
Abstract
To analyze the genetic structure and recombination characteristics of a newly discovered HIV-1 unique recombinant form (URF) isolated in Hebei Province, China, viral RNA was extracted from the plasma sample of the infected individual and reverse transcribed to cDNA. Two overlapping segments of the HIV-1 genome were amplified using a near-endpoint dilution method. Recombinant breakpoints were determined using RIP, jpHMM, and SimPlot 3.5.1 software. MEGA 6.0 software was used to construct a neighbor-joining phylogenetic tree. The near full-length genome sequence (8,862 bp) of a recombinant of CRF01_AE/CRF07_BC was obtained. The genome comprised at least seven overlapping segments, including four CRF01_AE and three CRF07_BC segments, with CRF01_AE as the backbone. A URF virus between CRF01_AE and CRF07_BC was amplified and characterized in this study. Parental viruses were homologous with HIV-1 strains prevalent among men who have sex with men in northern China and may originate from sexual transmission of local HIV-1 strains in Hebei Province.
Hebei is a northern province of China with a low HIV infection rate. 1 Upto October 31, 2020, 15,000 people in Hebei Province had been diagnosed with HIV-1/AIDS, and sexual contact was the main route of infection, especially homosexual transmission. The three main subtypes were circulating recombinant forms (CRF)01_AE (49.6%), CRF07_BC (29.7%), and subtype B (13.0%). 2 The cocirculation of the three dominant HIV strains among men who have sex with men (MSM) provides suitable conditions for forming new HIV-1 CRFs and unique recombinant forms (URFs). In addition, Hebei Province surrounds Beijing and Tianjin cities, and the convenient transportation network of the area promotes frequent communication and provides opportunities for dual or multiple infection in the MSM population. This has led to the emergence and prevalence of new recombinant strains, such as CRF123_0107, and CRF01_AE/CRF07_BC, in MSM in recent years. 3 –6
This study analyzed the characteristics of the near full-endpoint gene sequence of a HIV-1 sample from a MSM infection case in Baoding city and characterized the recombination breakpoints and patterns to understand the features of HIV recombination in Hebei Province and provides clues and references for guiding the prevention and control of AIDS in this area.
The study subject was a 28-year-old man who lived in Baoding, Hebei Province, via homosexual transmission-infected HIV. The plasma sample was from the patient named BDD028A, who was diagnosed as HIV-1 positive in September 2022 with a baseline CD4+ T lymphocyte count of 184 cells/μL and a viral load of 58,200 copies/mL by the Baoding CDC. This research was approved by the Medical Ethics Committee of the Baoding People's Hospital with a protocol number of 2019-03. Written informed consent was acquired from the subject before blood sample collection.
According to the manufacturer's instructions, virus RNA was extracted from 140 μL of the infected individual's plasma sample (BDD028A) using the QIAamp Virus RNA Mini Kit (Qiagen, Duesseldorf, Germany). For detailed information on the amplification and reaction systems of the near full-length genome (NFLG) sequence of BDD028A, please refer to our previous study. 5 The polymerase chain reaction products were purified and sequenced by Tianyi Huiyuan Bioscience & Technology, Inc. (Beijing, China).
The NFLG sequence was submitted to HIV-BLAST (https://www.hiv.lanl.gov/content/sequence/BASIC_BLAST/basic_blast.html) to analyze the subtype of BDD028A and similar sequences had been recorded. Additionally, the NFLG and subregion phylogenetic trees were constructed by Mega 6.0 using the neighbor-joining (N-J) method with 1,000 bootstrap replications based on the Kimura two-parameter model. The recombination pattern and breakpoints were characterized using the Recombination Identification Program (https://hiv.lanl.gov/contens/sequence/RIP/RIP.html), jpHMM, and SimPlot (v3.5.1.0).
