
Abstract
Continuous recombination and variation during replication could lead to rapid evolution and genetic diversity of HIV-1. Some studies had identified that it was easy to develop new recombinant strains of HIV-1 among the populations of men who have sex with men (MSM). Surveillance of genetic variants of HIV-1 in key populations was crucial for comprehending the development of regional HIV-1 epidemics. The finding was reported the identification of two new unique recombinant forms (URF 20110561 and 21110743) from individuals infected with HIV-1 in Tongzhou, Beijing in 2020–2022. Sequences of near full-length genome (NFLG) were amplified, then identification of amplification products used phylogenetic analyses. The result showed that CRF01_AE was the main backbone of 20110561 and 21110743. In the gag region of the virus, 20110561 was inserted two fragments from CRF07_BC, while in the pol and tat regions of the virus, 21110743 was inserted four fragments from CRF07_BC. The CRF01_AE parental origin in the genomes of the two URFs was derived from the CRF01_AE Cluster 4. In the phylogenetic tree, the CRF07_BC parental origin of 20110561 clustered with 07BC_N and the CRF07_BC parental origin of 21110743 clustered with 07BC_O. In summary, the prevalence of novel second-generation URFs of HIV-1 was monitored in Tongzhou, Beijing. The emergence of the novel CRF01_AE/CRF07_BC recombination demonstrated that there was a great significance of continuous monitoring of new URFs in MSM populations to prevent and control the spreading of new HIV-1 URFs.
Introduction
Acquired immune deficiency syndrome (AIDS) is a chronic infectious disease caused by human immunodeficiency virus (HIV) infection of CD4+T lymphocytes, resulting in a decline in the humoral immunity functions. According to newly published data from the Joint United Nations Program on HIV/AIDS (UNAIDS) in 2022, 85.6 million people have been infected with HIV-1 globally since the first case of AIDS was detected. 1 The rapid evolution and genetic diversity of HIV-1 were caused by high rates of replication, base mismatching, and recombination. 2,3 The predominant strain currently circulating in the world derived from HIV-1 group M, which has evolved into 10 subtypes including A-D, F-H, and J-L. 3 Simultaneously, the high rate of recombination among the different subtypes had developed 145 circular recombinant phenotypes (CRFs) (https://www.hiv.lanl.gov/content/sequence/HIV/CRFs/crfs.comp) and a significant number of unique recombinant forms (URFs). In China, an increasing number of infections living with HIV and the complexity of HIV-1 subtypes made it difficult to prevent and control the spread of the virus. 4 The fourth national HIV-1 molecular epidemiological survey in 2016 revealed a rapid increase in the variety of HIV-1 subtypes, which had become a priority for AIDS prevention and control in China. Multiple HIV-1 subtypes are prevalent in China, such as CRF01_AE, CFR07_BC, CRF08_BC, subtype B, and so on. 5–6 Since the 1990s, Subtype B had been more popular than others, then subtypes CRF01_AE and CRF07_BC replaced it gradually as changing of transmission routines and floating of popuation. 7–8 With a growing genetic heterogeneity of HIV-1, 9 it is possible that more and more URFs could be generated in future, so there is a necessity to conduct the surveillance of novel CRF and URF of HIV-1 to prevent them spreading further in China.
