
Abstract
Our objective was to evaluate the liver toxicity of antiretroviral regimens including fosamprenavir plus ritonavir (FPV/r) 1400/100 mg once daily (QD) in HIV/hepatitis C virus (HCV)-coinfected patients. This was a prospective cohort study that included 117 HIV/HCV-coinfected patients who started FPV/r 1400/100 mg QD-based antiretroviral therapy (ART) and who neither had received a previous antiretroviral regimen containing FPV nor had a past history of virologic failure while receiving protease inhibitors (PI). The primary end point of the study was the occurrence of grade 3–4 liver enzymes elevations (LEE) within 1 year after starting FPV/r QD. Factors potentially associated with grade 3–4 LEE, including baseline liver fibrosis, were analyzed. Eleven (9%) patients had a grade 3–4 LEE during the follow-up, resulting in an incidence of severe liver toxicity of 9% (95% confidence interval 4.1–14.6%). None of these cases led to FPV/r discontinuation. Baseline liver fibrosis could be assessed in 97 (83%) patients. Six of 71 patients (8%) with significant fibrosis had a grade 3–4 LEE versus 2 of 26 (8%) without significant fibrosis (p=1.0). Twenty (21%) patients had cirrhosis at baseline. There were no cases of LEE among cirrhotics. In conclusion, the incidence of severe liver toxicity after 1 year of therapy with FPV/r QD-based ART in HIV/HCV-coinfected patients is similar to what has been reported with other boosted PIs. In addition, the presence of significant fibrosis or cirrhosis was not associated with the emergence of liver toxicity. Thus, ART regimens containing FPV/r QD may be considered safe in HIV/HCV-coinfected patients, including those with cirrhosis.
Introduction
Fosamprenavir plus ritonavir (FPV/r) is one PI commonly used in daily clinical practice of HIV-infected patients. FPV/r 700/100 mg twice daily showed similar efficacy and safety as lopinavir/ritonavir in the KLEAN study. 8 However, there is little information regarding the liver tolerability of FPV/r in HIV/HCV-coinfected patients. In a subanalysis of KLEAN study, the incidence of grade 3 or 4 liver enzymes elevations (LEE) in HCV-coinfected patients was 8%. 9 In the SOLO trial, severe liver toxicity at 48 weeks occurred in only 3% of HIV-monoinfected patients, 10 although the frequency of grade 3 or 4 LEE in HCV or hepatitis B virus (HBV) coinfected patients was 24%. 11
In recent years, the dosage of FPV 1400 mg boosted with 100 mg of ritonavir once a day (FPV/r 1400/100 mg) has been increasingly used. This strategy showed noninferiority compared with 700/100 mg twice daily in naïve patients 12 and was as effective as atazanavir/ritonavir 300/100 mg in another study. 13 In fact, FPV/r 1400/100 mg QD is approved by the Food and Drug Administration and it is one of the alternative regimens for naïve patients recommended in Department of Health and Human Services (DHHS) guidelines. 14 In addition, pharmacokinetic studies have allowed using effective and safe dosage for patients with liver function impairment. 15 As a consequence, the use of FPV has become relatively common in daily clinical practice in recent years, particularly in the setting of HIV/HCV-coinfected patients. In addition to the advantages of once daily administration, low doses of ritonavir might enhance tolerability and reduce the risk of liver toxicity. For these reasons, FPV/r 1400/100 mg QD may theoretically be a good option for the treatment of HIV/HCV-coinfected patients. However, there is little information regarding its efficacy and liver safety in this setting, as the proportion of HCV-coinfected patients included in previous studies with FPV has been very low. 12,13
The objective of our study was to assess the liver safety of ART regimens containing FPV/r 1400/100 mg QD, including the possible influence of baseline liver fibrosis, in a cohort of HIV/HCV-coinfected patients.
Methods
Study design and patients
This was a multicenter prospective cohort study conducted in 11 hospitals in Andalusia, southern Spain. From July 2008 to January 2010, all HIV/HCV-coinfected patients with positive plasma HCV RNA who started an ART regimen containing FPV/r 1400/100 mg QD were included in this study, if they had not received a previous ART regimen containing FPV and had not a past history of virologic failure while on PIs. Patients were evaluated at 4 and 12 weeks after starting FPV/r and then at least every 12 weeks until completing 48 weeks of follow-up. In each visit, clinical examinations, as well as liver function tests and blood cell count determinations, were performed. Therapy against chronic HCV infection was allowed during follow-up.
