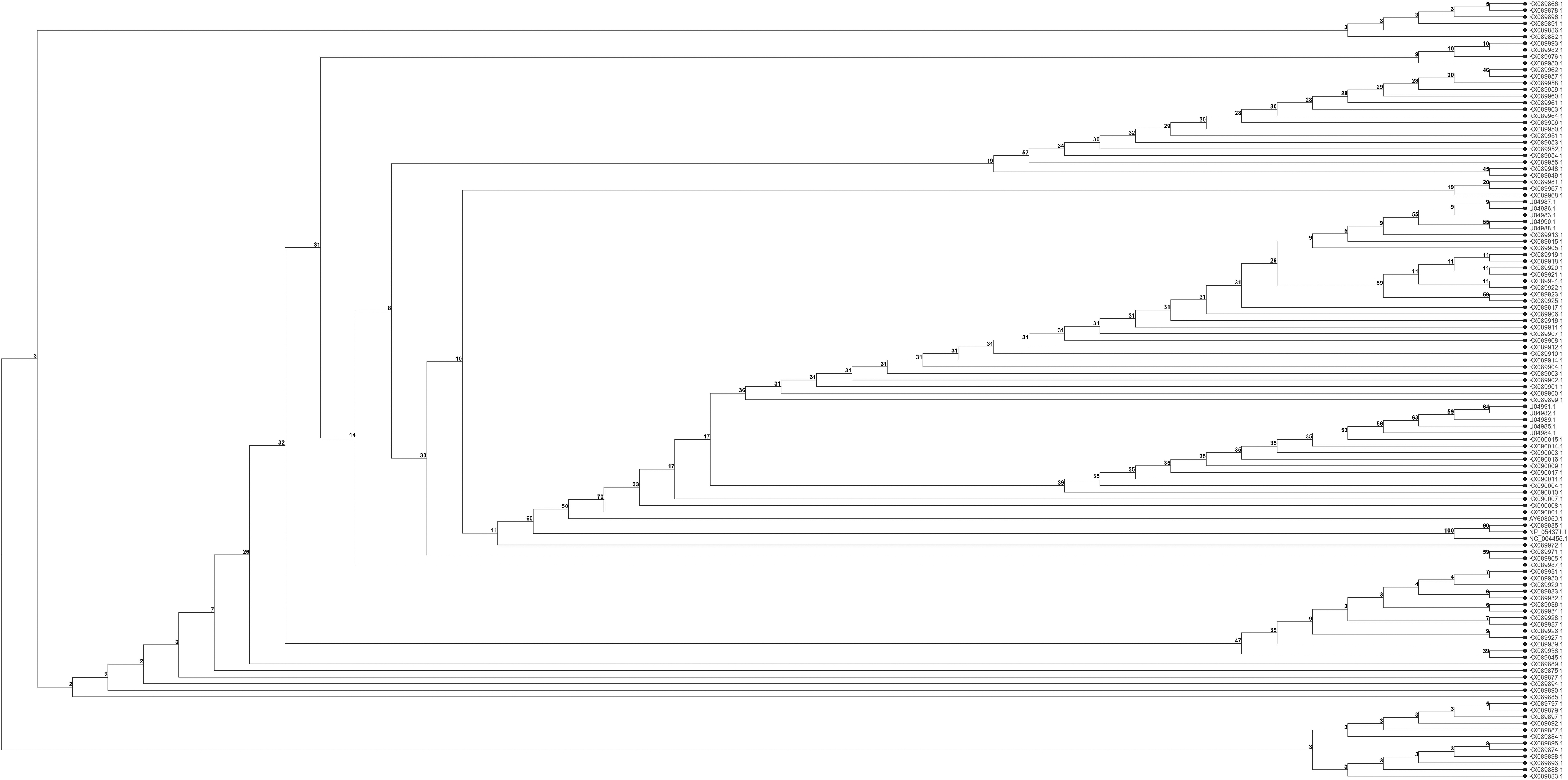
Phylogenetic tree based on Vpx protein sequences and the neighbor-joining method. The numbers at the forks show the number of occurrences of repetitive groups to the right out of 1,000 bootstrap samples. Vpx, viral protein x.