As the urban sub-center of Beijing, Tongzhou plays an important role in all aspects. Meanwhile, the favorable geographic and transportation location facilitated the transmission of the AIDS epidemic in the area to neighboring areas. Since the first case was reported in 1998 in Tongzhou, individuals infected with HIV-1 had amounted to 2,405 till 2023. A survey conducted among the MSM population in Beijing showed that 69.81%
The Tongzhou CDC collected plasma samples, along with demographic information, from two HIV cases (20110561 and 21110743). We extracted RNA using the MagNA-Pure LC Total Nucleic Acid Isolation Kit by MagNA-Pre LC 2.0 Instrument (Roche), followed by one-step reverse transcription with the PrimeScriptTM one-step RT-PCR Kit Version NO. RR055A) and nested PCR [TaKaRa Premix TaqTM (Ex-TaqTM Ver 2.0 plus dye) No. RR902A]. 10 Then, we sequenced the PCR product using the Sanger sequencing method of Beijing Biomed Gene Technology Co. 11 The sequencing files were analyzed using Sequencher 5.4.5 and CExpress 6.0 software to assess the quality of the original peak maps. The HIV database Quality Control tool was utilized for quality control to exclude sequences that contained >5% mixed bases and stop codons. Subtyping was performed using Comet, and we aligned the sequences 20110561 and 21110743 with the HIV-1 reference sequences of each subtype in MAFFT 7. The ends of the sequences were trimmed using BioEdit 7.2.5. We constructed Maximum-likelihood phylogenetic trees by Maximum-Likelihood using FAST Tree 2.1.10, with nucleotide substitution modeled as GTR+CAT, SPR = 4, and Shimodaira-Hasegawa test for calculating the branching nodes of the evolutionary tree. The constructed evolutionary tree was embellished using ITOL (iTOL: Interactive Tree Of Life [embl.de]). Further, we analyzed the recombination breakpoints using jpHMM and RIP tools (https://www.hiv.lanl.gov/content/sequence/HIV/HIVTools.html). And we constructed a Neighbor-Joining phylogenetic tree for each recombinant fragment using MEGA6.06 software to validate the genotype of each fragment and perform a homology analysis of each recombinant fragment. The aim was to obtain accurate recombination breakpoint results. The recombinant structural patterns of the virulent strains were mapped by Recombinant GenomeDrawingTool (https://www.hiv.lanl.gov/content/sequence/DRAW_CRF/recom_mapper.html). Amplification of NFLG sequences from plasma samples due to different subtypes in the ltr, gag, pol, and env gene regions. NFLG phylogenetic tree analysis showed that these sequences form a unique monophyletic cluster independent of other subtypes and CRFs (Fig. 1). Simplot v3.5.1 analysis showed that the NFLG sequence of 20110561 and 21110743 consisted of CRF01_AE and CRF07_BC (Fig. 2 and 3). The chimeric recombinant structures of two sequences obtained by the recombination identification program RIP with jpHMM were described as follows: ICRF01_AE(HXB2, 989-1230nt); II;CRF07_BC(HXB2, 1237-1867nt); IIICRF01_AE(HXB2,1891-9214nt), 20110561. ICRF01_AE(HXB2, 989-2658nt); IICRF07_BC(HXB2, 2662-4835nt); IIICRF01_AE(HXB2, 5036-5287nt); IVCRF07_BCHXB2, 52902-5788nt); VCRF01_AE(HXB2, 5801-6556nt); VICRF07_BC(HXB2, 6569-8423nt); VIICRF01_AE(HXB2, 8427-9214nt), 21110743 (Fig. 2). Neighbor-joining phylogenetic trees were constructed using the MEGA6.06 with 1,000 bootstrap replicates under the Kimura 2-parameter model. The results showed that parental origin of all CRF01_AE regions of the two NFLGs originated from the MSM-associated CRF01_AE Cluster 4 (Fig. 4). The CRF07_BC parental origin of 20110561 clustered with 07BC_N and the CRF07_BC parental origin of 21110743 clustered with 07BC_O (Fig. 4).
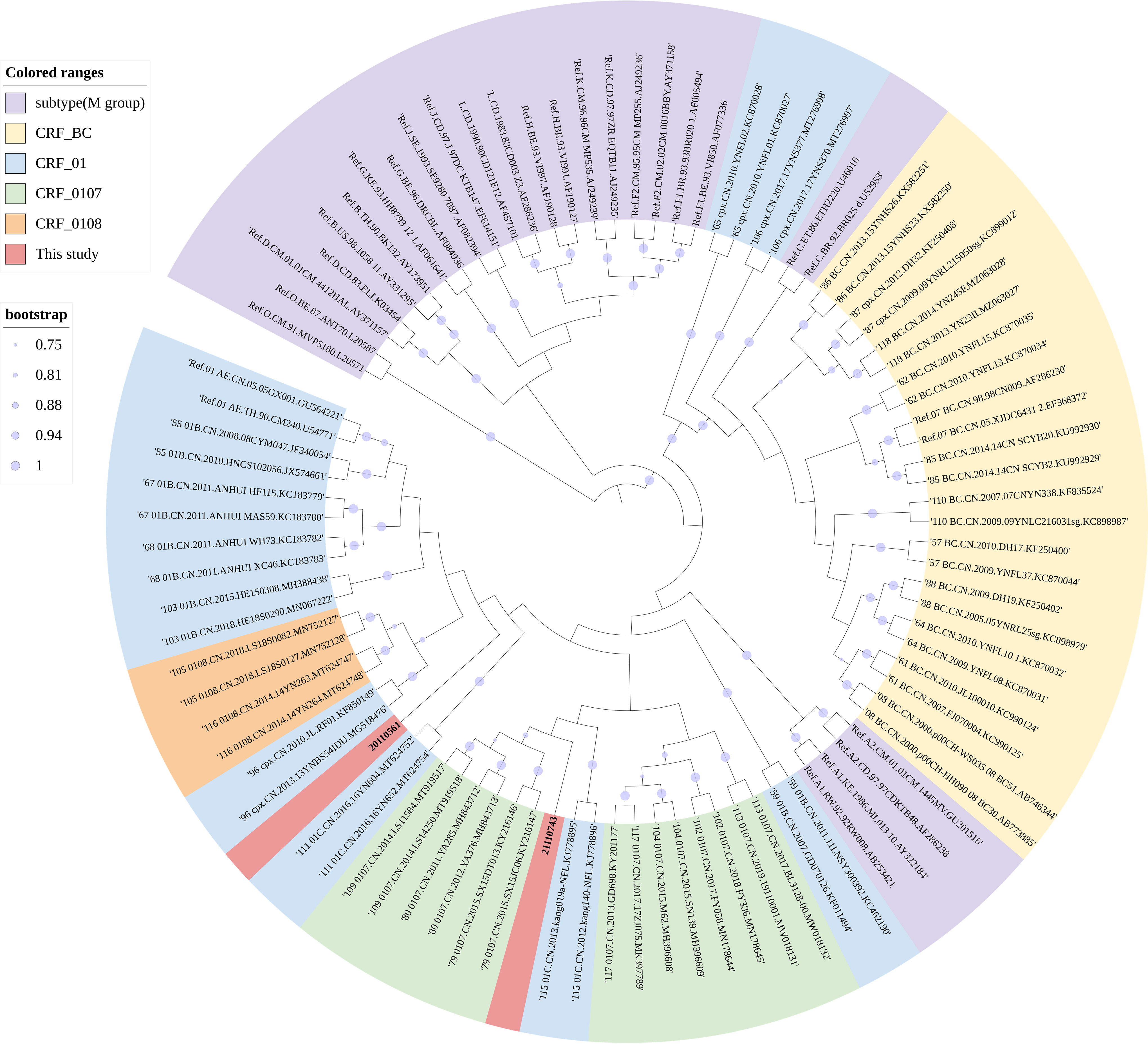