Definition of liver toxicity
Grade 3 or 4 elevations were defined as elevations of plasma aspartate aminotransferase (AST) or alanine aminotransferase (ALT) higher than or equal to 5 times above the upper limit of normality, when baseline levels were normal, or more than 3.5 times the baseline values, if they were abnormal. Grade 4 total bilirubin elevations were defined as elevations of bilirubin levels equal to or above 5 times the upper limit of normality.
Liver fibrosis assessment
A stepwise algorithm was used to assess liver fibrosis (Fig. 1). 5 Briefly, in patients with an available liver biopsy 1 year prior to or after starting FPV/r, the stage of liver fibrosis found in that biopsy was considered as the baseline liver fibrosis. In the absence of biopsy, liver fibrosis was assessed by transient elastography (TE), when it was available and performed 1 year before of after enrolment. For this study, patients showing liver stiffness (LS) values equal to or greater than 7.2 kiloPascals (kPa) were considered to bear significant fibrosis (≥F2 of the METAVIR classification), while patients with a LS above or equal to 14.6 kPa were considered as cirrhotics. In the case that neither a liver biopsy nor a TE evaluation was available, serum biomarkers were determined. First, baseline AST-to-platelet ratio index (APRI) was computed and, if it was greater than or equal to 1.5, significant liver fibrosis was diagnosed. In patients with a result of APRI below 1.5, Forns index was then computed. Values above or equal to 6.9 were considered indicative of significant fibrosis. In patients belonging to this group, in whom APRI value was lower than 1.5 and Forns index was below 6.9, the presence or not of significant fibrosis could not be ascertained.
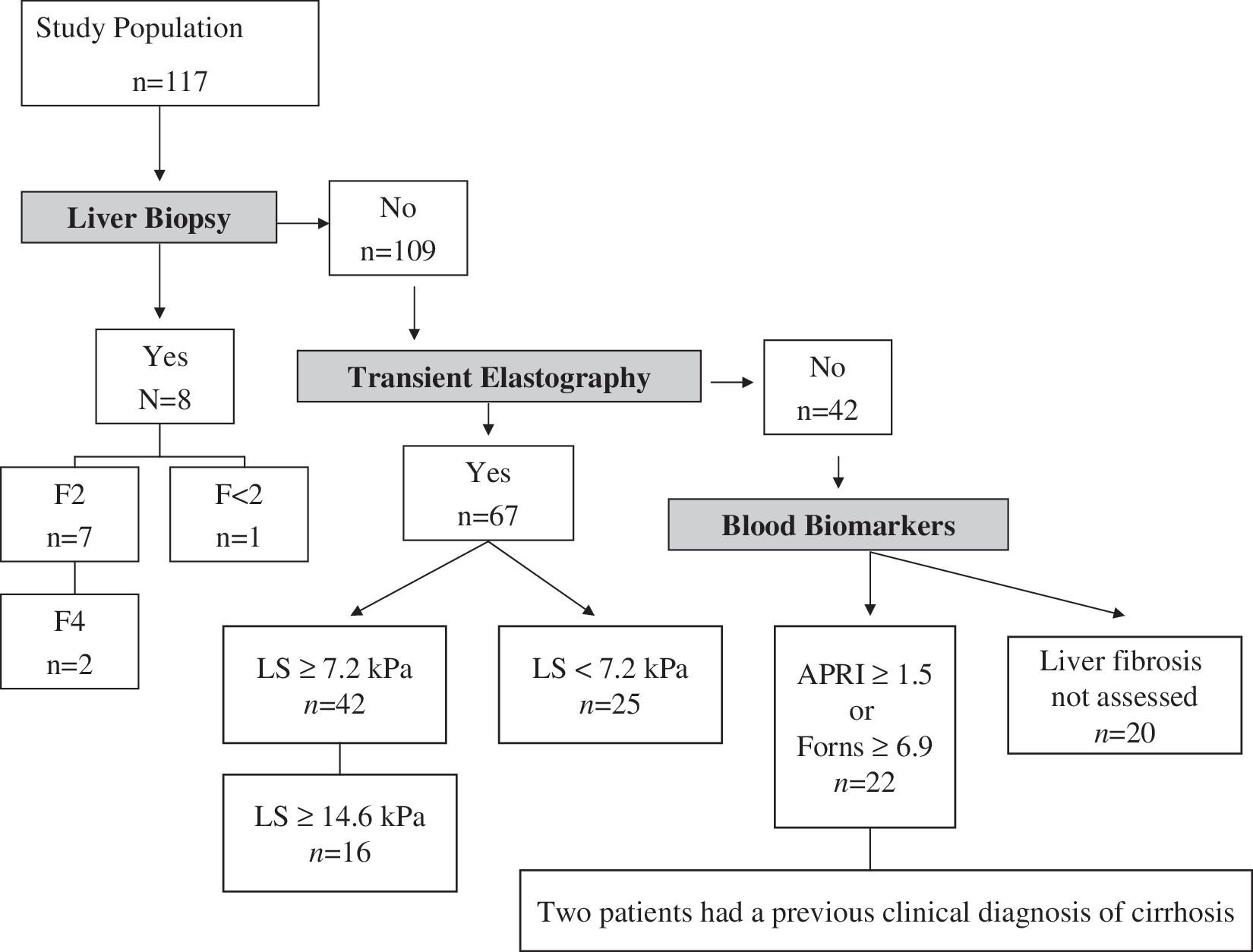
Algorithm for the assessment of liver fibrosis and number of patients classified in each step. In summary, liver fibrosis could be assessed in 97 patients: 71 bearing significant fibrosis (≥F2) and 26 having mild or absent fibrosis (F<2). Cirrhosis was present in 20 patients. LS, liver stiffness; kPa, kilopascals; APRI, AST-to-platelet ratio index.
