
Abstract
Introduction:
We are using big-data mining to develop computational models that predict whether previously uncharacterized compounds will or will not target important biological pathways. Mitochondria play essential life-sustaining roles, with their dysfunction linked to diverse pathologies.
Materials and Methods:
We built a mitochondrial inhibition model that combines molecular scaffolding and fingerprinting of a large database compiled primarily from in vitro high-throughput screening (HTS) data. We refined the model to include SMARTS profilers for known subtarget features (i.e., inhibition of Complexes I–V or uncoupling through protonophore action). For some of these, compound substructures capable of metabolic transformation to cyanide or hydrogen sulfide were identified and included based on potential for high in vivo toxicity despite lack of activity in cell-based platforms that appear to lack critical metabolic pathways and/or cell sensitivity.
Results and Discussion:
The model is comprehensive—the machine-learning component has high sensitivity (80.3%) and accuracy (79.4%), together with positive and negative predictive values of 60.9% and 90.7%, respectively. Model predictivity is limited by the heterogeneity of mechanistic mitochondrial targets, as well as sensitivity of HTS assays and domain of tested compounds. When applied to a database of human therapeutics withdrawn from the market due to liver injury, it identified all compounds demonstrated to target mitochondria.
Conclusions:
The model can be used in an integrated approach to complement early in vitro screening and as a covariate in Quantitative Structure Activity Relationship (QSAR) models for systemic toxicological end points.
Introduction
Mitochondria are organelles that play a central role in the homeostasis of most eukaryotic cells, including production of energy, control of apoptosis, 1 generation of reactive oxygen species, and subcellular compartmentalization for key metabolic enzymatic reactions. Approximately 90% of cellular adenosine triphosphate (ATP) is generated within mitochondria through the Krebs/tricarboxylic acid cycle and oxidative phosphorylation.2–4 Mitochondria also mediate steroid biosynthesis, 5 are involved in mammalian reproductive oocyte competence and embryo implantation, 6 and play a key role in providing the energy for neurotransmission. 7
Structurally, the mitochondrion contains an inner and outer matrix that allows for the development of a hydrogen ion-based transmembrane potential with an internal net negative charge. By reducing molecular oxygen and shunting electrons along a series of four polypeptide respiratory complexes (Complex I, II, III, and IV), the resulting potential difference allows for the driving of a proton importer (Complex V/ATP synthase) that generates ATP through phosphorylation of adenosine diphosphate. 8 Since mitochondria evolved from an endosymbiotic relationship between aerobic bacteria and early eukaryotic cells, they retain their own genome, which encodes 13 proteins that are components of the respiratory complexes. 9 These protein complexes are sensitive to inhibition by certain classes of compounds such as rotenone (Complex I), hydrogen cyanide and hydrogen sulfide (Complex IV), 10 and antimycin A (Complex III). 7
Given the unique structure of the mitochondrion, xenobiotics that target the respiratory complexes or act as uncouplers through a protonophoric mechanism can attenuate the mitochondrial membrane potential and impair the transport of electrons and downstream synthesis of ATP. 11 Protonophores uncouple the transmembrane potential by shuttling protons across the mitochondrial membrane. This can inhibit energy generation and induce apoptosis, which could lead to local cytotoxicity. 12 Substances that inhibit mitochondrial function are of interest to the pharmaceutical industry since this organelle represents an off-target toxicity that has resulted in the withdrawal of certain drugs (e.g., Tacrine) from the market.13,14 Likewise, the mitochondrial respiratory chain represents a potential target for cancer therapies, and the ability to rapidly identify substances that affect the respiratory complexes would be of value in the development of novel chemotherapeutic agents. 12
In addition to xenobiotic-mediated dysfunction, inherited mutations in mitochondrial DNA result in damage to many key organ systems (nervous system, liver, eyes, kidneys, etc.). 15 Mitochondrial dysfunction arising from mutations in PINK1 has been implicated in promoting neurodegeneration in conditions such as Parkinson's disease. 16
The mechanistic targets for most types of systemic mammalian toxicity are not well understood; thus toxicity is assessed through in vitro or in vivo screening. Given the central role played by mitochondria in eukaryotic biology, the development of a computational model to identify compounds that disrupt mitochondrial function would allow for its use in an integrated approach that complements early screening. The output of such a model could also be used as a covariate in Quantitative Structure Activity Relationship (QSAR) models for systemic toxicological end points.
Compounds that disrupt mitochondrial function share a defined set of structural motifs (scaffolds) that drive their biological function. Once identified, these scaffolds can be used to differentiate mitochondrial inhibitors from compounds that would not affect mitochondrial function. Previously, in silico scaffolding studies have been undertaken on select classes of compounds that disrupt mitochondrial function.17,18 In addition, a profiler for identifying substances that cause mitochondrial dysfunction through a protonophoric mode of action has been recently described by Enoch et al. 19 In addition, analysis of high-throughput in vitro assays for mitochondrial dysfunction 20 has been undertaken to predict the ability of novel compounds to target the mitochondrion. However, the scope of these investigations was limited, and negative (control) data were not incorporated within a computational prediction model. Thus, we sought to build a robust model to predict mitochondrial inhibition using two-dimensional (2D) structural characterization of both known inhibitors, as well as control compounds, from large datasets primarily derived from in vitro high-throughput screening (HTS) data.