ProtParam analysis
ProtParam results for Vpx are summarized in Table 2, which shows this protein is acidic (Theoretical pI: 6.4), with the highest percentage of acidic amino acids. The instability index defined the protein stability in a test tube showing that Vpx is an unstable peptide in experimental procedures. The aliphatic index is an indicator of protein thermostability that a relevantly low aliphatic index of Vpx predicted this protein is not thermostable.
ProtParam Results of the Vpx
Vpx, viral protein x.
Immunoinformatic analysis results
Suggested B cell epitopes are summarized in Table 3, and the combination results of B cell epitope prediction showed two major regions with the highest ability to be B cell epitopes, chosen as favorable epitopes: 5–31 and 30–60 amino acids. According to the predefined cutoff of the VaxiJen program, Vpx was confirmed as a probable antigen (overall prediction for protective antigen = 0.5339, model: virus and threshold: 0.4). The prediction of allergenic proteins by mapping of IgE epitope, SVM, and hybrid methods showed that Vpx is not an allergen protein.
The Results of B Cell Epitope Prediction for Vpx by Several Reliable Software
The bold values highlight the most promising B-cell epitope regions, obtained by combining the results from three different software.
Postmodification site prediction
Prediction of serine, threonine, and tyrosine phosphorylation sites by DISPHOS and NetPhos showed one common position (amino acid at position 2) in Vpx. In addition, glycosylation prediction was determined at a specific position (amino acid at position 26) for this modification process.
Secondary and tertiary structure
The SOPMA software results are presented in Figure 2, which displayed the pattern of four secondary structures, extended strand (3.57%), alpha helix (51.79%), random coil (42.86%), and beta-turn (1.79%). In addition, the SAMHD1 protein consisted of the alpha helix (47.92%), extended strand (9.42%), beta-turn (3.67%), and random coil (38.98%). However, the alpha helix and random coil were the most prominent secondary structures in both Vpx and SAMHD1.

