
Abstract
Patients with the genomic instability syndrome Fanconi anemia (FA) commonly develop progressive bone marrow failure and have a high risk of cancer. The prominent role of the FA protein family involves DNA damage response and/or repair. Oxidative stress, defined as an imbalance between the production of reactive oxygen species and antioxidant defense, is considered to be an important pathogenic factor in leukemia-prone bone marrow diseases such as FA. Cellular responses inducing resistance to oxidative stress are important for cellular survival, organism lifespan, and cancer prevention, but until recently, mammalian factors regulating resistance to oxidative stress have not been well characterized. Significant evidence supports excessive apoptosis of hematopoietic stem/progenitor cells, induced by stresses, most significantly oxidative stress, as a critical factor in the pathogenesis of bone marrow failure and leukemia progression in FA. In this brief review, we discuss the functional link between FA proteins and oxidative DNA damage response/repair, with emphasis on the implication of oxidative stress in the pathophysiology and abnormal hematopoiesis in FA. Antioxid. Redox Signal. 10, 1909–1921.
Introduction

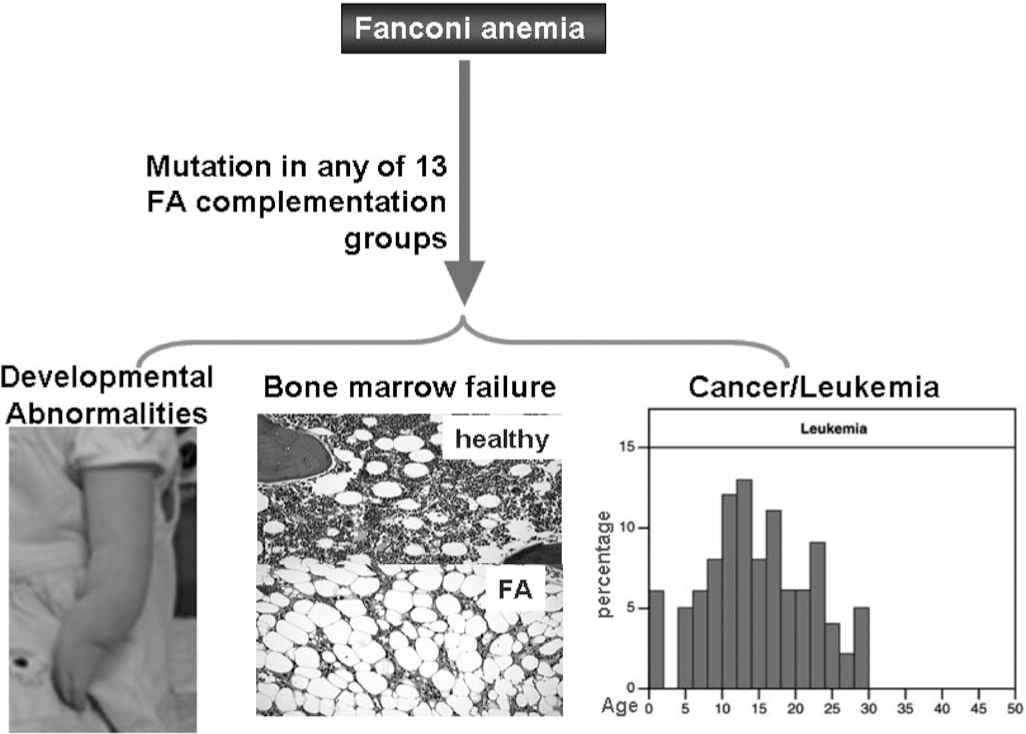
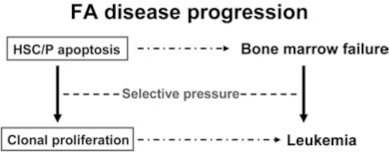
Since patients with FA uniformly develop BM failure and have high risk of progression to leukemia, FA proteins likely play specific roles in hematopoiesis by governing responses of hematopoietic cells to both genotoxic and cytotoxic stresses. Extensive studies indicate that chronic cytotoxic or genotoxic stresses differentially affect FA hematopoiesis by causing excessive apoptosis of hematopoietic stem cells and progenitors (HSC/P) (11, 12, 21, 31 –33, 42, 51, 52, 61, 71, 74 –76, 78, 79, 88, 100, 101, 103). Thus, the maintenance of HSC/P cells in bone marrow may require different functions of the FA proteins. This seems to be consistent with clinical outcomes and evolution of the disease: loss of FA functions causes excessive apoptosis of HSC/P in the early stage of FA, leading to BM failure. As the disease progresses, both apoptosis and genomic instability impose a selective pressure on FA HSC/P cells and promote the development of mutant clones leading to leukemia (Fig. 3).