We developed a hybrid approach that uses 2D cheminformatics techniques (molecular scaffolding and structural fingerprinting) coupled with a random-forest machine-learning/SMARTS profiling to predict mitochondrial inhibition. The basic procedure for scaffolding involves determination of the maximal common substructure of structurally similar clusters of molecules and was implemented within the open-source Konstanz information miner (KNIME) environment (an overview of a generic workflow for the models is shown in Fig. 1). In addition, a series of scaffolds that reflect substructures of known mitochondrial inhibitors were implemented as SMARTS patterns such that compounds containing these substructures were flagged as mitochondrial inhibitors and flagged for the specific submechanism(s). In such instances, our model was optimized for sensitivity such that any novel compounds initially characterized using the machine learning model as “negative” for mitochondrial inhibition but identified as “positive” using the SMARTS filter were labeled as positive in the model outcome. This is a conservative approach to avoid missing potentially disruptive compounds. To the best of our knowledge, our model is the first to predict both positive and negative outcomes for mitochondrial inhibition using scaffolding, fingerprinting, and machine-learning methodologies.
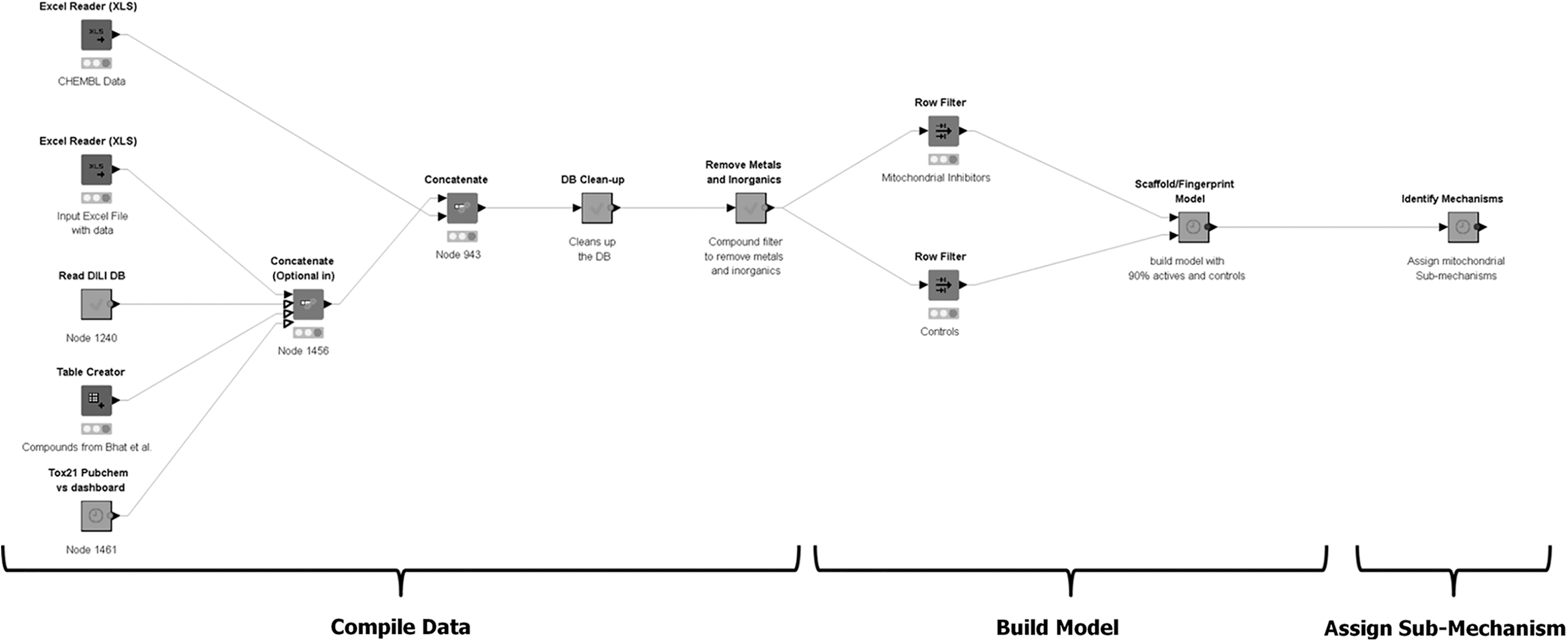
Development of the mitochondrial profiler depicting an overview of the modeling approach. Figure depicts the implementation of the mitochondrial profiler within a KNIME workflow. KNIME, Konstanz information miner.
