
Abstract
Many new circulating recombinant forms (CRFs) and unique recombinant forms (URFs) of human immunodeficiency virus type-1 (HIV-1) have been discovered in populations with multiple circulating HIV-1 genotypes. In this study, we report two novel URFs derived from two individuals who were HIV-1 positive in Hebei, China, who were infected through homosexual (BDD142) and heterosexual (BDD154) contact. Phylogenetic and recombinant analyses of the two NFLG revealed that they are second-generation recombinant strains originating from the CRF01_AE cluster 4/B and CRF01_AE cluster 5/B strains. The BDD142 viral genome consists of a subtype B fragment inserted into a CRF01_AE backbone, whereas the BDD154 virus genome consists of two subtype B fragments inserted into a CRF01_AE backbone. Prompt monitoring of molecular epidemiological shifts of HIV-1 within sexually transmitted populations and enhanced behavioral interventions targeting this group are imperative to mitigate the spread of HIV-1 effectively.
Introduction
Acquired immunodeficiency syndrome is a global epidemic caused primarily by human immunodeficiency virus type-1 (HIV-1). Marked characteristics of HIV-1 are its high mutagenicity and response to various environmental pressures by genome adaptation. 1 Recombination and mutation are important drivers of HIV-1 diversification, promoting evasion of host immune systems and resistance to antiretroviral drugs. 2 Currently, coinfections and the recombination of different HIV-1 genotypes have led to the emergence of 158 circulating recombinant forms (CRFs) (https://www.hiv.lanl.gov/components/sequence/HIV/search/search.html) and a considerable number of unique recombinant forms (URFs).
Hebei is a northern province of China with a low HIV prevalence. 3 In 2020, Lu et al. 4 reported the prevalence of HIV-1 in Hebei Province, with 98.9% of infections arising through sexual contact, among which 77.5% arose from men who have sex with men (MSM). The three main subtypes reported in Hebei Province were CRF01AE (49.6%), CRF07-BC (29.7%), and B subtype (13.0%). In 2016, Hebei Province reported the first discovery of URF (CRF01_AE/B), 5 and in the subsequent decade, many URFs (URFs_0107, URFs_01B, CRF01_AE/C, and CRF68_01B/CRF01_AE) 6 –9 and four new CRFs (CRF80_0107, CRF103_01B, CRF123_0107, and CRF159_01103) 10 –13 were successively reported. In this study, we identified two novel URFs derived from two individuals who were HIV-1 positive in Hebei, China, who were infected through homosexual (BDD142) and heterosexual (BDD154) contact. These two URFs were composed of subtypes CRF01_AE and B.
The two individuals who were HIV-1 positive (BDD142 and BDD154) were diagnosed with HIV-1 by the Center for Disease Control and Prevention of Baoding City, Hebei Province, China, in October 2022 and April 2023, respectively. BDD142 is a 47-year-old male infected through homosexual contact, with a baseline CD4+ T cell count of 38 cells/µL, whereas BDD154 is a 39-year-old female infected through heterosexual contact, with a baseline CD4+ T cell count of 498 cells/µL. The study was approved by the Medical Ethics Committee of Baoding People’s Hospital (protocol number: 2019-03). The patients signed written informed consent forms before sample collection.
