
Abstract
HIV remains a highly important public health and clinical issue despite many recent advances in attempting to develop a cure, which has remained elusive for most people infected with HIV. HIV disease can be controlled with pharmacologic therapies; however, these treatments are expensive, may have severe side effects, and are not curative. Consequently, an improved means to control or eliminate HIV replication is needed. Cytotoxic T lymphocytes (CTLs) play a critical role in controlling viral replication and are an important part in the ability of the immune response to eradicate most viral infections. There are considerable efforts to enhance CTL responses in HIV-infected individuals in hopes of providing the immune response with armaments to more effectively control viral replication. In this review, we discuss some of these efforts and focus on the development of a gene therapy-based approach to engineer hematopoietic stem cells with an HIV-1-specific chimeric antigen receptor, which seeks to provide an inexhaustible source of HIV-1-specific immune cells that are MHC unrestricted and superior to natural antiviral T cell responses. These efforts provide the basis for further development of T cell functional enhancement to target and treat chronic HIV infection in hopes of eradicating the virus from the body.
W
As is widely known, to date, there has only been a single reported case of cured chronic HIV infection in an adult (known as Timothy Brown), through bone marrow transplant from a donor lacking the normal gene for CCR5, which is a cell receptor required, in addition to CD4, for most strains of HIV to infect cells. However, the mortality rate of this procedure is about 15–20%, and matched bone marrow with this genetic profile is almost nonexistent for most ethnic groups, rendering this approach impractical for broader clinical applicability. The mechanism(s) of the success of this therapy is also not clear as it is likely that some combination of the myeloablation procedure, graft-versus-host response, and CCR5-deficient (HIV refractory) cellular reconstitution is responsible for the clearance of HIV infection. These unknowns also make repeating this cure approach very difficult.
Several recent studies have attempted to remove CCR5 from hematopoietic stem cells (HSCs) and/or deliver anti-HIV genes to protect cells from HIV infection in humans, but these studies face limitations due to unknowns regarding levels of transduced cell engraftment required to generate an HIV-resistant immune system. Further, attempts to treat individuals with myeloablation and allogeneic stem cells have resulted in short-term suppressed viral replication, followed by reemergence of the virus in the absence of ART. 1 In addition, while most transmitted strains of HIV utilize the CCR5 molecule as a coreceptor for infection, it is also well known that HIV can mutate and evolve in the host to utilize other coreceptors other than CCR5, including the CXCR4 molecule, thus limiting the protective effects of the lack of CCR5 expression on the cell. Thus, recapitulating the parameters that allowed Timothy Brown to clear HIV has proven difficult and the lack of successful ART to clear the virus reiterates a need for a therapeutic strategy to cure HIV infection.
The HIV-specific T cell response is a critical component in naturally controlling HIV viral replication following infection; however, due to a variety of reasons, including the insufficient generation of enough numbers and breadth of T cells and the maintenance of the functional T cell response during the infection, the T cell response is incapable of clearing the infection. CD8+ cytotoxic T lymphocytes (CTLs) partially control HIV in almost all infected persons, but eventually fail due to viral mutation, downregulation of viral antigen presentation, lack of CD4+ T cell help, and CTL clonal exhaustion and dysfunction. Since the first reports of HIV-1-specific CTLs in 1987, 2 it has become generally accepted that CTL antiviral activity is critical to immune containment of infection, although usually incomplete. Key evidence of this has been observed in the SIV-macaque model in studies that cannot be performed in humans where CD8 depletion in vivo results in loss of viral immune containment. 3 –6 In human HIV infection, there are observations of human leukocyte antigen (HLA)-associated footprints or evidence of preferential types of responses and specifically driven viral evolution in particular HIV epitopes. 7,8
CTLs play an important role in controlling acute HIV infection and in lowering viral loads and the pressure that they place on viral evolution [outside of the Envelope (Env) gene] is evident as it is mostly driven by these cells. 9 –13 The importance of CTLs in controlling viremia is additionally evident, in that the HLA class I locus is the strongest host genetic determinant of disease progression, 14 and CTLs have been observed to exert potent antiviral activity through killing of HIV-1-infected cells in vitro. 15,16 Thus, although ultimately unsuccessful in clearing the infection and in preventing disease progression, CTLs are a key immune mechanism for the clearance of HIV-1-infected cells in vivo. However, the ability of CTLs to control HIV in rare persons and their efficacy against other viruses indicate that overcoming these barriers should allow successful CTL-mediated control of HIV.