Materials and Methods
Databases
A database of mitochondrial inhibitors and control compounds (those that do not decrease the mitochondrial membrane potential) was compiled from the published literature17,21,22 and public databases such as CHEMBL23–25 and the ToxCast and Tox21 high-throughput screens for compounds that decrease mitochondrial membrane potential (APR_HepG2_MitoMemPot_24h_down and TOX21_MMP_ratio_down). 26 The Tox21 assay results were curated using the assay data in PubChem (assay ID 720637; revision 2.1) and using the Environmental Protection Agency (EPA) of the United States dashboard (accessed in October 2017). The ToxCast assay results were downloaded from the EPA dashboard. For each compound, its name, ID (CAS number of CHEMBL ID), and structure (encoded as SMILES strings) were imported into KNIME (ver. 3.6) along with a flag indicating whether it was active or negative in the indicated mitochondrial function assay (with 1 indicating that the compound was a mitochondrial toxicant and 0 indicating that it did not alter the mitochondrial membrane potential). Since our goal in developing this model is to identify mitochondrial inhibitors as a binary (yes/no) outcome, AC50 inhibition values from the ToxCast/Tox21 assays were not used.
For compounds that were duplicated across multiple sources, those that were flagged as mitochondrial inhibitors in any of the assessed data sources were marked as mitochondrial inhibitors in the final dataset to maximize the sensitivity of the profiler. Ultimately, 5961 compounds were included in the database with 1744 (29%) mitochondrial inhibitors and the remaining 4217 compounds (71%) serving as noninhibitory controls. The compiled datasets of active and inactive compounds used to build the model are available in Supplementary Tables S1 and S2, with the database of drugs that cause drug-induced liver injury (DILI) through mitochondrial dysfunction available in Supplementary Table S6. Experimentally derived pKa values for compounds were compiled using the OECD ToolBox (ver 3.5) and verified against values reported in ChemIDplus to ensure accuracy. Lipophilicity was assessed through calculation of logKow values using the KOWWIN module within Epi Suite (US EPA).
Model development
A model to predict the likelihood that a compound would inhibit mitochondrial function was built within the KNIME environment as described previously (Wijeyesakere SJ, Wilson DM, and Marty MS. Prediction of cholinergic compounds by machine-learning. Article under review for Comp Tox). Similar to the previous investigation, the outcomes of two sets of random-forest machine-learning models were used as covariates: (1) a scaffold-based model that used similarity of a test substance to scaffolds calculated from the active and control compounds and (2) a fingerprint model that used the Molecular Access Systems (MACCS) structural fingerprints. 27 When constructing the model, a random subset (10%) of the master database of active and control compounds was withheld as a test set, with the remaining compounds (90% of the dataset) used to train the model. The model was built five times with a different random subset of actives and controls (10% of the data) being withheld to test the model and derive the mean concordance statistics.
The supervised machine-learning classification algorithm used the default “Random Forest Learner (Classification)” node within KNIME. The random forest algorithm builds 100 slightly different subdecision trees from randomly sampled subsets of the training set data and merges their predictions together to make a consensus prediction regarding whether a compound is or is not a mitochondrial inhibitor. Each submodel classifies challenge (test set) compounds as positive or negative (i.e., classifies the assessed substance as a mitochondria inhibitor or not), with the final decision for each compound based on a “majority vote” of the ensemble output from the subdecision trees. For each decision, an information-gain criterion is used as the split attribute. There are no restrictions or preset limits on the number of splits in a tree (i.e., no limits on tree depth, allowing more information to be captured in the model). Models were built independently using the predefined MACCS fingerprints or derived 2D scaffolds. For example, in the fingerprint model, the model samples the fingerprints from the random subset of positive inhibitors and negative control noninhibitors pulled into each tree and then determines the best bits to use as split criterion to distinguish the inhibitors from noninhibitors.
For the development of the scaffold-based prediction model, the training set of active (mitochondrial inhibitors) and control (inactive) compounds (90% of the data) was scaffolded through the algorithm described above (actives and control datasets were scaffolded separately). The mitochondrial inhibitor and control scaffolds were then fingerprinted using the MACCS algorithm and compared to the MACCS fingerprints of the withheld active or control compounds within the training set for model building or test set for model evaluation. The Tanimoto similarities of the compounds to their matched mitochondrial inhibitor and control scaffolds were used as covariates within a random forest prediction model, with the inclusion of the molecular weight of the compound and a fragment complexity score (to account for the size and connectivity of the chemical structure). The fragment complexity metric was calculated using the Chemistry Development Kit (CDK) molecular properties node within KNIME.
The final hybrid scaffold/fingerprint-based model was constructed using predictions from the scaffold- and fingerprint-based models as sole covariates within a random forest classifier. The accuracy of the resulting model was tested against the reserved test set of compounds (10% of actives and controls) that were withheld from the training set. The hybrid model was built and queried five times, each time with a randomly selected training set (90%) and test set (10%) to generate statistics on model performance.
Several of the targets for mitochondrial inhibition include the specific respiratory protein Complexes I–V, as well as nonspecific uncouplers, that act through a protonorphoric mechanism. We mined the public literature for structural features associated with known inhibitors that act through inhibition of these targets and then encoded these alerts within a database as their respective SMARTS patterns (available in Supplementary Table S7).17,28–30 We implemented these alerts within our existing KNIME workflow such that if a compound was identified as mitochondrial inhibitor using the statistical model and fit one of these alerts, it would add a classification tag. To add further sensitivity to our KNIME workflow, we further modified it such that if a compound was not classified as a mitochondrial inhibitor using the statistical model but was flagged by one of these alerts, the activity call was overridden and the compound was classified as a potential mitochondrial inhibitor using one of these specific mechanisms.