Viral RNA was extracted from the plasma (280 μL) of each subject using the QIAamp Viral RNA Mini Kit (Catalog no. 52904, Qiagen, Hilden, Germany) in accordance with the manufacturer’s instructions. PrimeScript™ III Reverse Transcriptase (Code No. TCH0 13, TaKaRa Biotechnology, Dalian, China) was used to reverse transcribe the RNA into 3′ and 5′ half-molecule cDNAs using the primers 1.R3. B3R: 5′-ACTACTTGAAG CACTCAAGGCAAGC TTTATTG-3′ and 07Rev8: 5′-CCTART GGGATGTGTACTT CTG AACTT-3′. Nested polymerase chain reaction (PCR)s were performed using TaKaRa PrimeSTAR® GXL DNA Polymerase (Code No. R050A, TaKaRa Biotechnology, Dalian, China) to amplify two regions of the near-full-length genome (NFLG). The primers used for PCR have been reported previously. 13 The PCR conditions for both rounds were 98°C for 20 seconds, followed by 30 cycles of 98°C for 10 seconds, 55°C for 15 seconds, and 68°C for 6 minutes, and a final extension step at 68°C for 10 minutes. Amplification products were detected using 1.0% agarose gel electrophoresis, and amplified products of the expected size were purified from the corresponding electrophoretic bands and sequenced using Sanger sequencing technology by Tianyi Huiyuan Bioscience & Technology Inc. (Beijing, China). Standard reference sequences for HIV-1 subtypes were downloaded from the HIV database (https://hiv.lanl.gov/components/sequence/HIV/search/ search.html). The two NFLG sequences were aligned with subtype reference sequences and CRFs from China using MAFFT v7.0 and manual editing with ClustalW in Bio-Edit 7.0. The NFLG