Secondary structure of Vpx and SAMHD1 predicted by SOPMA. For Vpx: Blue: alpha helix (51.79%), purple: random coil (42.86%), red: extended strand (3.57%), and green: beta turn (1.79%). For SAMHD1: Vpx: blue: alpha helix (51.79%), purple: random coil (42.86%), red: extended strand (3.57%), and green: beta turn (1.79%). SAMHD1, sterile alpha motif and HD domain 1.
The results of the 3D structure evaluation for Vpx and SAMHD1 are summarized in Supplementary Data 1. The models were refined by Galaxyrefine online software. The best-refined models for both Vpx and SAMHD1 (Fig. 3) were regarded for validation analysis using various programs: ERRAT, Ramachandran, ProSA, and QMEAN. The ERRAT generates the overall quality factor, which is the number of side chains (nonbonded interactions) formed between pairs of different atomic types and models that the overall quality factor value greater than 85 is considered to be qualified. PROCHECK tool provides the Ramachandran plot checks the stereochemical quality of the protein structure; among various proposed models, the one with more residues in the Ramachandran-favored region and fewer residues in the Ramachandran-disallowed region is a better model for further analysis.
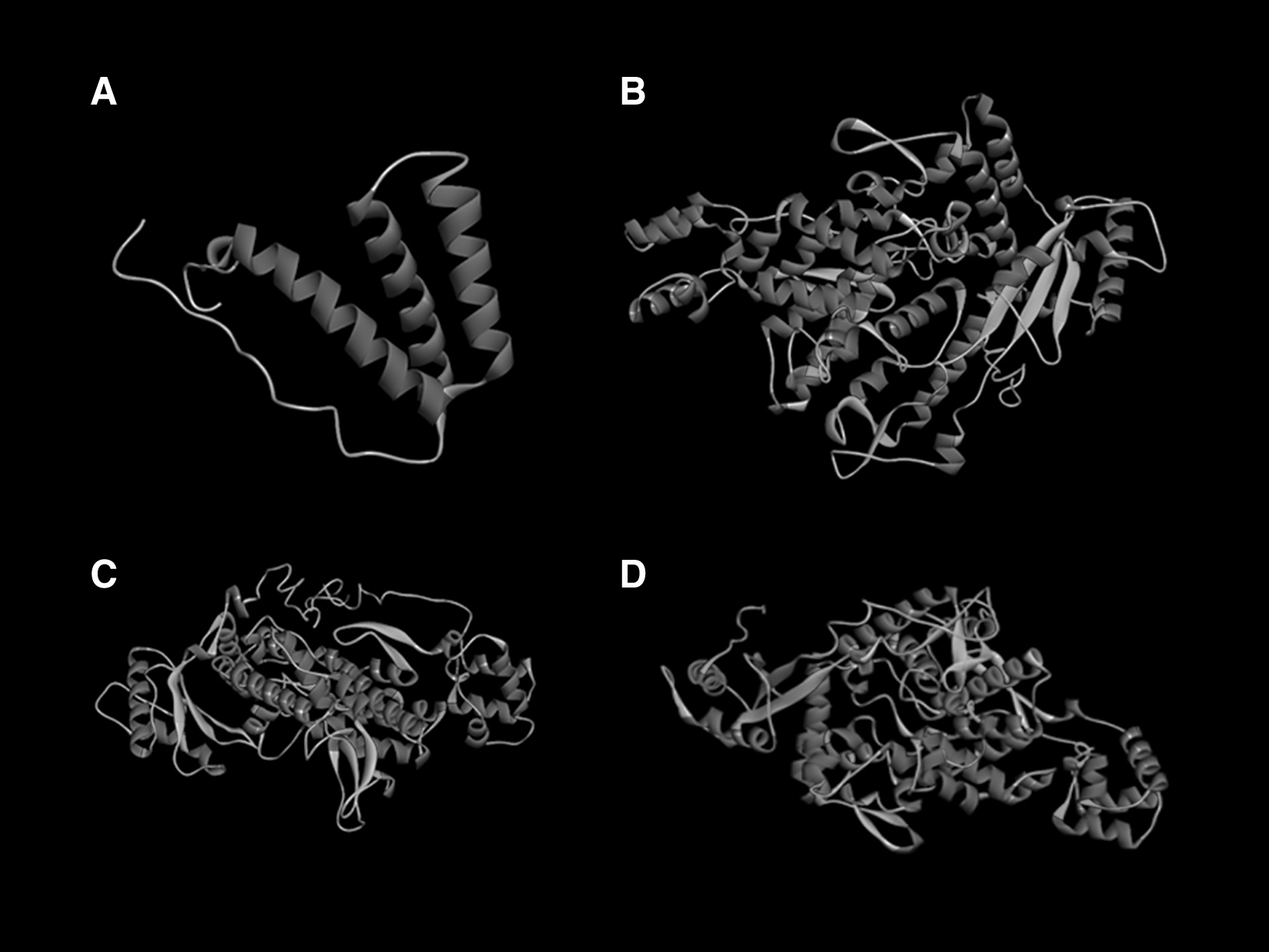
The final tertiary structure of Vpx
ProSA-Web server predicts the Z-score, which reflects the overall quality of the model, and a positive Z-score indicates that the input structure has erroneous or problematic parts. 16 Another validation server QMEAN estimates how comparable the model is to experimentally derived structures of similar size. QMEAN Z-scores around zero illustrate good agreement between the experimental and the proposed model structure. 17 Finally, the best-qualified models were applied for the docking analysis.
Docking results
Docking analysis showed high energy values between Vpx and all SAMHD1 isoforms and also displayed the role of various amino acids in the interaction between Vpx and SAMHD1 proteins. From our data, the highest energy values belonged to SAMHD1 isoform 2. The list of amino acid residues at the interaction sites of the docking analysis is summarized in Table 4. Figure 4 illustrates the interactions between Vpx and all three SAMHD1 isoforms. The higher energy values belonged to HM803689-cm (−729), NP_758889-and (−703), and P19508-sm (−612), and the lowest energy was for P05917-mac (−305).