The rational for this further modification was threefold: (1) certain substructures such as cyanide or hydrogen sulfide can exist as coordination complexes with metal ligands that undergo facile dissociation in aqueous biological environments thus releasing compounds known to inhibit Complex IV but that would not be identified in a machine-learning model that uses overall structural similarity; (2) Cytochrome P-450-mediated metabolism of organic compounds to toxic forms of cyanide or hydrogen sulfide can occur, 28 but is poorly represented in existing metabolism models or in knowledge-based prediction models. Therefore, we compiled a set of compound features that represent the scope of substructures we have encountered with the potential to undergo metabolism to toxic forms of cyanide or hydrogen sulfide and translated these as SMARTS patterns; and finally (3) there was not an existing database of compounds that act through a protonorphoric mechanism that can uncouple the proton gradient responsible for the electrochemical potential gradient within the mitochondria. We mined the literature for compounds known to act in this manner and captured their essential generic structural features as SMARTS patterns for any parent compound potentially able to function in this manner. The SMARTS patterns associated with these modes of action are detailed in Supplementary Table S7.
Statistical analyses of machine learning model
Concordance analysis between the in vitro outcomes of a compound and associated predictions from the mitochondrial profiler was undertaken to determine the sensitivity, specificity (SP), and balanced accuracy (BA) of the profiler, as well as the negative predictive values (NPVs) and the Youden index. The BA was used in lieu of a general accuracy measurement as the analyzed dataset contained unequal numbers of active and inactive compounds. 31 This statistics was calculated as follows:
All statistical analyses were undertaken in KNIME (ver. 3.6) and GraphPad Prism (ver. 6.0) with statistical significance determined at an alpha level of 5%. All analyses were repeated five times by rebuilding the model each time with a randomly selected training set (90%) and a randomly selected test set (10%), with the mean ± standard error of the mean of these five trials depicted in plots.
Data availability
All data used to construct the model and those supporting the findings of this study are available within the article and associated Supplementary Tables S1, S2, S3, S4, S5, S6, S7.
Results
Database development
We focused on compounds that alter mitochondrial membrane potential to train our model. Compounds that alter the membrane potential and associated controls were obtained primarily from public ToxCast and Tox21 databases (downloaded through the online EPA ToxCast/Tox21 dashboard or PubChem), which contain high-throughput in vitro analyses of diverse compound libraries. 26 The ToxCast and Tox21 cell-based assays assessed mitochondrial membrane potential in HepG2 cells through fluorescent reporters 24 hours post-treatment with the test materials. The ToxCast assay used the Mito Red fluorophore, which is oxidized to the fluorescent form in healthy mitochondria, to assess inhibition, whereas the Tox21 assay used the membrane-potential sensor Mito MPS, which aggregates in polarized environments, yielding a differential fluorescence signal. The Tox21 assay was coupled with a cell viability counter screen to assess cytotoxicity of the test substances. The ToxCast/Tox21 assay results represent a diverse set of tested compounds (∼2000 for ToxCast or 8000 for Tox21) composed of industrial chemicals, pesticides, pharmaceuticals, consumer products, and food additives. To ensure broader coverage of potential mitochondrial inhibitors, additional compounds that target mitochondrial respiration were curated from CHEMBL 23 and the published literature (statistics associated with the compiled database is detailed below).
The compound names, ID (CAS number of CHEMBL ID) and structure (encoded as SMILES strings), were imported into KNIME (ver. 3.6). For compounds that were duplicated from these sources, those that were flagged as mitochondrial inhibitors in any of the data sources were considered positive in the final dataset. This was done to improve model sensitivity and minimize the likelihood of a “false negative” prediction. Following compilation of the mitochondrial inhibitors from the Tox21 assay (using the EPA online dashboard), results for the same assay dataset but reported in the NCBI PubChem portal (PubChem assay ID 720637) were examined to understand the inclusiveness of the Tox21 database. When the active substances from these two datasets were compared (following removal of compounds with missing/invalid compound structures), 857 compounds were reported as active in the Tox21 mitochondrial membrane potential disruption assay within the EPA dashboard versus 1005 compounds within the PubChem dataset. When comparing these datasets, 405 compounds (shown in Supplementary Table S1) were identified as active in the PubChem dataset but not within the EPA iCSS dashboard, and 274 substances were flagged as active in the EPA iCSS dashboard dataset, but not in PubChem. These variances are likely due to differences in the algorithm used to process the raw mitochondria inhibition data. All were included as mitochondrial inhibitors within our final dataset to optimize our model for sensitivity, minimizing the incidence of “false negatives” and enhance the precision with which compounds can be ruled out as mitochondrial inhibitors.
The resulting dataset comprised 5961 compounds of which 1744 (29%) were mitochondrial inhibitors, with the remaining 4217 compounds (71%) serving as inactive (negative) controls. The compiled datasets are available in Supplementary Tables S2 and S3.