An increasingly popular approach to treat infections or malignancies has involved the redirection or reprogramming of the immune response through genetic manipulation of immune cells that allow the targeting of these cells to specific antigens of interest. 17 There are currently a number of clinical trials in place that involve the redirection of T cells to target a variety of different types of malignancies; 18 however, there remains a strong desire to tap into the potential of redirecting T cells to target HIV infection.
A popular strategy to enhance host immunity is to genetically modify peripheral blood cells with an antigen-specific molecularly cloned T cell receptor (TCR) or a chimeric TCR-based molecule containing a ligand receptor binding domain that can redirect cells to target specific antigens. In the case of HIV infection, antigen-specific TCRs from HIV-reactive T cells obtained from infected individuals have been identified, cloned, and used to modify peripheral cells from the same patient. 19 –22
While attempts to modify peripheral T cells with HIV-specific TCRs have been largely experimental and ineffective therapeutically thus far, genetically modified CD8 cells have in fact exhibited enhanced and polyfunctional immune responses against viral antigens in vivo and these cells have been demonstrated to have an increased ability to control HIV infection. 21,22 A potential benefit of this approach is due to the specificity of the TCR, which would likely limit issues with tolerance or self-reactivity. However, an inherent limitation to this approach is the absolute requirement for a specific HLA molecule to present antigen to the T cell, limiting each TCR to persons with the right HLA type. Further, HIV has evolved means to counteract HLA presentation to CTLs and thereby limit their efficacy. Perhaps a more important caveat for engineered TCR-based gene therapy is the ability of HIV to escape HIV-specific T cell responses by mutation. Thus, there are significant advantages and disadvantages with this approach.
Another approach for immune engineering is to utilize a chimeric antigen receptor (CAR) specific for HIV in place of TCRs. HIV-specific CARs have an antigen binding domain specific for HIV and an internal TCR signaling domain (Fig. 1). When they bind the target antigen, which occurs directly without the need for presentation by HLA molecules, they trigger cellular activation similar to that triggered by TCR ligation. Thus, this approach could be used in HIV-infected persons of any HLA type. Antitumor CARs have been safely utilized in peripheral T cells and have produced antitumor responses in the treatment of malignancies. 17 For HIV infection, only one CAR has been previously tested in clinical trials. This reagent involved the use of a CAR containing the extracellular and transmembrane domains of CD4 fused to the CD3-ζ signaling domain. Activation of modified T cells occurs following CD4 binding viral gp120. This protein was expressed in modified human peripheral T cells using a gamma-retroviral vector in peripheral T cells and evaluated in three clinical trials 23 –25 more than 20 years ago. Treatment was well tolerated and safe, but these studies were all confounded by the concurrent administration of combination ART, although it appeared in one trial that tissue viral replication was reduced. Further, a significant problem with this approach was that the reinfused gene-modified T cells were premorbid and likely dysfunctional due to massive ex vivo expansion and persisted only at low levels. Both approaches using a molecularly cloned HIV-specific TCR and an HIV-specific CAR are currently under investigation, and each has its potential benefits and drawbacks in the development of an effective protocol to enhance antiviral T cell responses.