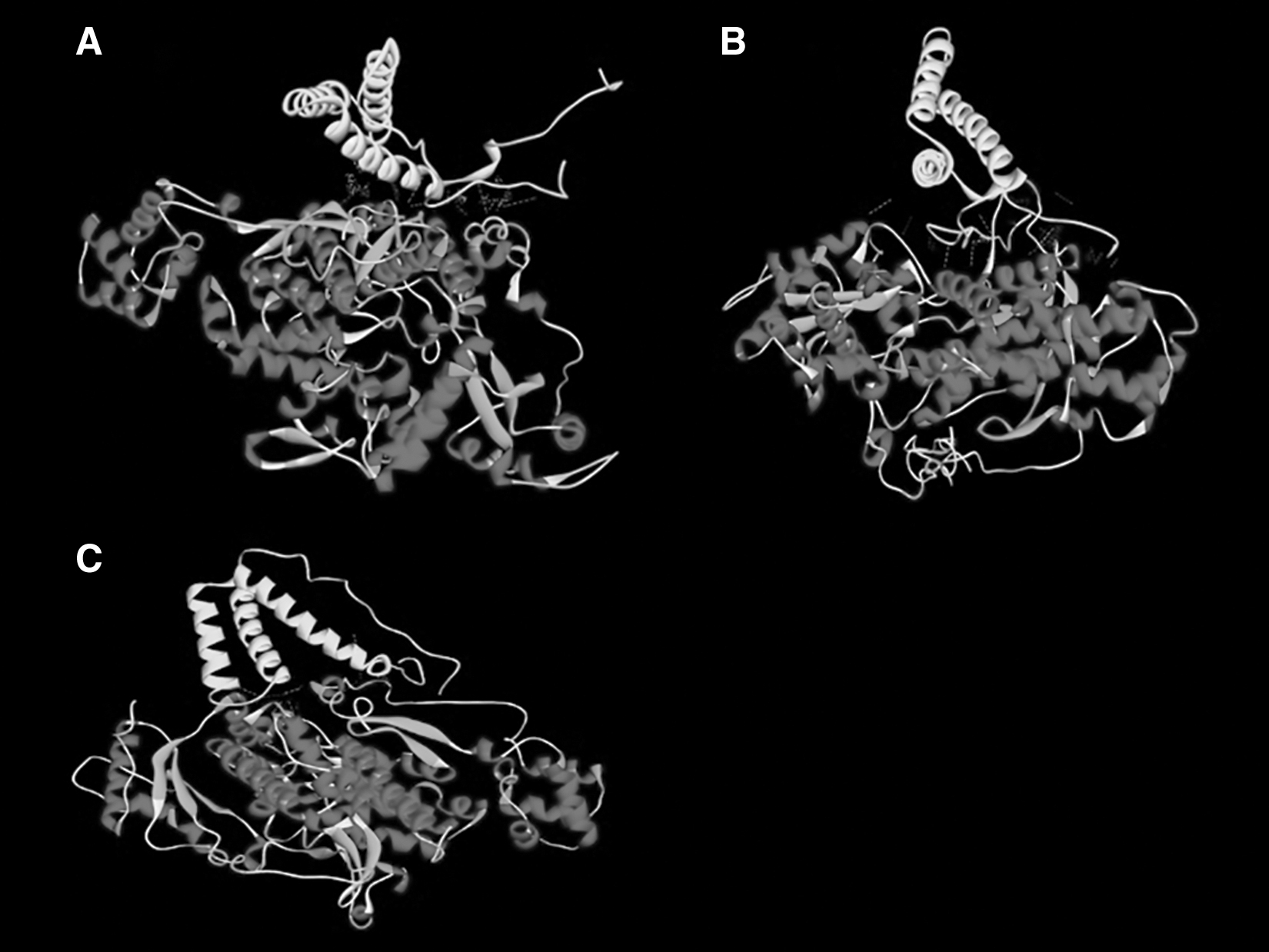
The interaction between Vpx protein and SAMHD1 isoform 1
The Interaction Sites of the Docking Between Four Vpx Proteins and Three Isoforms of SAMHD1
The interaction sites for three isoforms of SAMHD1 are summarized in one column for each Vpx.
Red-capped mangabeys SIV.
Mandrill SIV.
Macaques SIV.
Cynomolgus SIV.
SAMHD1, sterile alpha motif and HD domain 1; SIV, simian immunodeficiency virus.
Discussion
The present study used a bioinformatic approach to predict the interactions between SIV Vpx and SAMHD1.
Molecular docking methods are powerful tools that enable researchers to reveal the interaction between SIV proteins and host factors to predict the evolutionary relationship between virus and host cell factors that is helpful to recognize the viral behavior. The higher docking score represented better binding affinity that could be attributed to the presence of H bonds that exhibited a strong type of interaction, which indicated the strong attachment of Vpx with SAMDH1 to suppress SIV functions that would contribute to the promising treatment outcome.