Within the final dataset of mitochondrial inhibitors, 217 compounds (detailed in Supplementary Table S4) were differentially active in the ToxCast and Tox21 assays (i.e., they induced a decrease in the mitochondrial membrane potential in one assay but not the other). Interestingly, carminic acid (CAS 1260-17-9) was identified as decreasing the mitochondrial membrane potential in the ToxCast assay but not in the Tox21 assay, while 2-aminoanthraquinone (CAS 117-79-3) was positive in the Tox21 assay (as deposited in both the EPA dashboard and PubChem) but not the ToxCast screen. Both these compounds contain an anthraquinone moiety, which is known to inhibit respiratory Complex I. 32 The reason for this discrepancy is unclear, but in the case of 2-aminoanthraquinone may be due to processing and curve fitting of the ToxCast binding data. In the case of carminic acid, the differential results between the ToxCast and Tox21 assays may be due to differences in the fluorescent reporters used to assess the mitochondrial membrane potential.
It is noteworthy that both carminic acid and 2-aminoanthraquinone were not flagged for cytotoxicity in either the Tox21 viability counter screen or the ACEA Biosciences, Inc. (ACEA) cytotoxicity assay within ToxCast (ACEA_T47D_80hr_Negative; ACEA BioSciences, Inc., San Diego, CA). The Tox21 viability screen used measurement of cellular ATP levels as a cell viability marker, while the ToxCast screen measured loss of electrical impedance in T47D cells. While these cytotoxicity results may appear discrepant with the ability of these compounds to impair mitochondrial function, it is likely due to a combination of effects such as the manner in which baseline values for the cytotoxicity data were determined. Preferential reliance on aerobic glycolysis (known as the “Warburg effect”) in the cell lines used in these assays [HepG2 cells (Tox21 assay) or T47D cells (ToxCast ACEA cytotoxicity assay)] may allow for cell survival and proliferation in the absence of oxidative phosphorylation (reviewed in Ref. 33 ).
Analysis of protonophores in the mitochondrial inhibitors
Protonophores represent a class of compounds that uncouple the transmembrane potential within the mitochondrion by shuttling protons across the mitochondrial membrane. The loss of this proton gradient can inhibit energy generation, leading to cell death. Biochemically, in order for a compound to act as a protonophore, it must be capable of protonating and de-protonating at physiological pH and be capable of gaining access to the mitochondrion. By analyzing our database of mitochondrial inhibitors, we sought to understand differences in the pKa and lipophilicity of protonophores by each compound class.
Our database contained 78 protonophores with published pKa values (Supplementary Table S5). As seen in Figure 2A, these compounds had a mean pKa of 8.06 ± 0.31, a value close to physiological pH (7.4). Further analysis of the pKa values of these compounds by class revealed marked differences in average pKa values, with alkylphenols displaying the highest pKa values (8.97 ± 0.31) followed by the halogenated phenols (pKa = 7.23 ± 0.35) and the nitrophenols (pKa = 5.50 ± 0.82). Similar trends were observed when analyzing the lipophilicity of protonophores, with alkylphenols being the most lipophilic (logKow = 3.38 ± 0.18) followed by the halogenated phenols and nitrophenols (logKow = 3.29 ± 0.18 and 2.23 ± 0.52, respectively) (Fig. 2B). Thus, when considering whether a substance could act as a mitochondrial inhibitor through a protonophoric mechanism, consideration of its ionizability (pKa) and lipophilicity (logKow) could aid in excluding hydrophilic compounds, as well as strong acids and bases from consideration through this mode of action. While pKa and logKow considerations were not included in the prediction algorithm, consideration of these factors by trained subject matter experts in mitochondrial biology can help identify the likelihood of a predicted protonophore exerting its effects through this mechanism.