We finally obtained a 8,862 bp-NFLG sequence (HXB2: 751–9,613) of BDD028A. The N-J tree showed that BDD028A forms a monobranch, suggesting that BDD028A may be an URF (Fig. 1). The recombinant breakpoints analysis revealed that BDD028A was composed of seven mosaic gene fragments, including four CRF01_AE subregions (I, III, V, and VII) and three CRF07_BC regions (II, IV, and VI), with seven recombinant breakpoints (Fig. 2). Recombinant analysis (Fig. 2) indicated that themosaic genome structure of the NFLG of BDD028A is ICRF01_AE (HXB2, 790–2435nt), IICRF07_BC (HXB2, 2436–3384nt), IIICRF01_AE (HXB2, 3385–3787nt), IVCRF07_BC (HXB2, 3788–420nt5), VCRF01_AE (HXB2, 4206–6397nt), VICRF07_BC (HXB2, 6398–8516nt), and VIICRF01_AE (HXB2, 8517–9411nt). Subregion phylogenetic analysis was constructed to further determined the genetic origins of each fragment. Segments I, III, V, and VII were clustered with CRF01_AE cluster 4; segments II, IV, and VI were clustered with CRF07_BC (Fig. 3).
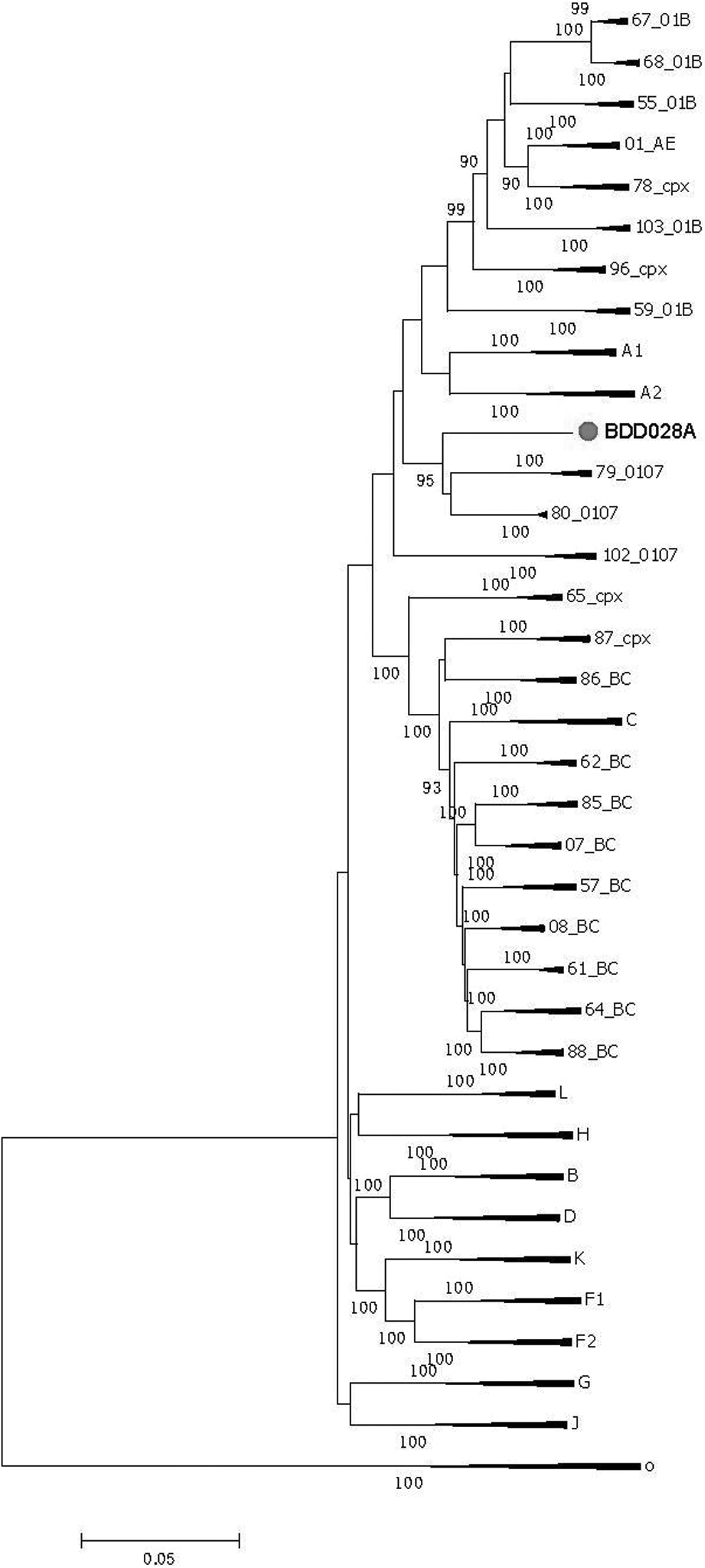
Phylogenetic tree based on the NFLG sequence of BDD028A. The standard subtype reference sequences were downloaded from the Los Alamos National Laboratory HIV Database (www.hiv.lanl.gov). The neighbor-joining phylogenetic tree of BDD028A (8,862 bp, red-filled circle) was constructed based on the NFLG sequence using MEGA 6.0. The stability of each node was assessed using bootstrap tests with 1,000 replicates, and only bootstrap values ≥90% are shown at the corresponding nodes. The scale bar represents a 5% genetic distance. NFLG, near full-length genome.
