
Abstract
Although HIV-1 infection has now become a treatable chronic condition and not the deadly illness it once was, the costs of that treatment are substantial, and each infection prevented saves both financial and other costs. In China, the most predominant subtypes are CRF07_BC, CRF01_AE, and CRF55_01B, and the various second-generation recombinants are produced from the recombination between these subtypes. HIV full-length genome sequences can provide important information on their epidemiology. In this study, we identified two unique recombinant forms (URFs) designated as JLCC230106 and XJWQ230011, which are composed of CRF01_AE/CRF07_BC and CRF07_BC/CRF55_01B, respectively. Phylogenetic and recombinant analyses utilizing near-full-length genome (NFLG) confirmed that these URFs originated from CRF01_AE/CRF07_BC and CRF07_BC/CRF55_01B strains. The emergence of novel recombinants is increasing the genetic diversity of HIV in China. This information can be shared with clinicians, human behavior specialists, or public health policymakers and used as an aid in discovering which methods are best or most cost-effective in combating the spread of HIV.
Introduction
Since the discovery of HIV in the 1980s, a significant number of HIV-1 subtypes have been reported continuously. To date, 160 distinct HIV-1 subtypes, including circulating recombinant forms (CRFs), have been reported, and over 60 of them originate from China (https://www.hiv.lanl.gov/components/sequence/HIV/crfdb/crfs.comp). Among the prevalent subtypes in China, CRF07_BC, CRF01_AE, and CRF55_01B represent the three most dominant variants, collectively accounting for 74% of HIV-1 new infections in 2022. 1
HIV-1 CRF01_AE was initially identified among sex workers and injection drug users (IDUs) in the provinces of Guangdong, Guangxi, and Yunnan during the period of 1996–1997. Subsequently, it disseminated rapidly to other provinces along the southeastern coast. 2 In contrast, CRF07_BC emerged in northwestern China among IDUs along a drug trafficking route throughout the 1990s. 3 Furthermore, CRF55_01B experienced significant growth from 2005 to 2009, following its emergence in Shenzhen, and its expansion accelerated markedly after 2010. This particular strain began to spread to other provinces in 2007, and post-2010, CRF55_01B demonstrated a trend of rapid dissemination and epidemic expansion from the Guangdong-Shenzhen region to additional provinces across China. 4
The extensive prevalence of various subtypes of HIV facilitates recombination between them. In recent years, second-generation recombinants have emerged frequently, and most of them are CRF01_AE/CRF07_BC recombinants. The initial identification of the CRF79_0107, which was combined with the CRF01_AE/CRF07_BC recombinant, was documented in 2017, 5 followed by a rapid emergence of 22 additional cases within an 8-year period. No recombinants of CRF07_BC/CRF55_01B had been reported until 2024; however, in 2024, six CRF07_BC/CRF55_01B recombinants were identified from men who have sex with men (MSM). (https://www.hiv.lanl.gov/components/sequence/HIV/crfdb/crfs.comp).
In this study, we identified two URFs that comprise CRF01_AE/CRF07_BC and CRF07_BC/CRF55_01B, respectively, in Jilin province and Xinjiang province, northern China.
Materials and Methods
Donors XJWQ230011 (Urumqi, Xinjiang Province) and JLCC230106 (Changchun, Jilin Province) are 26 years old and 19 years old, respectively. Both were infected with HIV-1 through MSM. Additional information about the patients is provided in Table 1. The studies involving human subjects were approved by the Institutional Review Boards of the National Center for AIDS/STD Control and Prevention, Chinese Center for Disease Control and Prevention (X140617334).
Demographic Characteristics of the Participants with HIV-1 Infection
The NFLG sequences of two HIV-1 isolates were successfully obtained from plasma samples using a two-half-molecule approach. 6 The two NFLG sequences were then aligned with reference sequences and circulating recombinant forms (CRFs) in China using Aliview (version 1.28) and manually adjusted with BioEdit (version 7.2.5.0). Additionally, phylogenetic trees of the NFLG sequences and subregional trees were constructed using the maximum likelihood (ML) method with IQ-TREE 2 software. The recombinant breakpoints of the sequences were identified using the online Jumping Profile Hidden Markov Models (jpHMM), the Recombinant Identification Program (RIP 3.0), and SimPlot 3.5.1. The final recombination pattern for each recombinant strain was illustrated using the Recombinant HIV-1 Drawing Tool (https://www.hiv.lanl.gov/content/sequence/DRAW_CRF/recom_mapper.html).