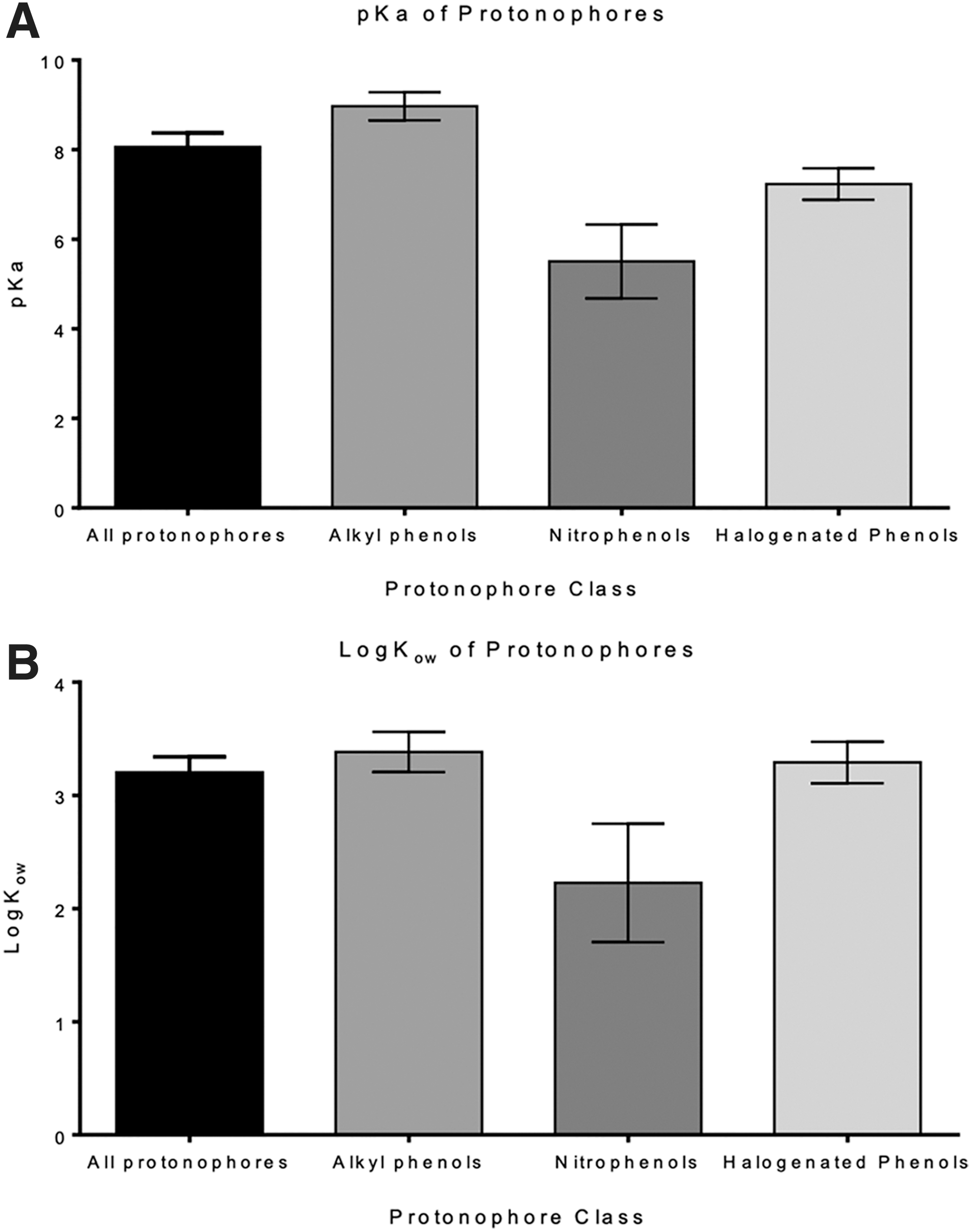
Analysis of physicochemical properties of protonophores. Figure shows the mean pKa values
Development of a profiler for mitochondrial inhibitors
Our mitochondrial profiler uses the output of two machine learning models together with a series of scaffolds that reflect substructures of known mitochondrial inhibitors that we implement as SMARTS patterns to ensure matching substances are flagged as mitochondrial inhibitors and the specific submechanism(s). We iteratively and randomly tested our profiler on the reserved data (amounting to 174 structures comprising 10% of the original database of mitochondrial inhibitors) and inactive control compounds to assess the ability of our model to predict the mitochondrial inhibition potential of novel compounds (i.e., those not present within the training set). This was repeated five times, with a different random subset of compounds being withheld as the test set. Our model showed high sensitivity (80.3%), BA (79.4%), and specificity (78.4%), together with a Youden Index of 0.59, highlighting the ability of the profiler to classify test substances as potential mitochondrial inhibitors or not (Table 1).
Concordance Analysis of the Described Mitochondrial Profiler Comprising a Hybrid Scaffold/Fingerprint-Based Random-Forest Machine-Learning Model
Data represent the average of five independent runs where a random subset of compounds comprising 10% of the dataset was withheld for assessing the models.
BA, balanced accuracy; NPV, negative predictive value; PPV, positive predictive value.
In addition, the high sensitivity and corresponding high negative predictive value for the model (90.7%) (Table 1) highlight the precision with which a compound can be ruled in as a mitochondrial inhibitor or the confidence with which a negative call can be attributed to a test substance not being a mitochondrial inhibitor. The lower positive predictive value (60.9%) is due, in part, to the biological complexity of the mitochondrion, as well as our model being optimized for sensitivity such that “false negative” predictions are minimized. Taken together, these findings suggest that the described profiler can be used to accurately identify mitochondrial inhibitors, as well as rule out compounds that are unlikely to target the mitochondrion. Thus, in the absence of any experimental data on an as yet uncharacterized compound or mechanism, this model can be used to rule a compound in or out as a mitochondrial agent with high accuracy.
Given the biological complexity of the mitochondrion, and the heterogeneous nature of the targets for xenobiotic interaction, we sought to better understand the limitations of our fingerprint-based profiler by asking how the size of the training set affects its predictive power. We partitioned the master dataset of mitochondrial inhibitors such that 1%–90% of the mitochondrial agents were randomly withheld (in 1% increments) within the training set used to build the fingerprint-based model, with the remaining compounds used to test the model. As described earlier (Wijeyesakere SJ, Wilson DM, and Marty MS. Prediction of cholinergic compounds by machine-learning. Article under review for Comp Tox), given the time needed to build the individual hybrid scaffold/fingerprint-based models, the fingerprint-based model was deemed to be sufficiently robust for use in understanding the impact of dataset size on model sensitivity.