Phylogenetic tree analysis. A Neighbor-joining phylogenetic tree of 20110561 (8202 bp) and 21110743 (8231 bp) was constructed based on the NFLG sequences using Mega6.0. Bootstrap tests with 1,000 replicates assessed the stability of each node, and Bootstrap Value below 75 is no markings, while a value between 0.75 and 1.0 is displayed in five sizes of purple circles. Pure subtypes are represented with a light purple background, B and C subtype recombination with a yellow background; B, C, and CRF_01AE recombination with a light blue background; CRF_0107 are represented with a light green background, and CRF_0108 are represented with a light orange background.
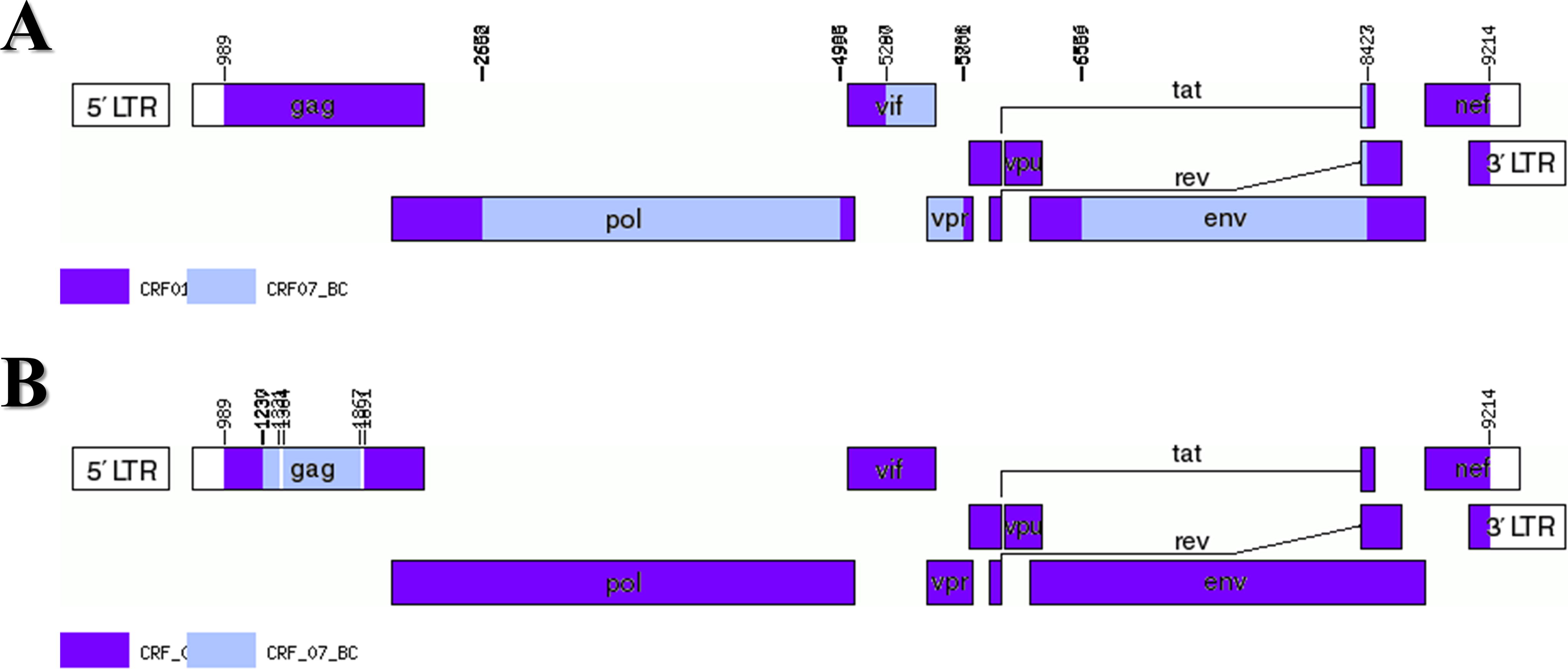
Genome maps of the NFLG of 21110743
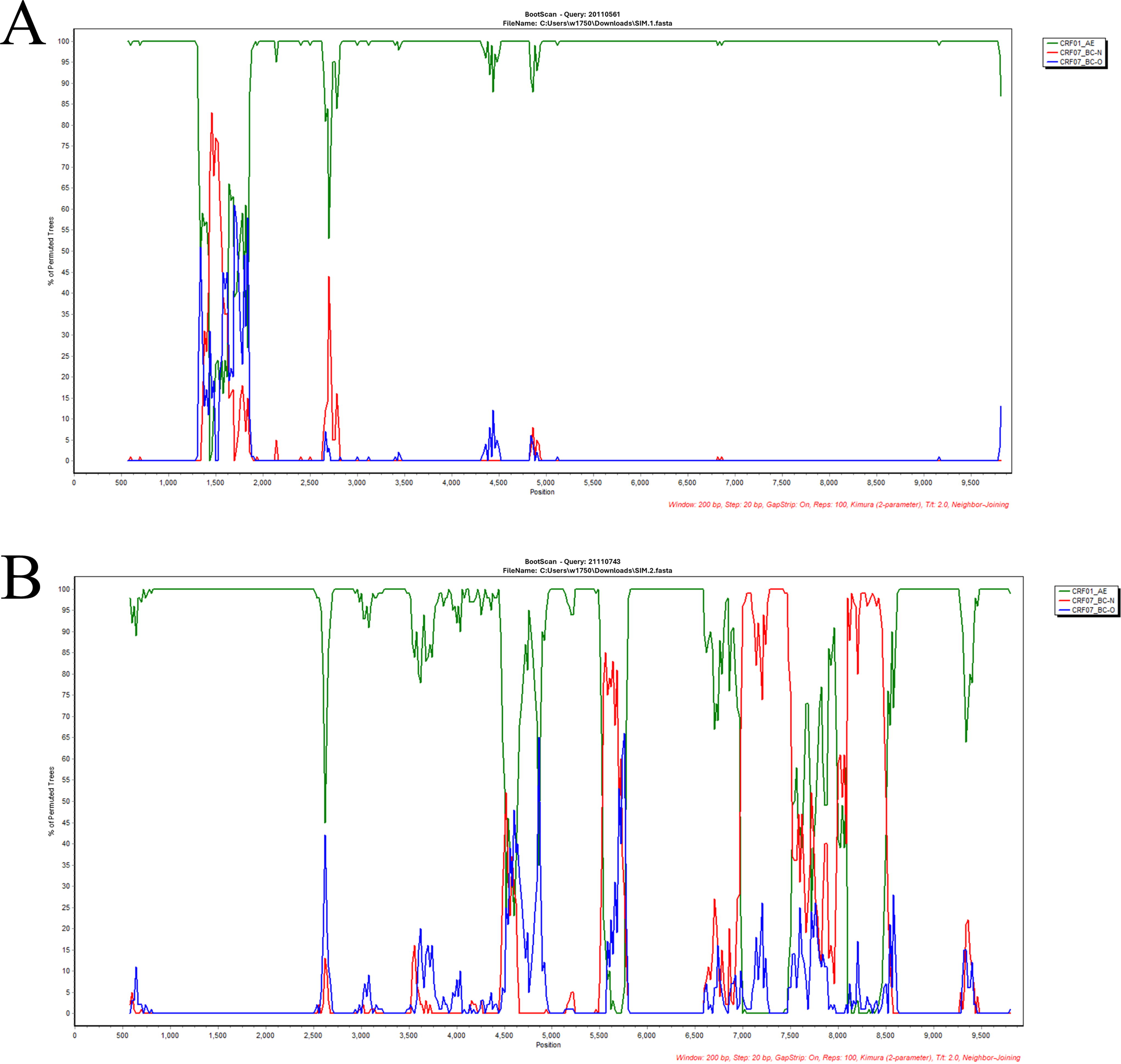
Bootscanning plot (up panel) and similarity plot (below panel) of 20110561 and 21110743. These analyses were performed using SimPlot v.3.5.1 with subtypes CRF07_BC and CRF01_AE as reference sequences. The bootscan window was 200 bp with a step size of 20 bp.

Subregion phylogenetic tree. The subregion tree analyses of 20110561 and 21110743 were constructed by Mega6 through the Neighbor-joining method with 1,000 bootstrap replications. The 20110561 and 21110743 are labeled with the red solid circle (). Bootstrap values ≥75% were shown at the corresponding nodes. The scale bar represents 5% genetic distance.
In this study, two new recombinant strains of HIV-1 (20110561 and 21110743) were isolated from MSM in Tongzhou. The recombination patterns of the two NFLG sequences differed significantly from the URF sequences published in open reports and papers. Previous recombination breakpoints in URF mostly occurred in structural genes such as gag, pol, and env gene regions. However, the NFLG sequence reported in this study (21110743) with CRF01_AE subtype as the genomic backbone showed a segment was inserted from CRF07_BC subtype in the regulatory gene region (vif-vpu) (about 600 