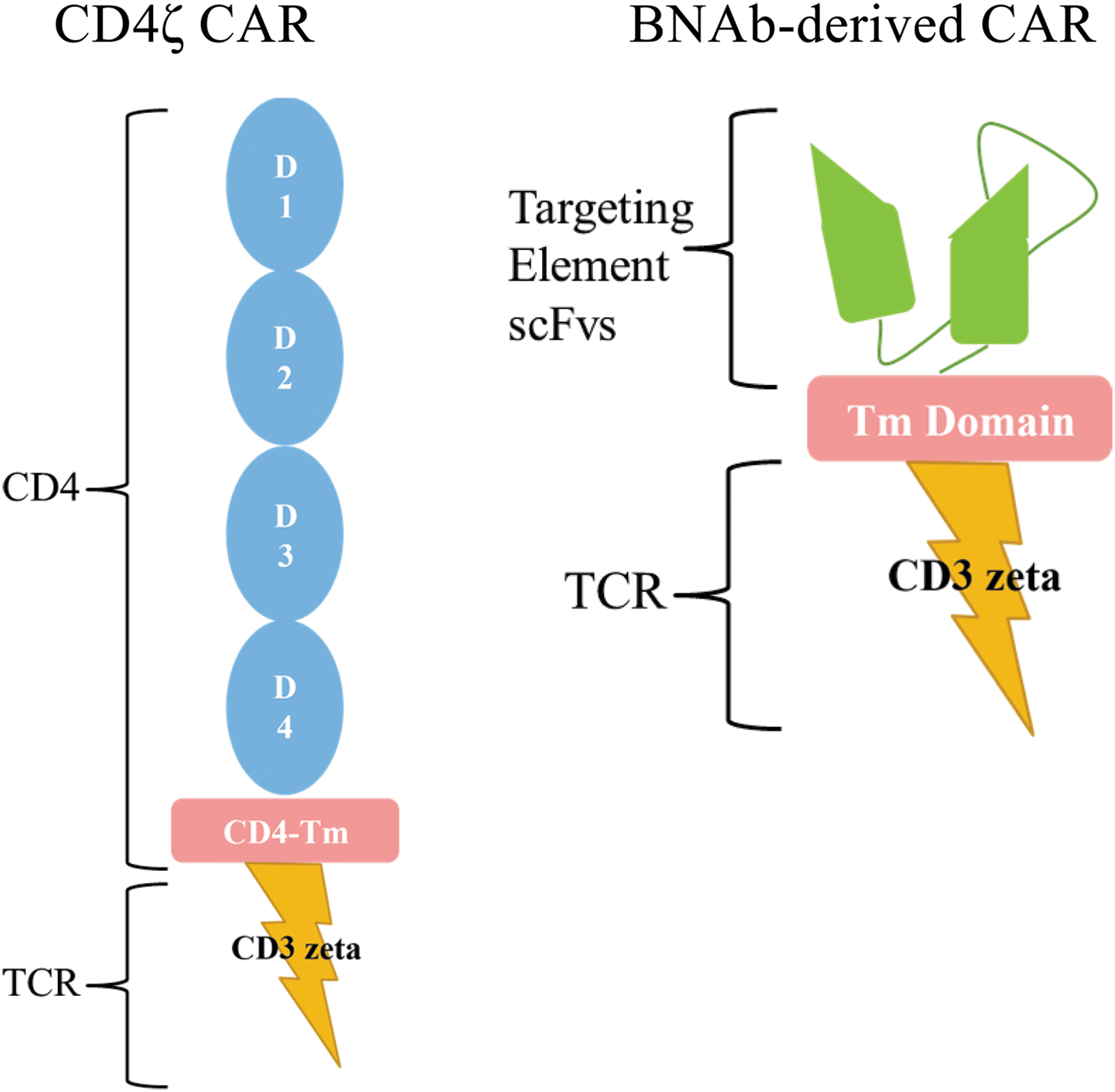
HIV-specific chimeric antigen receptors (CARs). The left figure depicts the CD4ζ CAR, which comprised the entire natural extracellular domain [domains (D) 1–4] and transmembrane domain of the CD4 molecule fused to the CD3ζ signaling domain. The right figure depicts a broadly neutralizing antibody (BMAb)-derived CAR, which contains an HIV-recognizing extracellular single-chain variable fragment (scFv). The scFv is a fusion protein of the variable regions of the heavy (VH) and light chains (VL) of an HIV-specific neutralizing antibody that is usually linked together with a short linker peptide and is the targeting element of the CAR. This is fused to some sort of a transmembrane domain (for instance, of the CD8 molecule) as well as the CD3ζ signaling domain. Additional signaling domains from costimulatory molecules can also be included in CAR constructs to increase the potency of the signal (not shown).