FA Hematopoiesis
The most important clinical features of FA are hematological. Children with FA often develop pancytopenia during the first few years of life. Complications of BM failure are the major causes of morbidity and mortality of FA, and 80% of FA patients die from BM failure (3, 6, 22, 45, 47, 53). In addition, FA patients have increased susceptibility of developing myelodysplasia (MDS) or acute myeloblastic leukemia (AML) (3, 6, 13, 22, 41, 95). They have also been known to frequently develop clonal chromosomal abnormalities in the BM progenitor cells in the later stage of the disease (3, 47, 95). In fact, certain clonal cytogenetic abnormalities, such as monosomy 5 and monosomy 7, are common in MDS and AML occurring secondary to treatment with alkylating agents and in children with FA who have evolved to MDS and AML (58, 83, 102). Thus, FA has been proposed as a genetic model system for studying these hematological malignancies (3, 13, 95).
Increasing evidence indicates progressive BM failure in children with FA results from excessive apoptosis and subsequent failure of the HSC compartment. The first evidence that FA BM cells demonstrate increased apoptosis was from a report that demonstrated CD34+ cells from children with FA expressed high levels of the death receptor Fas (11, 72). Subsequently, many laboratories have reported FA BM cells are hypersensitive to a variety of extracellular biological apoptotic cues, including interferon-γ (IFN-γ) and tumor necrosis factor α (TNF-α) (19, 21, 31, 42, 51, 52, 71, 74 –76, 78 –81, 86, 88, 101, 103). The most compelling evidence comes from studies of the functions of FANCC in apoptotic responses of hematopoietic cells, largely because FANCC was the first FA gene cloned and the Fancc knockout mouse was the first murine model of FA generated. For example, suppression of FANCC expression represses clonal growth of normal erythroid and granulocyte-macrophage progenitor cells and disruption of the Fancc gene in mice renders hematopoietic progenitor cells hypersensitive to the pro-apoptotic effects of IFN-γ and TNF-α (21, 31, 42, 51, 71, 74 –76, 78, 79, 88, 101, 103). Conversely, overexpression of FANCC suppresses apoptosis in human hematopoietic progenitor cell lines (27, 28), in CD34+ cells from FA patients with FANCC mutations (101) and in HSC/P cells from Fancc knock-out mice (79, 103). These data suggest deregulation of apoptotic responses in hematopoietic cells may account at least in part for the nearly universal development of BM failure in children with inactivating FA mutations. However, the mechanism by which FA proteins modulate apoptotic responses and the downstream effector molecules involved are mostly unknown.
Results from BM culture assays have also demonstrated defective hematopoiesis in FA (3, 22, 95). Bone marrow cells from FA patients also show altered expression of certain growth factors and cytokines, such as reduced expression of interleukin-6 (IL-6) and granulocyte-macrophage colony-stimulating factor (GM-CSF) but increased secretion of mitotic inhibitor TNF-α (14, 19, 81, 82, 86, 90). These alterations may change the BM microenvironment (for instance, leading to factor deprivation or constant exposure to mitogenic inhibitors) and cause deregulation of cellular homeostasis. Studies of FA patients have demonstrated that BM from FA patients has decreased frequency of colony-forming progenitor cells, as well as a reduction in colony size (18, 29). These data suggest loss of FA gene function results in reduced survival or proliferation of lineage restricted progenitors and ultimately injury to the progenitor cell compartment. Using a murine model of FA complementation group C (Fancc), Haneline et al. (32, 33) convincingly demonstrated that Fancc −/− BM cells had a profound decrease in repopulating ability, and complementation with a retroviral vector encoding the normal human FANCC could restore Fancc −/− mouse BM stem and progenitor cell growth and proliferation in vivo. These data are consistent with damage to the stem cell compartment in FA.