The fingerprint-based random forest learner was iteratively run with the restricted training set (1%–90% of active compounds and 90% of the controls). Following each run, we tested the concordance of the resulting models using the reserved compounds. As seen in Figure 3, 65% of the mitochondrial inhibitors were needed in the training set to yield a model sensitivity >70%. This is in contrast to our previous findings with the cholinergic system, which represents a set of three homogenous targets in which ≤12% of actives were needed to yield sensitivity values in excess of 90% (Wijeyesakere SJ, Wilson DM, and Marty MS. Prediction of cholinergic compounds by machine-learning. Article under review for Comp Tox). Despite the diverse nature of discreet molecular targets within the mitochondrion, our model is able to demonstrate high accuracy and sensitivity and will likely be improved with additional in vitro data on compounds that inhibit the mitochondrial membrane potential (Table 1).
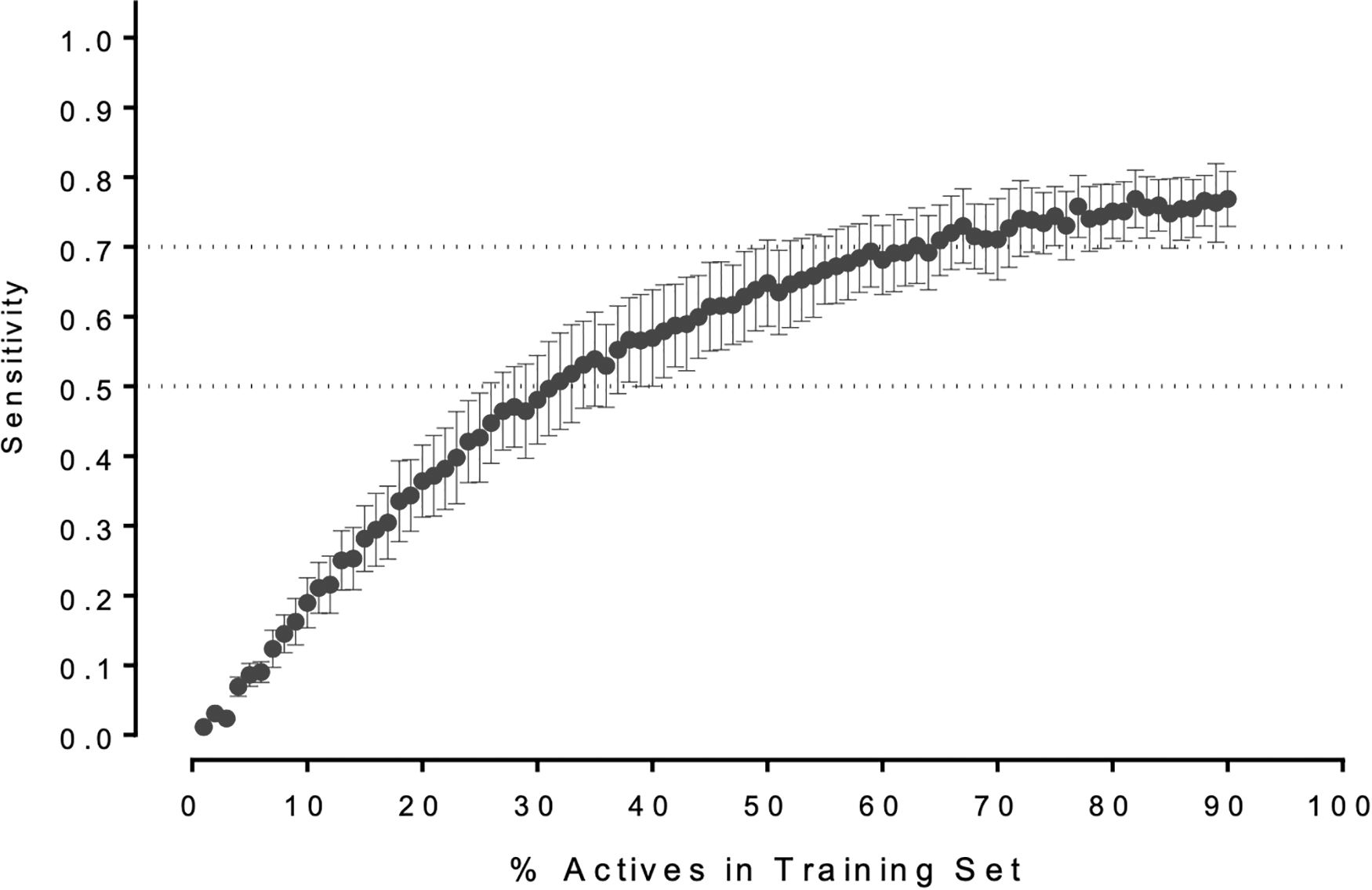
Performance of the supervised machine-learning models is dependent upon the size of the training set. Plots depict the change in the sensitivity as a function of the fraction of the known mitochondrial toxicants present within the training set (together with 90% of the compiled controls). Plots depict the mean sensitivity ± SEM for five independent measurements.