The two NFLG sequences were submitted to the Stanford Drug Resistance Database (https://hivdb.stanford.edu/hivdb/by-sequences/), and the HIVdb program was utilized to assess drug resistance-related mutations. The Ge-no2pheno (coreceptor) 2.5 online tool was used to predict the use of coreceptors by the corresponding strains according to the sequence of V3 loop amino acids, and the false positive rate was set to 10.00%.
Results and Discussion
The NFLG sequences derived from JLCC230106 and XJWQ230011 were 8,893 bp and 9,029 bp in length, respectively, spanning from the gag region to the U3 region of the 3' long terminal repeat. These two sequences formed a unique monophyletic branch with a bootstrap value of 95% and 85%, respectively, indicating a distant relationship to all known HIV-1 CRFs, as determined by phylogenetic analysis of the NFLG (Fig. 1). The mosaic structures were further analyzed using RIP 3.0, jpHMM, and BootScanning analyses individually. As shown in Figure 2, JLCC230106 is a recombinant of CRF01_AE/CRF07_BC, while XJWQ230011 is a recombinant of CRF07_BC/CRF55_01B. The subregions of the two NFLG sequences are as follows: JLCC230106: ICRF01_AE: HXB2, 633-1283; IICRF07_BC: HXB2, 1284-6252; IIICRF01_AE: HXB2, 6253-9601; XJWQ230011: ICRF55_01B: HXB2, 633-1309; IICRF07_BC: HXB2, 1310-2622; IIICRF55_01B: HXB2, 2623-4113; IVCRF07_BC: HXB2, 4114-4428; VCRF55_01B: HXB2, 4429-4697; VICRF07_BC: HXB2, 4698-4904; VIICRF55_01B: HXB2, 4905-9081; VIIICRF07_BC:HXB2, 9082-9479. The subregion trees were also constructed for each fragment (Fig. 3).
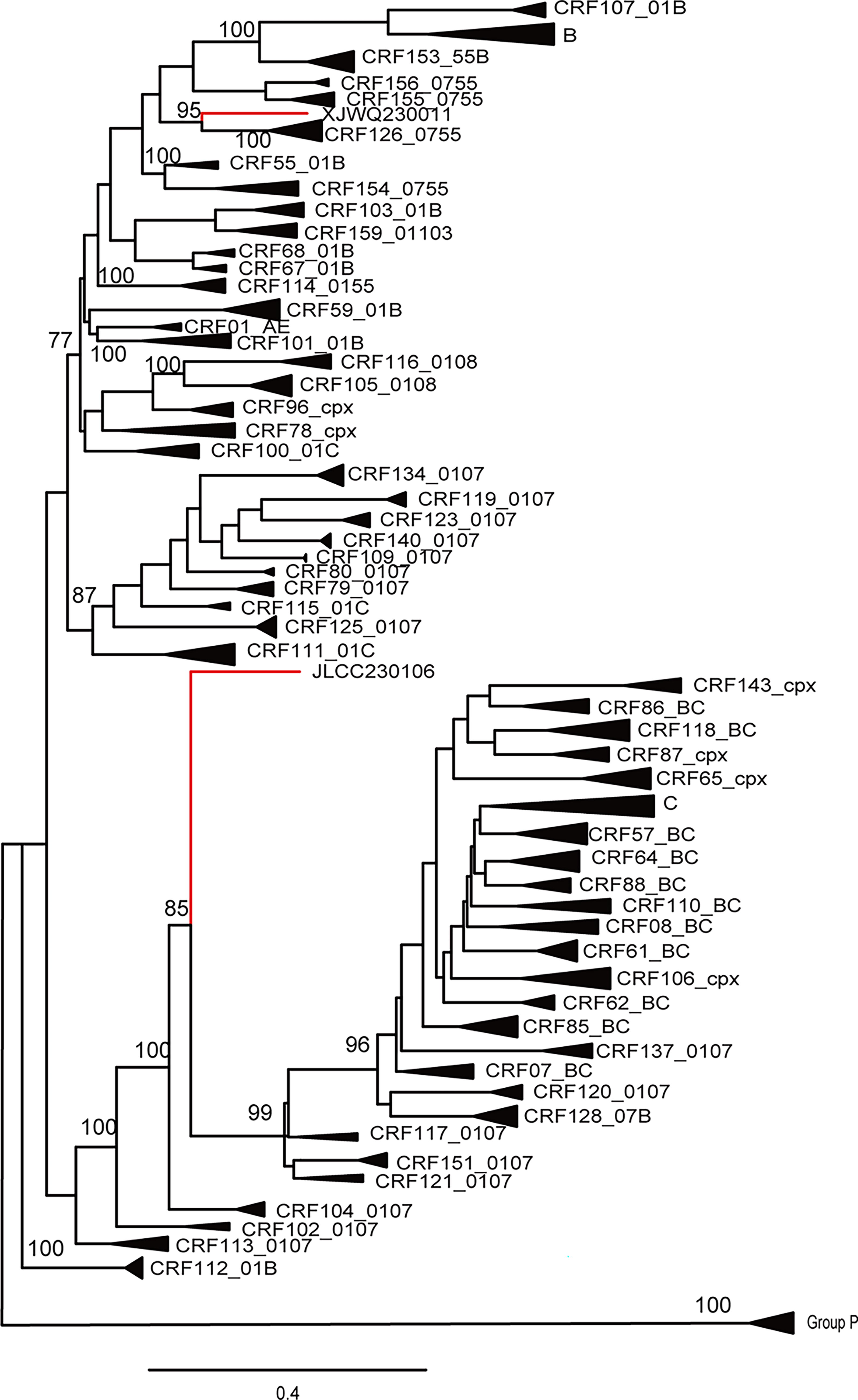