FA Oxidant Hypersensitivity
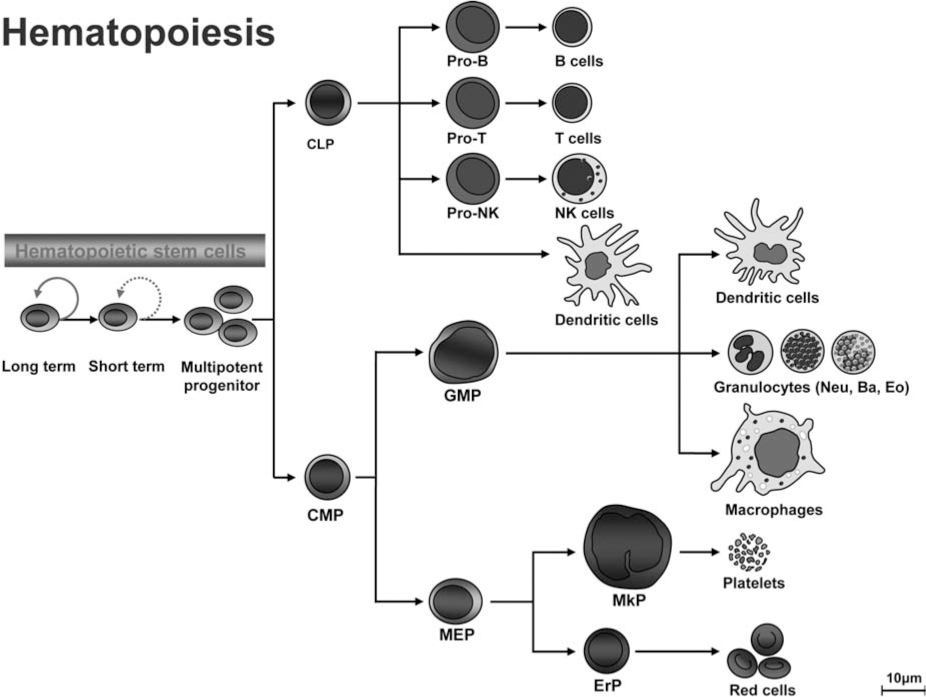
Like other somatic stem cells, hematopoietic stem cells (HSCs) maintain life-long hematopoiesis in the bone marrow via their ability to self-renew and to differentiate into all blood lineages (Fig. 4). These HSCs are essentially required for the hematopoietic homeostasis. During differentiation, long-term hematopoietic stem cells (LT-HSCs) transit through short-term (ST)–HSCs and committed progenitor stages, which are characterized by restricted lineage potential and reduced self-renewal capacity. This is followed by ultimate differentiation into mature myeloid, erythroid, or lymphoid lineages. Molecular mechanisms controlling self-renewal and cell-fate decisions within the hematopoietic system remain poorly defined. In this context, HSC do not only need to replenish peripheral blood cells of all lineages, but also have to keep their pool relatively constant. There is good evidence that certain extrinsic cues provided in a special environment, the HSC-niches, essentially take part in regulating the HSC pool in vivo and might also be involved in leukemogenesis. Hematopoietic cells are exposed to various reactive oxygen species (ROS), which are routinely generated during metabolic or inflammatory process. ROS stimulation induces a variety of responses in hematopoietic cells, including cellular proliferation and growth inhibition (50, 70, 87, 108). Like cells from other tissues, hematopoietic cells have developed several mechanisms to prevent the damage induced by oxidative stress. Antioxidant enzymes, including superoxide dismutases (SODs), catalase, glutathione peroxidases, and peroxiredoxins, can directly eliminate ROS. Other cellular enzymes can function to repair DNA damage induced by ROS in hematopoietic tissues.