phylogenetic tree was constructed by FastTree v2.1.10 based on the approximate maximum likelihood method with the general time reversible model and adjusted with Figtree v1.4.4. Online jpHMM and SimPlot 3.5.1 software were then used for similarity mapping and bootscan analysis to identify the recombination breakpoints.
The NFLG phylogenetic tree (Fig. 1) showed that the two sequences formed a monophyletic branch separate from other subtypes and CRFs. JpHMM and boot scanning analyses indicated that BDD142 and BDD154 are composed of three interlaced mosaic gene segments, including two CRF01_AE subregions (I, III) and the 1B region (II), with two recombinant breakpoints relative to the HXB2 coordinate (Figs. 2 and 3). The recombinant mosaic structure of the two sequences is described as follows: I CRF01_AE (HXB2, nt 774–2,340), IIB (HXB2, 2,341–5,157 nt) and IIICRF01_AE (HXB2, 5,158–9,602 nt) for BDD142 (Fig. 3A), and ICRF01_AE (HXB, 2,774–2,122 nt), IIB (HXB2, 2,123–5,152 nt) and IIICRF01_AE (HXB2, 5,153–9,457 nt) for BDD154 (Fig. 3B). Subregional phylogenetic analysis revealed that the CRF01_AE region of BDD142 descends from the CRF01_AE cluster 5 lineage (Fig. 4A), whereas the CRF01_AE region of BDD154 descends from the CRF01_AE cluster 4 lineage (Fig. 4B). The subtype B regions for both URFs were clustered within the northern China subtype B lineage (Fig. 4A and 4B).
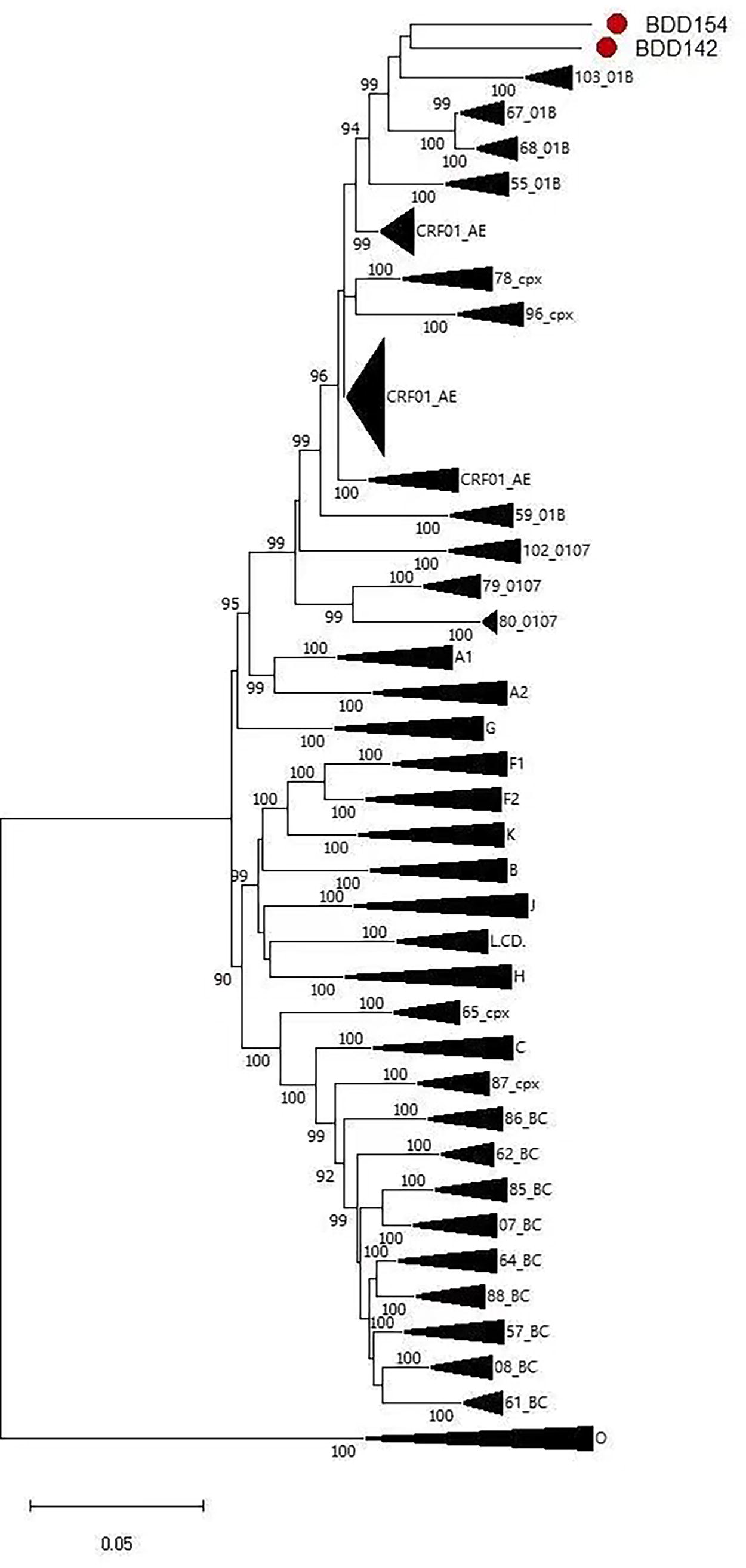