Statistical analysis
The outcome variable of the study was the emergence of grade 3 or 4 LEE within the 48 weeks after starting FPV/r 1400/100 mg QD. Analyses were done according to an intention-to-treat approach. Additionally, on-treatment analyses were also performed. Those patients who received therapy against HCV infection during follow-up were censored for these analyses at the moment of starting such a therapy.
The efficacy of FPV/r 1400/100 mg QD was also evaluated. For this purpose, the proportion of patients with undetectable HIV viral load (<50 copies per milliliter) at 48 weeks was computed. Analyses on efficacy were performed on an intention-to-treat basis, with missing patients or prematurely discontinuations considered as failures. In addition, on-treatment and last observation carried forward analyses were performed. For the latter, HIV viral load at the last visit while on FPV/r was used in those patients who were lost to follow-up or prematurely discontinued FPV/r.
The possible risk factors for the development of grade 3–4 LEE were analysed, including baseline liver fibrosis. Continuous variables are expressed as median (Q1–Q3) and categorical variables as numbers (percentages). The Student's t test was used for comparisons between continuous variables if a normal distribution was followed and the Mann-Whitney U test if not. Categorical variables were compared with the χ2 test or the Fisher's test when appropriate. The Wilcoxon test was used to compare values of continuous variables at different time points. The variables associated with grade 3–4 LEE with a p<0.2 in the univariate analysis were entered in the multivariate analysis. Logistic regression models with grade 3–4 LEE as the dependent variable were performed. Associations with a p<0.05 were considered significant. The adjusted odds ratio (AOR) and the respective 95% confidence intervals (CI) were also calculated. The statistical analysis was carried out using the SPSS 14 Statistical Software Package (SPSS Inc., Chicago, IL).
Ethical aspects
The study was designed and conducted following the Helsinki Declaration. The ethics committee of the Hospital Universitario de Valme approved the study. All the participant subjects gave their written consent to participate in the study.
Results
Characteristics of the study population
One hundred seventeen patients fulfilled the inclusion criteria for the study. The main baseline features of the study population are summarized in Table 1. The main reasons for prescribing FPV/r were starting a first ART combination and toxicity of previous ART (Table 1). One hundred eight patients received a nucleoside reverse transcriptase inhibitors (NRTI) backbone, consisting of tenofovir plus emtricitabine in 80 (68%) patients and abacavir plus lamivudine in 28 patients (24%). In 9 patients (8%) FPV/r was prescribed along with other antiretroviral drugs. During follow-up, 15 patients (13%) received therapy against HCV with pegylated interferon plus ribavirin. Baseline liver fibrosis could be assessed in 97 (83%) patients (Fig. 1). Of them, 71 (73%) harbored significant fibrosis. In 94 patients (80%), the presence of cirrhosis could be assessed, 20 (21%) of them were considered to bear liver cirrhosis.
Median (Q1-Q3).
Available in 109 patients.
IDU, injection drug users; HBsAg, hepatitis B virus surface antigen; ART, antiretroviral therapy; HCV, hepatitis C virus; FPV/r, fosamprenavir/ritonavir.