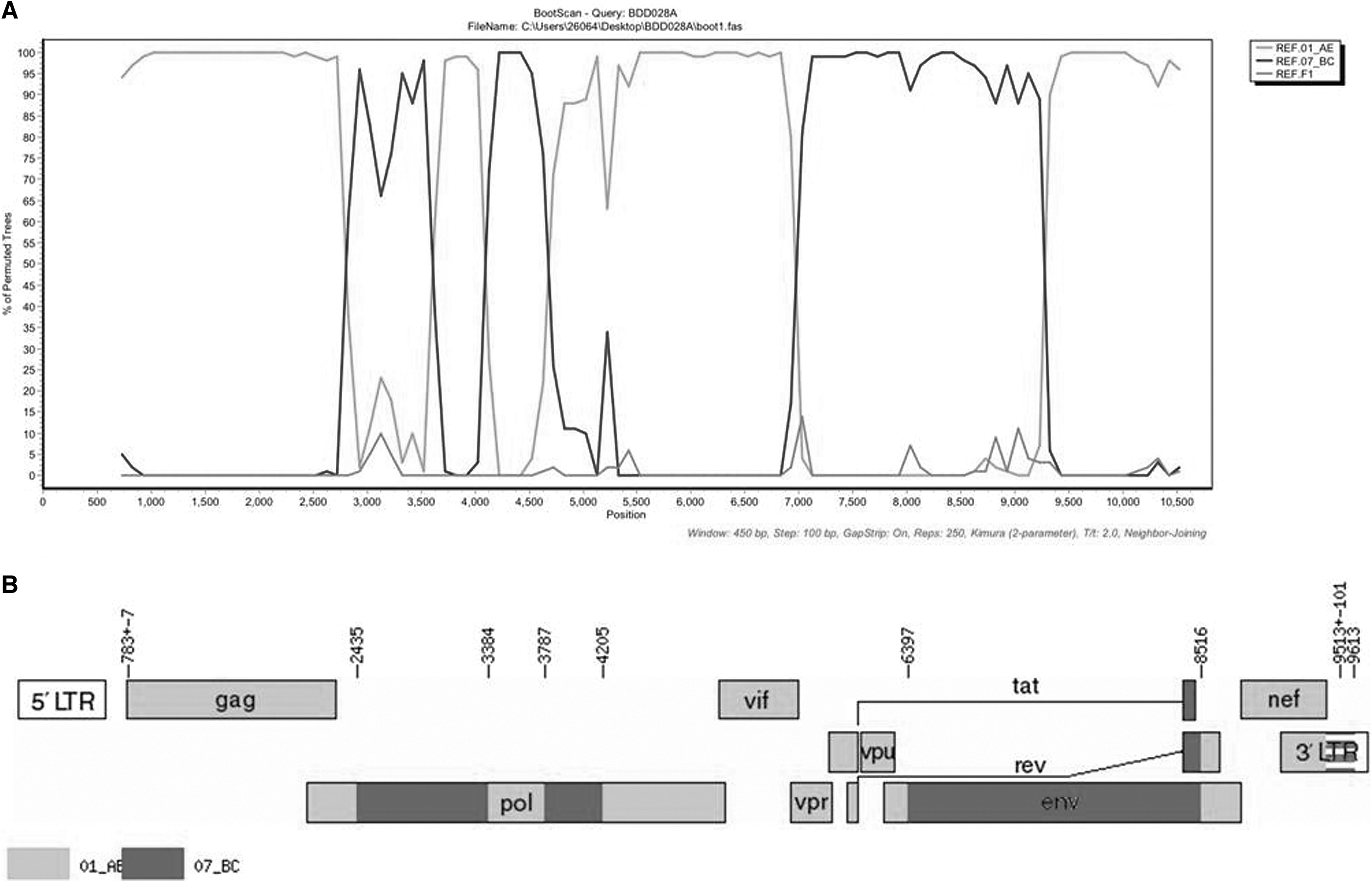
Recombination breakpoints in the near-full-length genome sequence of BDD028A.

Subregional phylogenetic trees of the novel CRF01_AE/CRF07_BC identified. Trees were constructed using the neighbor-joining method with 1,000 bootstrap replicates using MEGA 6.0. Bootstrap values ≥90% are shown at the corresponding nodes. The scale bars indicate a genetic distance of 5%. Each segment of BDD028A is marked by a red-filled triangle.
Genetic diversity and gene mutation are the important characteristics of HIV-1 that enables transmission, while genetic recombination among different subtypes of HIV-1 strains is an important source of HIV-1 evolution. 7 The distribution of HIV-1 genotypes has clear population and regional characteristics. 8 Therefore, the identification of URFs is significant in understanding the relationship among different transmission routes and subtypes and finding the possible bridge population characteristics.
At present, CRF01_AE and CRF07_BC are the predominant intersubtype recombinants among sexually active populations, especially among MSM in China. 9 In China, CRF01 HIV-1 was discovered to evolve at least seven to eight genetic clusters, each with a unique geographical distribution and prevalence pattern. CRF01_AE clusters 4 and 5 are mainly distributed among the MSM population in northern China, including Beijing and Tianjin. 