The phylogenetic tree with the two NFLG sequences. The phylogenetic tree was constructed by FastTree v2.1.10 based on the approximate maximum likelihood method with the general time reversible model and adjusted with Figtree v1.4.4. Only bootstrap values >90% are shown in the corresponding nodes. The scale bar represents a 5% genetic distance.
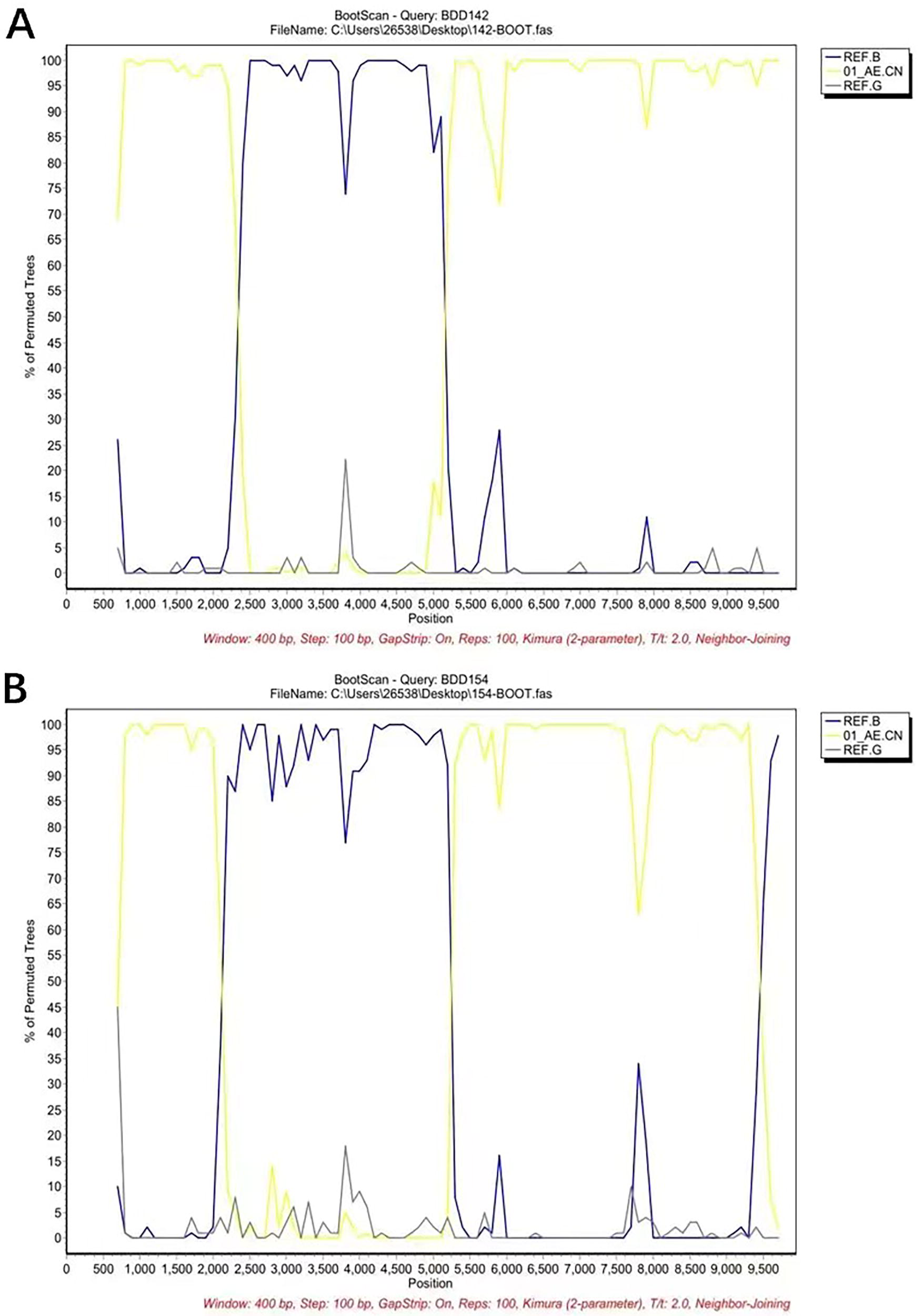
Bootscan results of the novel CRF01_AE/B have been identified.
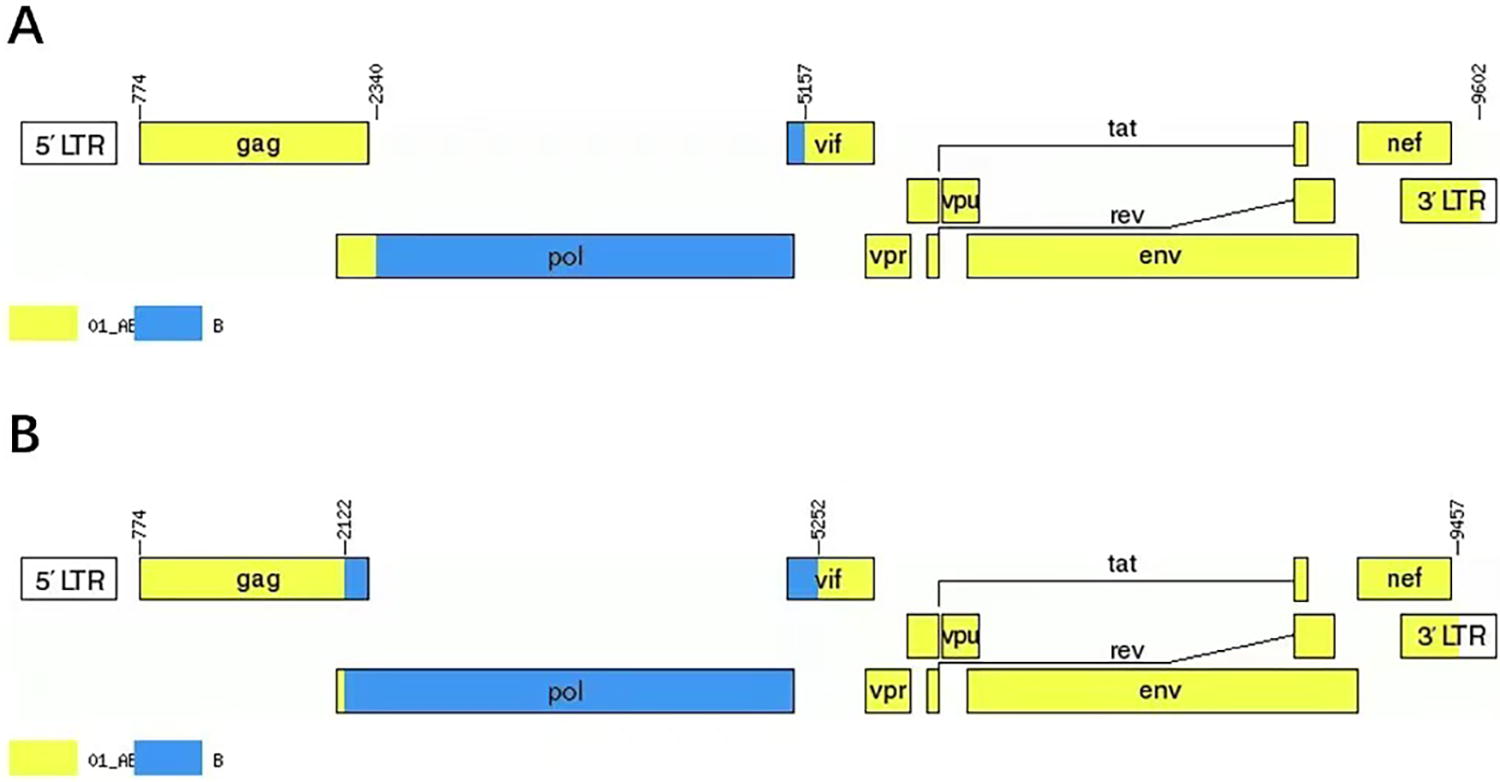
Genetic map of

The subregion trees of
Prior to 2005, the primary mode of HIV-1 transmission in Hebei Province was blood-borne, with subtype B being the dominant strain. 5 However, as the transmission shifted toward sexual contact, particularly among MSM, CRF01_AE has emerged as the leading strain in Hebei, with subtype B now ranked third. 14 According to relevant research, 15 the widespread dissemination of CRF01_AE formed seven CRF01_AE clusters with different geographic distributions and epidemic patterns in China. Among them, CRF01_AE clusters 4 and 5 were prevalent among MSM in northern cities. In this study, phylogenetic analysis of BDD142 revealed that the recombinant fragments within the CRF01_AE subtype can be traced back to cluster 5 of CRF01_AE strains. Similarly, phylogenetic analysis of BDD154 indicated that the recombinant fragment in the CRF01_AE subtype clustered with cluster 4 of CRF01_AE. Given that BDD154 is female, dual infection can be confidently excluded. Thus, this URF was most likely transmitted directly through sexual contact. This finding suggests that some URFs may have already circulated in a limited area within Hebei Province.