Identification of compounds that cause DILI or identification of novel mitochondrial inhibitors
DILI is a major cause for withdrawal of pharmaceuticals from the market (reviewed in Ref. 34 ). Given the central role of mitochondria in energy production and the high energy demands of hepatocytes, we asked if our profiler could identify hepatotoxic drugs that exert their effect through mitochondrial dysfunction. To this end, we compiled a database of 27 compounds that are known to cause DILI through a mitochondria-mediated mechanism (Supplementary Table S6). Our mitochondrial profiler was able to flag all compounds in the DILI database as mitochondrial inhibitors, highlighting the utility of our profiler in rapidly and cost-effectively identifying these compounds without the need for physical samples or in vitro tests.
In addition, a recent publication described a tiered approach using a battery of high-throughput screening assays on the Tox21 chemical library to identify potential mitochondrial toxicants. This work resulted in the identification of four “high activity,” structurally diverse compounds that were previously not well characterized for mitochondrial effects. 35 Our profiler flagged all four compounds, demonstrating its utility in aiding the rapid identification of mitochondrial inhibitors.
The model statistics and results described above encompass the scope of our machine learning model for mitochondrial inhibition coupled with SMARTS filters to identify specific submechanisms for mitochondrial inhibition. Implementation of the SMARTS filter is expected to have increased the sensitivity at the expense of generating additional false positives. We endorse such an approach because follow-up laboratory screening can be implemented within an integrated approach to better understand the extent to which such compounds might inhibit mitochondria. Two example classes of compounds where this was necessary include those with embedded subfragments that can be metabolized to cyanide or hydrogen sulfide, which were not adequately flagged in the in vitro machine learning model but are known to show potential high in vivo toxicity.
Discussion
Despite the biological complexity of the mitochondrion, and the heterogeneous nature of the targets within the organelle, we developed a computational model that identifies mitochondrial inhibitors with high sensitivity, specificity, and accuracy. Using this profiler, we were able to identify compounds that induced liver injury through mitochondrial inhibition, as well as novel mitochondrial toxicants recently identified through in vitro screening. Given the central role of the mitochondrion in cellular homeostasis, we propose that our profiler could be important for development of therapeutic or efficacious compounds (i.e., chemotherapeutics, biocides, and so on), as well as in identifying the mechanisms responsible for inadvertent or intentional overexposure to any class of compound, including air pollution, illicit and nonillicit pharmaceuticals, solvent vapors, inhaled vapors/mists/aerosols, agrochemicals, and so on, knowledge of which might aid in therapeutic interventions or limit inadvertent exposure to mitochondrial inhibitors.
As with any computational tool, when implementing this model, it is important to highlight the need of oversight by subject matter experts trained in mitochondrial biology. This is consistent with our strategy of developing approaches in which scientists with extensive training in various biological disciplines manage the implementation of screening methods for those disciplines across a variety of in silico, in vitro, and in vivo platforms.
When considering the diversity of inactive (control) substances, it is important to note that the ToxCast/Tox21 dataset represents a library spanning select categories of compounds such as pesticides (most compounds tested in ToxCast Phase I), 36 compounds of regulatory interest (ToxCast Phase II), 36 or those targeting nuclear receptor and cell stress pathways (Tox21). Thus, while the ToxCast/Tox21 dataset provides a readily accessible source for negative (control) in vitro data, it is not an entirely random sampling of the known chemical space. This represents a potential area of improvement for our model that can be rectified through future publicly available high-throughput in vitro studies covering a broader region of available chemistries.
To further advance the science in understanding the role of metabolism in promoting the release of cyanide from alkyl or aryl nitriles, we are conducting collaborative research to develop and implement an in vitro screening model for such substances. We are hopeful that the outcome of this research can eventually be used in a HTS manner to generate data to retrain and refine our mitochondrial inhibition model. Importantly, we initially found that our statistical model did not adequately identify the compounds that could generate toxic forms of cyanide or hydrogen sulfide following metabolic activation, a finding that was rectified through the addition of SMARTS filters for nitrile-containing substructures (Supplementary Table S7).
By compiling both positive and negative data, our model is unique in that in addition to predicting potential mitochondrial inhibition, it is able to assign a compound as not being a likely mitochondrial toxicant. Mitochondria are complex organelles with numerous approaches used to assess their inhibition, thus any mechanistic profiler may be potentially limited by the submechanisms or compound classes in scope for the assays used to assess mitochondrial function. Within the initial stages of a tiered approach toward testing novel compounds, our workflow can serve as a valuable tool in rapidly screening potential mitochondrial agents, thereby aiding in lead optimization and product safety evaluations.
Footnotes
Acknowledgment
The authors thank Dr. Matthew LeBaron for his critical review of the article and helpful conversations.
Author Contributions
S.J.W. and D.W. contributed to the conception of the hypothesis, design of the experiments, and analysis of the data. T.A., A.P., and D.K. curated the datasets used to build the model. S.J.W. setup and ran the computational analyses to collect data to build the predictive models. S.J.W., D.W., D.K., and S.M. contributed to the writing of the article.
Author Disclosure Statement
The authors are or were employees of the Dow Chemical Company and declare that they are not aware of any conflicts of interest.
Funding Information
No external funding sources were involved in this research endeavor.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.