While the manipulation and modification of peripheral blood T cells have certain benefits (including the ease of obtaining high numbers of cells and their ability to be manipulated ex vivo), there are several disadvantages (including the manipulation process causing cellular dysfunction and potentially low levels of engraftment of the genetic modification and persistence of genetically modified cells). The use of HSCs would provide for potential improvements over peripheral T cell modification, including the long-term maintenance of functional gene-modified effector T cells of both CD4+ and CD8+ lineages. A stem cell-based gene therapy approach using a molecularly cloned HIV-specific TCR or CAR would allow proper thymic selection of modified cells and exclusion of endogenous TCR surface expression, eliminating the risk of generating self-reactive TCR through mispairing. 26 A stem cell-based gene therapy approach would allow for long-lived continuously renewable immunity capable of continuously generating cells that develop into HIV-targeting T cells that could potentially overcome the barriers that inhibit the eradication of HIV from the body.
We have previously demonstrated that we can engineer HIV-specific T cell responses utilizing a stem cell-based gene therapy approach with a molecularly cloned T cell receptor (TCR) that targets HIV. 27 –29 We determined that the modification of a human HSC with a lentiviral vector containing an anti-HIV TCR can direct the differentiation of mature, polyfunctional HIV-specific T cells in human thymic tissue in vivo in the SCID-hu mouse, a humanized mouse model that recapitulates human T cell development and thymic selection. 27 These cells, carrying the transgenic anti-HIV TCR, survived hematopoietic differentiation and thymopoiesis and developed into cells capable of killing HIV-infected cells ex vivo. This initial proof of concept demonstrated for the first time that a human TCR could be used in this manner to direct human T cell differentiation. Interestingly, this study demonstrated the importance of HLA-matched tissue as tissues that were not matched to the TCR specificity of HLA-A*0201 (the HLA usage of that particular TCR) did not allow T cell differentiation past the immature stage of T cell development, presumably through the absence of positive selection.
Follow-up studies to this demonstrated that an HIV-specific TCR is capable of reducing HIV replication in vivo. 28 In this approach, we utilized the surrogate humanized bone marrow, fetal liver, and thymus (BLT) mouse model to demonstrate that human CD34+ HSCs can be genetically modified with a lentiviral vector containing a molecularly cloned TCR specific to HIV and subsequently develop into mature fully functional CTLs that reconstitute the peripheral immune system. 28 We further demonstrated that these HIV-specific CTLs could effectively lower viral loads following HIV infection. These cells underwent normal developmental processes, including their maturation into T cells in the human thymus, and responded to HIV infection in vivo in a highly active and normal manner. These studies demonstrated that the modification of an HSC with HIV-specific TCR and the subsequent development of HIV-specific T cells was a feasible approach and was biologically possible in producing immune cells that were capable of lowering viral loads and killing HIV-infected cells. However, this technology is again somewhat limited as it requires TCRs that match the individual's HLA molecules due to the process known as MHC restriction.
As mentioned above, CARs may be superior to TCRs as they bypass the need for MHC restriction. The inherent limitations to using a molecularly cloned TCR are the absolute requirement for a specific HLA molecule to present antigen to the T cell, limiting each TCR to persons with the right HLA type as well as the fact that HIV has evolved means to counteract HLA presentation to CTLs and thereby limit their efficacy. To overcome these issues associated with HLA restriction of an HIV-specific TCR, we explored the notion of utilizing an HIV-specific CAR in a stem cell-based approach.
The initial studies in this investigation used the aforementioned CD4ζ CAR as a prototype anti-HIV CAR. 30 –32 The use of this CD4ζCAR has important advantages, in that it has been found to be a safe reagent in multiple long-term clinical trials with over 500 patient years of clinical safety data 33,34 and this is strong evidence that it does not induce cytokine storms that have been an unwanted element with other CAR-based approaches in treating malignancies. 35,36 It is also unlikely to generate escape variants of HIV envelope as the loss of CD4 binding of an escape variant will likely have a dramatic effect on viral fitness.