10,11 Subregion tree analysis of BDD028A in this study suggested that the genetic origin of the CRF01_AE regions was the CRF01_AE cluster 4 which is related to the MSM, and that CRF07_ BC regions were from CRF07_BC which is common in MSM in Northern China. Recombination is a major mechanism whereby HIV-1 rapidly evolves, increasing its fitness, for example, by acquiring drug resistance or by evading immune surveillance. Frequent recombination also makes the molecular diagnosis, treatment, and vaccine development of HIV-1 more difficult. 12 –15
The increasing of HIV-1 recombinant forms has enriched the diversity of HIV-1 prevalent in Hebei province, suggesting that the molecular monitoring of HIV-1 diversity is important in this region.
Sequence Data
The nucleotide sequence of the near full-length genome of BDD028A has been deposited in the GenBank database under accession number OQ366386.
Footnotes
Acknowledgment
Ethics statement
The studies were reviewed and approved by the Medical Ethics Committee at the People's Hospital of Baoding 2019-03. Written informed consent of BDD028A in this study was provided by the patient.
Authors' Contributions
H.G., B.L., and W.F. designed the study. H.S., B.L., and Y.L. amplified and spliced the sequences. H.G., and W.F. analyzed the data. H.G., B.L., and Y.L. wrote the manuscript. All authors read and agreed to the published version of the manuscript.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.