The patient disposition at 48 weeks is depicted in Fig. 2. Nineteen patients (16%) were lost to follow-up while 76 patients (65%) completed 48 weeks on FPV/r-based ART. In 20 patients (17%), FPV/r was prematurely discontinued. The reasons for discontinuations are shown in Fig. 2. Two (2%) patients died during follow-up.

Patient disposition at the end of the study. FPV/r, fosamprenavir/ritonavir.
There were no relevant differences between the main baseline characteristics of patients who completed or not 48 weeks of therapy with FPV/r (Table 1). Patients who started therapy against HCV infection were censored at the date of initiating such a therapy and were considered as noncompleters in the on-treatment analysis for the primary outcome variable, the emergence of grade 3–4 LEE.
Changes in CD4 cell count and plasma HIV-RNA load
The median (Q1–Q3) CD4 cell count at baseline and at week 48 were 266 (157–423) and 392 (257–568), respectively (p<0.0001). Thirty-four (29%) patients had plasma HIV viral load less than 50 copies per milliliter at baseline. On an intention-to-treat basis, considering missing patients or prematurely discontinuations as failures, 66 (56%) patients achieved undetectable plasma HIV viral load at 48 weeks of therapy with FPV/r. On treatment analyses included 76 patients that continued on FPV/r therapy at 48 weeks. With this approach, the proportion of patients that achieved virologic suppression was 87%. Finally, an analysis with the last observation carried forward was performed. Ninety-four patients (80%) had undetectable HIV viral load using this approach.
Liver toxicity
Eleven patients developed a grade 3–4 LEE during follow-up. None of these episodes led to FPV/r discontinuation. Thus, the incidence of severe liver toxicity was 9% (95% CI 4.1–14.6) in an intention-to-treat basis. On treatment analyses included 65 patients that completed 48 weeks of therapy with FPV/r, after excluding those patients who started therapy against HCV infection during follow-up. Of them, 9 (13.8%) had a grade 3–4 LEE during follow-up. Using this approach, the incidence of severe liver toxicity was 13.8% (95% CI 5.4–22.2).
Nine of the 11 patients who developed a grade 3–4 LEE during follow-up had a high ALT or AST at baseline. In these patients, ALT or AST elevations increased more than 3.5 times the baseline levels but did not exceed more than 5 times these levels. LEE occurred in the first 6 months after starting FPV/r in 6 patients. Liver enzymes returned to baseline levels after 3 months in 5 patients while decreased but persisted slightly higher than baseline in the remaining 6 patients. None of the episodes of grade 3–4 LEE led to FPV/r discontinuation.
There was no association between the degree of preexisting liver fibrosis and the emergence of severe liver toxicity. Namely, 6 of 71 patients (8%) with baseline significant fibrosis had a grade 3–4 LEE versus 2 of 26 (8%) without significant fibrosis (p=1.0; Table 2). Univariate analyses of the predictors of grade 3–4 LEE are presented in Table 2. Multivariate analyses did not find any independent predictor or grade 3–4 LEE. There were no cases of grade 4 total bilirrubin elevations.
Available in 109 patients.
Liver fibrosis could be assessed in 97 patients.
Liver cirrhosis could be assessed in 94 patients.
LEE, liver enzymes elevation; AOR, adjusted odds ratio; CI, confidence interval; IDU, Injection drug user; ART, antiretroviral therapy; TDF/FTC, tenofovir/emtricitabine; ABC/3TC: abacavir/lamivudine; FPV/r: fosamprenavir/ritonavir.
Liver safety of FPV/r in cirrhosis
Twenty patients (21%) had liver cirrhosis at the moment of starting FPV/r. In three of them cirrhosis had previously developed decompensations. The remainder harbored Child-Pugh-Turcotte class A cirrhosis. After 48 weeks, 15 of 20 patients with baseline cirrhosis were on FPV/r therapy. Of them, 13 had undetectable HIV viral load. Thus, the proportion of patients who achieved undetectable HIV viral load at 48 weeks was 65% on an intention-to-treat basis and 87% when on-treatment response was considered, respectively. Two patients discontinued FPV/r due to gastrointestinal side effects and two patients were lost to follow-up. There were no cases of grade 3–4 LEE among cirrhotics. One patient with previous decompensated cirrhosis, who suffered from an episode of ascites, jaundice, and hepatic encephalopathy in the week before FPV/r was initiated, died after 2 weeks of starting FPV/r due to spontaneous bacterial peritonitis.