Our results indicated that in SIV, different amino acids are involved in the interaction regions. However, it seems that the interaction site in almost all sequences is from amino acids 4 to 90. It must be noted that for SIV-from three amino acids (23, 26, and 86) are the main elements of the interaction site and that for SIV-and three amino acids (19, 23, and 89), and SIV-mac amino acid 13, and a region from 80 to 88 are the main components, and finally for SIV SM, four amino acids 83, 84, 88, and 90 are the main components of the interaction site.
A conserved motif (HHCC motif) in Vpx was suggested previously, which is located from amino acid 36 to 90, and it was suggested to be the main region of interaction and degradation of SAMHD1, similar to these data in our study almost all interaction sites included this motif, which confirmed the vital role of this motif in Vpx function to degrade SAMHD1. 18
The docking energy value between Vpx and SAMHD1 showed that the highest energy value belonging to Siv-mac has the highest energy value and may have the strongest interaction with SAMHD1.
Various molecular mechanisms or biological processes are involved in the pathogenesis of infectious diseases, and various genes and molecules of infectious agents have interactions with different targets in the host cell, among which the processes and factors involved in resistance against the host's immune system or antibiotic drugs are more important. 19 –22
The present study results showed that other regions of SAMHD1 have interacted with Vpx, compared with previously suggested regions (590–626) in SAMHD1. 23
Fregoso et al. showed that Vpx form SIV-mac needs the C-terminus of SAMHD1 for interaction and degradation. However, in contrast with Fregoso's results, our prediction showed that in the interaction between Vpx-mac and SAMHD1, two regions (26–31 and 134–139) and two amino acids 425 and 429 are important in the interaction. 24
Yu et al. determined the importance of a change (M68K) in Vpx protein, which disrupts Vpx's ability to counteract SAMHD1. 25 Contrary to Yu's findings, amino acid 68 was K in all the studied sequences. The present study showed that this amino acid is not involved in the interaction between Vpx and SAMHD1. Hence, further research is needed to determine the impact of amino acid 68 Vpx function changes.
A study conducted by Wang et al. showed that five amino acids (threonine 619, leucine 575, lysine 577, arginine 578, and lysine 579) in SAMHD1 are partially or fully required in restricting Vpx. In contrast with Wang's results, we did not find any of these positions in our prediction. This may be related to different SAMHD1 isoforms, which were used in the two studies. 26
According to Schwefel et al., a Vpx region from amino acid 1 to 90 interacts with SAMHD1 and has been shown to have substitutions in this region in different Siv strains (SIVsm, SIVmac, SIVrcm, SIVmnd). 27
Akin to Schwefel's results, our prediction showed that all amino acids involved in the interaction sites are located in this region. In addition, similar substitutions were found in our prediction.
Miyakawa et al. studied the role of phosphorylation in Vpx function. They determined a PIM family of serine/threonine kinases that are involved in the phosphorylation of Vpx and the promotion of Vpx-mediated SAMHD1 counteraction. 28 They suggested that phosphorylation at Ser13 has a significant impact on the stability of Vpx interaction with SAMHD1 and proteolysis of SAMHD1. Similar to our prediction, the ser13 was found by Miyakawa et al. in 2019, which showed that phosphorylation of this position has a great impact on the interaction of Vpx with SAMHD1 and promotes proteolysis of SAMHD1, and it can be suggested that inhibition of the PIM kinases can reduce the Vpx ability to degrade SAMHD1. 28
One of the features that were assessed was the protein properties of Vpx such as theoretical pI (Table 2) that is beneficial for modeling the behavior in the environment and physical/chemical treatment processes. Furthermore, the average GRAVY, which assessed a grand average hydropathy of Vpx, with negative scores conforming to Vpx, inquired moderately the hydrophilic property and also suggests the possibility of better interaction of Vpx with water. Attributing to the short half-lives of Vpx, which is dependent on various factors such as surface charge, molecular weight, and size, this protein degrades fast in Escherichia coli, humans, and yeast, which is highly applicable for protein expression. However, Hosseini and Hanke et al. expressed Vpx in E. coli and showed the stability of this protein in this host. 29,30
In addition, Zhang et al. expressed Vpx in yeast and Hofmann et al., Fujita et al., and Berger et al. showed the stability of expressed Vpx in 293T cells. 31 –33 These data confirmed the substantial potential of the three mentioned hosts to express Vpx. Inconsistency between the results of this report and previous studies maybe due to the effects of various unknown factors that impact the half-life of the protein in vitro which are excluded from the bioinformatics analysis.