A neighbor-joining phylogenetic tree was constructed for JLCC230106 and XJWQ230011 based on the NFLG sequences using IQ-TREE 2. Red line denotes study subject.
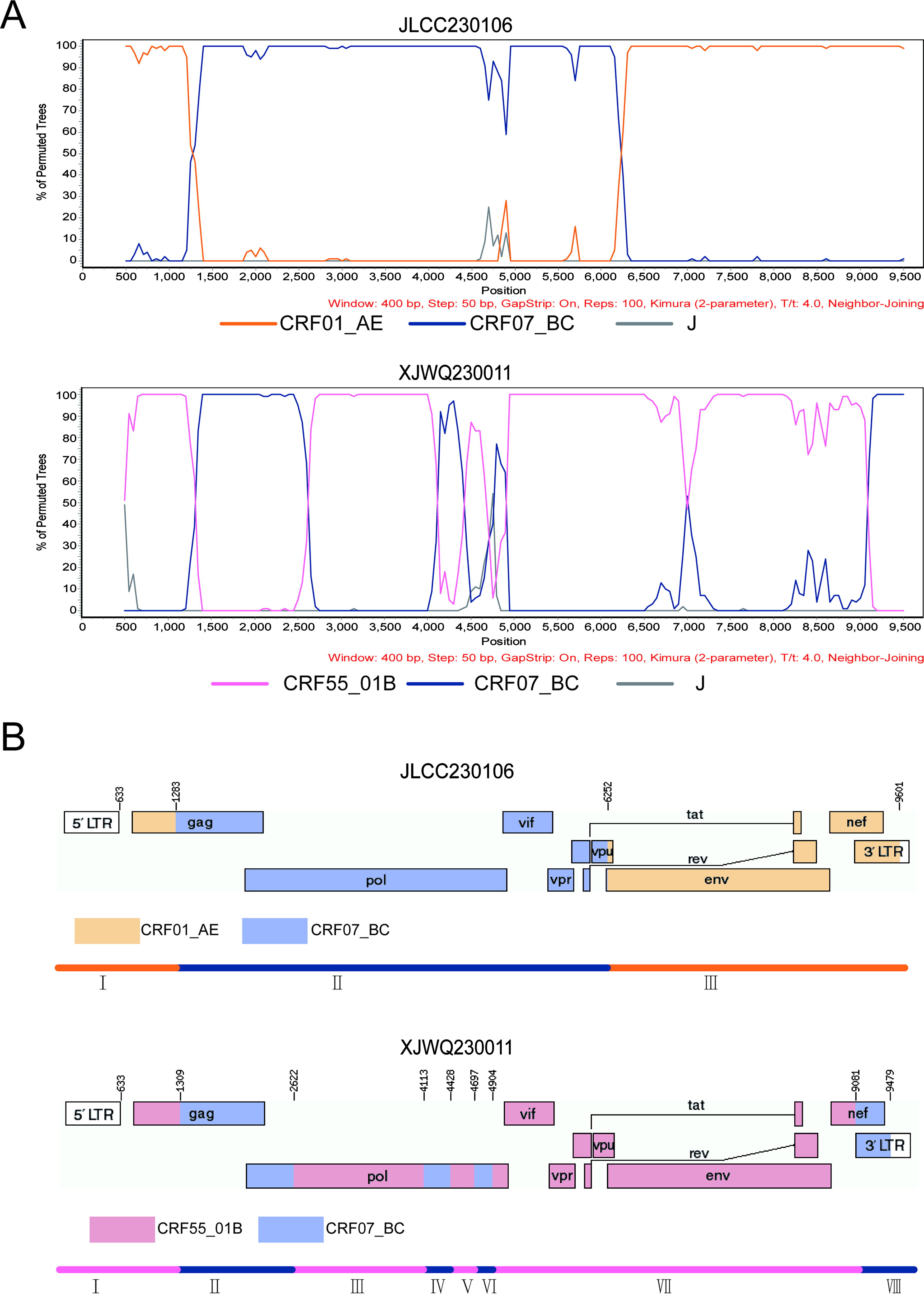
Bootscanning analyses

Subregion tree analysis of two HIV-1 URFs. Phylogenetic analyses of different segments of JLCC230106
Drug resistance analysis indicated that the two NFLGs did not exhibit drug resistance-related mutations in the protease and integrase regions. However, in XJWQ230011, V179D (a non-nucleoside reverse transcriptase inhibitors-related mutation) presented in the reverse transcriptase region. This mutation conferred intermediate resistance to efavirenz, emtricitabineand nevirapine, as well as potential resistance to rilpivirine. In JLCC230106, D67N (a nucleoside reverse transcriptase inhibitor-related mutation) presented in the reverse transcriptase region. This mutation resulted in the development of intermediate resistance to zidovudine (AZT) and stavudine, characterized as low-level resistance. Both sequences are predicted to use CCR5 as a coreceptor.
In this study, we identified two HIV-1 URFs from Jilin province and Xinjiang province. In recent years, the HIV-1 diversity prevalent in China has gradually increased, particularly with the continuous emergence of second-generation recombinants, including CRFs and URFs. 7 –9 This trend may be attributed to rapid economic development and increased population mobility within China, which have facilitated the HIV transmission. 4 The occurrence of superinfection with different viral subtypes in the same geographic area is a critical precursor for viral recombination. 