Discussion
This study shows that the incidence of severe liver toxicity during the first year of therapy with ART combinations including FPV/r 1400/100 mg QD in HIV/HCV-coinfected patients is similar to what has been reported with other boosted PIs. 3 –5,7 In addition, the presence of significant fibrosis or cirrhosis does not seem to increase the risk of liver toxicity. Thus, ART regimens containing FPV/r 1400/100 mg QD may be considered safe in HIV/HCV-coinfected patients, including those with cirrhosis.
This is the first study to our knowledge that has evaluated the liver safety of regimens containing FPV/r 1400/100 mg QD in HIV/HCV-coinfected patients. The incidence of grade 3 or 4 LEE associated with ART based on FPV/r in our study was 9%, a rate that is in the range of what has been reported with other boosted PI. 3 –5,7 Remarkably, none of these episodes led to FPV/r discontinuation, confirming that this dosing schedule of FPV/r is well tolerated in HIV/HCV-coinfected patients. On the other hand, this study provides data regarding the efficacy of this strategy in the HIV/HCV-coinfected population. The efficacy of FPV/r 1400/100 mg QD was previously assessed in three studies. 12,13,16 However, only one of these studies included 11 patients carrying HCV chronic infection. 12 Our results demonstrate that FPV/r 1400/100 mg QD is also efficacious in HIV/HCV-coinfected patients. In fact, among 76 patients that continued on FPV/r after 48 weeks, 66 (87%) of them showed HIV viral load suppression.
We did not find any independent predictor of the emergence of severe liver toxicity with the use of FPV/r, including baseline fibrosis. Previous studies suggested that the presence of significant fibrosis may increase the risk of liver toxicity with NNRTI 6 or with some PI, such as nelfinavir. 17 In our study, there were not differences in the frequency of grade 3–4 LEE between patients with or without significant liver fibrosis at baseline. This observation is in agreement with recent evidence on ART-related liver toxicity, 3,5,7 that also do not find any influence of baseline liver fibrosis in the development of severe liver toxicity with efavirenz or boosted PI-based ART. Interestingly, 9 of the 11 episodes of grade 3–4 LEE occurred in patients harboring high baseline liver enzymes, although this association did not reach statistical significance, probably due to the low number of grade 3–4 LEE events. Finally, there were no episodes of severe liver toxicity among patients harboring HCV genotype 3. The role of HCV genotype 3 in the risk of liver toxicity of ART in HIV/HCV-coinfected patients has been a matter of controversy, with some studies suggesting an increased risk in HIV-infected patients also infected by HCV genotype 3. 18,19 Although multivariate analyses do not find an independent association of HCV genotype with the risk of grade 3–4 LEE, we cannot definitely rule out a possible role of HCV genotype, as our study might have not enough power to detect such an association.
Liver cirrhosis is currently a point of major concern for clinicians who care for HIV/HCV-coinfected patients. Studies based on transient elastography have reported that liver cirrhosis is present in, at least, 19–24% of HIV/HCV-coinfected patients 20,21 and in 42% of those also infected by hepatitis B virus. 20 Thus, this is a real challenge for health care providers attending HIV-infected patients in areas with a high prevalence of viral hepatitis coinfections. In addition, end-stage liver disease due to HCV progresses faster in HIV-infected patients and has a dramatically high mortality. 22,23 Previous studies have suggested that ART may enhance survival in this setting. 23 –25 However, there still a little evidence on the safety of most of antiretroviral drugs in patients with impaired liver function. Some reports have shown that the plasma levels of lopinavir/ritonavir or efavirenz may increase in patients with cirrhosis, 26 –28 although its clinical relevance is not completely known. In addition, most of recommendations regarding dose reductions in this setting are based on poor data and expert opinions as pharmacokinetics studies are few for most of antiretroviral drugs. 14 A previous study has evaluated the plasma levels of FPV/r in patients with cirrhosis. 15 Based on these data, FPV 700 mg BID or 1400 mg QD boosted with 100 mg of ritonavir is recommended for patients with stage A Child-Turcotte-Pugh cirrhosis. Regarding patients harboring stage B Child-Turcotte-Pugh cirrhosis, pharmacokinetics studies have demonstrated that reducing doses of FPV/r to 700/100 mg QD in these individuals might not be recommended, as plasma levels achieved with this doses does not guarantee virologic efficacy. Alternatively, a strategy consisting of administration of fosamprenavir at 450 mg twice daily plus ritonavir at 100 mg once daily has been suggested. 15 In our study, liver tolerance of FPV/r 1400/100 mg QD was good among 20 patients with cirrhosis, including three patients with previously decompensated cirrhosis. Liver cirrhosis did not influence the emergence of severe liver toxicity, as there were no episodes of grade 3–4 LEE among cirrhotics. After 48 weeks, 15 were still on FPV/r and 13 of them, maintained complete viral suppression. In absence of clinical trials comparing specific combinations of ART in patients carrying stage A or B Child-Turcotte-Pugh cirrhosis, our results suggest that FPV/r 1400/100 mg QD may be a good option for these patients.