We and others have done extensive testing of the antiviral function of CD8+ T cells transduced with CD4ζ CAR, finding that transduced cells are capable of killing HIV-infected cells and suppressing viral replication 30 –32,34,37 and (unlike the HIV-specific TCR) that this activity is independent of HLA-I molecules. 38 CARs can also function in CD4+ T cells to act as HIV-1-specific helper cells. IL-2 and IFN-γ are induced in CD4ζ CAR-transduced CD4+ cells when cocultured with infected cells. Interestingly, possibly one reason that antiviral efficacy was minimal in previous trials with the CD4ζ CAR was that it was erroneously thought that CD4ζ CAR-expressing cells were not infected by HIV. 34 We and others tested whether HIV can infect CD8+ cells expressing the CD4ζ CAR molecule and determined that CD8+ cells, which now express CD4 through transduction of CD4ζ CAR, are susceptible to HIV infection. 30,39 In this regard, genetic engineering with CD4ζ CAR alone could be rendered useless if CTL can now be infected by HIV. CD4 can also be induced on CD8+ cells following TCR stimulation, allowing infection and killing of the CTL. 40,41 Further, destruction of developing or supporting immune cells such as engineered CD4+ helper cells would limit effectiveness of CTLs. Thus, we have combined CD4ζ CAR with anti-HIV reagents to confer protection from HIV infection.
We introduced two shRNAs, one that downregulates CCR5 and one that downregulates HIV expression by targeting the LTR region into the CD4ζ-expressing vector. Using this combinational vector, we successfully diminished the susceptibility to HIV infection in vitro and in vivo through the CD4ζ CAR molecule. 30 To test a proof of concept that HSC transduced with a CAR would proceed successfully through thymopoiesis, we transplanted HSCs modified with a lentiviral vector containing the CD4ζ CAR into the humanized BLT mouse. We observed the efficient development in the peripheral blood of mature T cells expressing the lentiviral vector marker gene (green fluorescent protein) and the CD4ζ gene (Fig. 2A). We saw reduced viral loads in animals that received the CD4ζ CAR when compared with control animals (Fig. 2B). We also saw significant expansion and effector/memory cell differentiation of CD4ζ CAR cells following HIV infection, indicating full functional responses of these cells. 30 Interestingly, those cells that expressed the highest levels of the transgenic CD4ζ CAR appeared to have shut down their endogenous TCR expression, indicating that the transgenic CD4ζ CAR became the sole TCR on these cells, likely by activating the allelic exclusion machinery in these cells.

Following HIV infection in these animals, we found that levels of cells expressing HIV and viral loads were significantly diminished in animals expressing the CAR versus control animals. We are currently investigating and have made significant progress in the use of this CAR in the nonhuman primate model, where modification of HSCs and subsequent T cell development appears to be well tolerated and safe in early studies (not shown). In summary, we have established an efficient system to closely examine the development of genetically engineered HIV-specific T cells from HSCs in vivo and demonstrated the feasibility of the use of molecularly cloned HIV-specific TCR and CARs in this approach.
Recently, we and others have developed CARs that utilize antigen-binding components derived from the sequences of HIV-specific broadly neutralizing antibodies (BNAbs) specific for a variety of HIV epitopes. 32,42 Some of these CARs also contain additional signaling sequences, which provide costimulatory capability upon ligand binding, to achieve a more robust activation event upon ligation with the target epitope. Upon assessment in a suite of assays, each of these CARs is functional; however, efficiency varies with each ligand-binding moiety, each assay used, and the virus strain being employed. Importantly, Liu et al. 42 recently reported that a CAR utilizing the ligand binding domain of VRC01, a broadly neutralizing anti-HIV antibody, reduced viral rebound following removal of antiretrovirals in an in vitro latency model. Thus, these molecules are effective at depleting HIV-expressing cells and reducing virus replication in vitro, but they have not yet been tested in in vivo models through a stem cell-based approach. These latter studies are in progress with several of these BNAb-derived molecules in our laboratory. Importantly, having several reagents that effectively target a variety of HIV epitopes, when used in combination, may decrease the ability of the virus to escape from this type of approach.
In summary, a variety of CARs specific against HIV are under development and pre-clinical testing. These reagents may be useful in peripheral cell or stem cell-based approaches and have considerable potential for use in shock and kill as well as long-term immune surveillance strategies.
Footnotes
Acknowledgments
This work was funded by grants from amfAR ARCHE awards (grant nos. 108688-54-RGRL, 108929-54-RGRL); the NIH, grant no. AI078806 (SGK); the UCLA Center for AIDS Research (CFAR), grant no. P30AI28697; the California Center for Regenerative Medicine, grant no. TR4-06845; and the UC Multi-campus Research Program and Initiatives, California Center for Antiviral Drug discovery (CCADD).
Author Disclosure Statement
No competing financial interests exist.