bp). The two URF source subtypes and populations were consistent. Therefore, the strains CRF01_AE and CRF07_BC were co-prevalent among MSM. Cluster 4 was the primary cluster for CRF01_AE, which has higher levels of X4 phagocytosis than cluster 5. As the viral infection progresses, there was the possibility that viral tropism would shift from R5 to X4 phagocytosis. CRF07_BC was widely spread in China due to migration for economic, cultural, and technological. The emergence of these new complex recombinant forms had increased the diversity of HIV-1 prevalence in the region. Therefore, it is necessary to focus on monitoring the evolution of the HIV-1 gene at the molecular level in Tongzhou. Strengthening infection control measures in MSM populations to reduce HIV-1 infection in other at-risk groups.
Footnotes
Sequence data
The two NFLG sequences reported in this study have been submitted to the GenBank database under accession numbers PP968711-PP968712.
Author Disclosure Statement
No competing financial interest exist.
Authors’ Contributions
C.W.: Experimental operation, data analysis; X.G.: Article writing, experimental operation, data analysis; L.L.: Provide experimental samples; J.G.: Sequence assembly; J.Z.: RNA extraction, Inform informed consent; Y.F.: Provide HIV reference sequences; Z.L.: Contact the sampler; A.T.: Article correction; J.W.: Article correction; X.L.: Article correction; H.L.: Article correction, laboratory procedure; L.L.: Provide experimental conditions.
Funding Information
The financial support in this study derived from the National Key Research and Development Program of China under Grant numbers (2022YFC2304900, 2022YFC2305202), Science & Technology Plan Projects of Beijing Tongzhou District under project number (KJ2022CX078), and the National Natural Science Foundation of China under Grant number (82173583), so we expressed a sincere gratitude to them.