10 Given that CRF01_AE and CRF07_BC are the most prevalent subtypes, many second-generation recombinants are new strains derived from CRF01_AE and CRF07_BC. 11,12 Conversely, CRF55_01B, a more recently identified strain, has exhibited low pathogenicity coupled with high transmissibility, leading to its rapid spread and the emergence of numerous second-generation recombinants, including CRF55_01B, within the current year. 13 –15 The emergence of these second-generation recombinants may complicate the genomic structure of HIV-1, potentially posing significant challenges to HIV prevention and control efforts, as well as to the development of effective treatments and vaccines. Therefore, it is crucial to monitor the novel HIV-1 subtypes, particularly in the MSM population, to formulate more effective prevention strategies.
Footnotes
Acknowledgments
Thank all the personnel from Xinjiang Uygur Autonomous Region Center for Disease Control and Prevention and Jilin Center for Disease Control and Prevention who participated in this research.
Sequence Data
The gene sequences of JLCC230106 and XJWQ230011 were deposited in the GenBank with the accession numbers PQ568110 and PQ568111, respectively.
Authors’ Contributions
Z.W., H.X., and LJ.L.: Designed the study; H.L. and Q.G.: Performed the experiments; H.L., Q.L., and Q.G.: Participated in sample storage, sequences assembly; H.L. and Y.F.: Providing HIV reference sequences and data analysis; H.L. and Z.W.: Participated in article writing and correction.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
Supported by the National Key Research and Development Program of China (NO. 2022YFC2305201).