ProtParam results indicated that the VPX should be stable in the different hosts, which is in line with previous results, however, we predicted that this protein might be unstable in a tube.
Postmodification translations (PMTs) of proteins play a critical role in the attachment of small proteins or functional groups such as phosphorylation and glycosylation to the exact residue that changes the structure and charge of the protein. The viral infection uses PMTs to enhance protein virulence properties and antigenicity; plus, PMTs have a role in interferon response, solubilization, and viral replication would increase, which has a significant role in viral pathogenesis. For that reason, host cells deregulate PMTs during the production of the viral proteins to control the virus replication, stimulate immune response pathways, and suppress the synthesis of the viral protein to eradicate the virus. One of the most widespread protein modifications, which is a prerequisite for proper folding and progeny formation of viral proteins, is glycosylation. To suppress viral infections, the improvement of antiviral drugs, which sites in viral proteins, may offer an ideal chance for clinical therapies.
This study defined glycosylation sites and according to the data, the effect of the glycosylation on Vpx and SIV pathogenesis is not described clearly; thus, experimental studies are needed to elaborate on this issue. Phosphorylation prediction for Vpx suggested some residues suitable for phosphorylation that may be vital for the interaction of Vpx with cellular factors. From our findings, the most suggested site for Vpx phosphorylation modification was S2. It can be suggested that preventing phosphorylation at such a position may exhibit more viral infectivity. On that account, interfering with the phosphorylation of Vpx may cause lower viral replication and SIV pathogenesis. All in all, Vpx postmodifications are crucial for SIV pathogenesis; thus, using a bioinformatic server to unveil the underlying mechanisms, PMT sites, and pathways can suggest new viral inhibitors. Therefore, bioinformatic studies are essential to disclose PMT issues for efficient therapeutic approaches.
A vital step in designing novel peptide vaccines involves B cell epitope identification. An accurate prediction of B cell epitopes requires reliable software for predicting the immune response and antibodies for several biotechnology and clinical applications. 34 BepiPred-2.0 is a famous software for predicting B cell epitopes from antigen sequences. It is based on a random forest algorithm on epitopes annotated from antibody–antigen protein structures and data derived from solved 3D structures, and on a large collection of linear epitopes downloaded from the IEDB. 35 BcePred used various residue properties commonly used in B cell epitope prediction, which used 1,029 nonredundant B cell epitopes achieved from the Bcipep database and an equal number of nonepitopes randomly selected from the SWISS-PROT database. 34
The accuracy of prediction based on properties varies between 52.92% and 57.53%. ABCpred is a freely available software to predict B cell epitopes, which used the recurrent neural network method and showed more than 67% accuracy. 36
In the present study, we used all three software tools to predict the most reliable B cell epitopes. Finally, the results showed two major B cell epitopes (5–31, 30–60), which were common among the used software. Compared with Fujita et al., which showed different domains in Vpx protein, the first B cell epitope is located on a region known as the SAMHD1 binding site and the second one is located in two major helices 1 and 2, which has unknown function. 1 As the first B cell epitope is not located in the helix structure, it can stimulate the humoral system better compared with the second B cell epitope.
Conclusion
Overall, the present study findings could confirm the possible interaction between Vpx and SAMHD1 proteins. This interaction plays a vital role in SAMHD1 degradation and reveals some interaction sites between Vpx and SAMHD1 proteins. In addition, this study indicated some significant properties of Vpx, which could be useful to express this protein. This could reveal its ability to induce the immune system.
Availability of Data and Materials
Data that support the findings of this study are available from the corresponding author (Dr. Ava Hashempour) upon reasonable request.
Confirmation Statement
All authors confirmed that their research is supported by the Shiraz HIV/AIDS Research Center, which is primarily involved in research.
Footnotes
Authors' Contributions
Z.H. was responsible for methodology and software. B.D. was responsible for formal analysis and writing—original draft. A.H. was responsible for supervision, funding acquisition, and writing—review and editing.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was funded by Shiraz University of Medical Sciences, Shiraz, Iran (Grant No. 11988).
Supplementary Material
Supplementary Data 1
Supplementary Data 2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.