Our study has a few limitations. First, the proportion of patients lost to follow-up was 16%. This rate, although relatively high, probably reflects real conditions of follow-up of a prospective cohort of HIV/HCV-coinfected patients, as patients were not selected to receive therapy with FPV/r due to other reasons than physician criteria. Besides, comparisons between patients who did or did not complete 48 weeks of therapy with FPV/r do not show significant differences, suggesting that relevant bias may have not occurred. Second, liver fibrosis could not be assessed in all patients. Nevertheless, most of 80% patients had available a liver fibrosis assessment, being improbably the occurrence of relevant biases. Third, liver fibrosis was assessed by different methods. However, algorithms combining transient elastography and serum biomarkers, as the one used for this study, have been previously validated and have shown a high accuracy to predict liver fibrosis and cirrhosis in HIV/HCV-coinfected patients. 29,30
In summary, ART combinations including FPV/r 1400/100 mg QD have a good liver tolerance in HIV/HCV-coinfected patients, including those with significant liver fibrosis. Besides, the efficacy and safety of this dosing schedule in patients harboring liver cirrhosis seems very good. Thus, ART regimens containing FPV/r 1400/100 mg QD may be considered safe in HIV/HCV-coinfected patients, including those with cirrhosis.
Footnotes
Acknowledgments
This study has been partly supported by grants from the Spanish Health Ministry (ISCIII-RETIC RD06/006 and FIS EC07/9014). J.A.P. is the receptor of an intensification grant from the Fundación Progreso y Salud of the Consejería de Salud de la Junta de Andalucía (Reference AI-0021).
Author Disclosure Statement
Nicolás Merchante reports having received lecture fees from Abbot Laboratories. He has received research support from Gilead, Boehringer Ingelheim, GlaxoSmithKline and Bristol-Myers Squibb Pharmaceuticals. Luis F. López-Cortés has received unrestricted funds for research and honoraria for speaking at symposia organized on behalf of Abbott Laboratories, Bristol-Myers Squibb, GlaxoSmithkline, Gilead Sciences, Janssen-Cilag España, Merck Sharp & Dohme España, Roche Pharma SA., and ViiV Healthcare Pharmaceuticals. Marcial Delgado-Fernández reports having received consulting fees from ViiV Laboratories. Dolores Merino reports having lecture fees from Jansen, Bristol-Myers Squibb, Merck Sharp & Dome, Abbot, Boehringer Ingelheim and Gilead Pharmaceuticals. Mohamed Omar reports having received lecture fees from GlaxoSmithKline Pharmaceuticals. Antonio Rivero has received lecture fees and grants for research from Abbott, Boehringer & Ingelheim, Bristol-Myers Squibb, Gilead, GlaxoSmithKline, Janssen-Cilag, Merck Sharp & Dohme, Pfizer, Roche and Schering-Plough Pharmaceuticals. Juan Macías has been an investigator in clinical trials supported by Roche, Bristol-Myers Squibb and Abbot Pharmaceuticals. He has received lecture fees from Roche, Gilead, Boehringer Ingelheim and Bristol-Myers Squibb, and consulting fees from Boehringer Ingelheim Pharmaceuticals, Bristol-Myers Squibb, Merck Sharp & Dome and Schering-Plough Pharmaceuticals. Juan A. Pineda reports having received consulting fees from GlaxoSmithKline, Roche, Bristol-Myers Squibb, Abbot, Gilead, Pfizer, Merck Sharp & Dome, Jansen and Boehringer Ingelheim Pharmaceuticals. He has received research support from GlaxoSmithKline, Roche, Bristol-Myers Squibb, Schering-Plough, Abbot, Pfizer, Merck Sharp & Dome, Jansen and Boehringer Ingelheim Pharmaceuticals and has received lectures fees from GlaxoSmithKline, Roche, Abbot, Bristol-Myers Squibb, Boehringer Ingelheim, Pfizer, Merck Sharp & Dome, Jansen and Schering-Plough Pharmaceuticals. The remaining authors have no conflict of interest.