In this study, we identified two novel URFs from two individuals who were HIV-1 positive in Hebei, China, who were infected through homosexual and heterosexual contact. Given the MSM population’s predisposition to multiple sexual partners, bisexual behavior, and unprotected sexual practices, they are considered a high-risk group for HIV-1 infection and serve as a “bridge population” for HIV-1 transmission. Therefore, it is imperative to promptly monitor the molecular epidemiological shifts of HIV-1 within the MSM population and enhance behavioral interventions among this group to control the spread of HIV-1 effectively.
Footnotes
Acknowledgments
The authors thank Liwen Bianji (Edanz) (www.liwenbianji.cn) for editing the English text of a draft of this article.
Sequence Data
The nucleotide sequences of BDD142 and BDD154 have been deposited in the NCBI GenBank database under accession numbers PQ099878 and PQ099879, respectively.
Authors’ Contributions
Z.Z., Y.Z. and H.S. designed the study. J.J., S.C. and W.F. acquired the sequences. S.C. and H.S. analyzed and interpreted the data. Z.Z., Y.Z., and J.M. wrote the manuscript. All authors read and agreed to the published version of the manuscript.
Ethics statement
The studies involving human participants were reviewed and approved by The People’s Hospital of Baoding, Baoding Ethics Committee. The patients/participants provided written informed consent to participate in this study.
Author Disclosure Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding Information
This study was supported by the research project of Hebei Provincial Health Commission (No. 20240874).