Studies on pathophysiological mechanisms of oxidative stress responses in stem cell diseases such as aplastic anemia (AA) have been very instructive and provide insights into the function of normal hematopoietic stem cells and their self-renewal capacity. One of the well-studied AA disease models is FA. There is strong evidence that FA cells are intolerant of oxidative stress. This was first suggested by the observation that cultured FA cells are vulnerable to oxygen-induced chromosomal aberrations (38). The finding was later confirmed by two other groups that showed FA fibroblasts and primary bone marrow cells grow better under hypoxic conditions than in ambient air (8, 85). Over the last decade, FA oxidant hypersensitivity has been documented in many studies using primary and immortalized cell cultures as well as ex vivo materials from patients (5, 8, 11, 27, 30, 43, 73, 77, 84).
While FA murine models do not recapitulate some of the major FA clinical manifestations such as BM failure and leukemia, hematopoietic cells from FA knockout mice exhibit extreme oxidant sensitivity. Saadatzadeh et al. (84) show that primary hematopoietic progenitors and murine embryonic fibroblasts (MEFs) isolated from Fancc−/− mice exhibit hypersensitivity to oxidative stress generated by hydrogen peroxide (H2O2). Furthermore, pretreatment with antioxidants selenomethionine or N-acetylcysteine (NAC) preferentially enhanced the survival of Fancc−/− cells. Mechanistically, the authors found that H2O2 induced overactivation of the serine-threonine kinase apoptosis signal-regulating kinase 1 (ASK1) in Fancc−/− cells, which was correlated with elevated H2O2-induced apoptosis in these mutant cells. Interestingly, ASK1 has been shown to be an important kinase involved in oxidant- and TNF-α-induced apoptosis (36), and Fancc−/− cells are uniquely hypersensitive to TNF-α (31, 78, 87, 107). It is therefore a plausible hypothesis that the TNF-α-oxidant–ASK1 pathways may play an important role in the observed functional deficits of Fancc−/− hematopoietic stem/progenitor cells, including reduced reconstitutional ability and proliferative potential, diminished self-renewal, and increased apoptosis. In addition, mice with combined deficiencies of the antioxidative enzyme, Cu/Zn superoxide dismutase, and Fancc genes demonstrated a defective hematopoiesis (30). Although not displaying development defects or increased chromosomal aberrations typical of FA, Fancc−/− Sod−/− mice had a phenotype of bone marrow hypocellularity, which is not detected in single mutant mice. Furthermore, hepatocytes from Fancc−/− Sod−/− mice exhibited a zonal pattern of microvesicular steatosis. All of the observations indicated the altered redox state of the hematopoietic progenitors in these mice was responsible for an impairment of cell proliferation or survival.
To dissect the relationship between oxidative damage and BM failure in FA, we have recently demonstrated that oxidative stress generated by repeated cycles of hypoxia-reoxygenation leads to excessive oxidative DNA damage and premature senescence in Fancc−/− bone marrow hematopoietic stem/progenitor cells (106, 107). These studies suggest that stress-induced senescence may be a novel mechanism underlying hematopoietic stem cell depletion in bone marrow failure diseases, including FA. More recently, we showed that ROS induce hematopoietic suppression in Fancc−/− mice exposed to bacterial toxin lipopolysaccharide or pro-inflammatory cytokine TNF-α (4, 65, 87, 108). TNF-α-induced senescence correlates with the accumulation of ROS and oxidative DNA damage, and pretreatment of TNF-α-injected Fancc−/− mice with a ROS scavenger significantly reduces oxidative base damage, DNA strand breaks, and senescence. Furthermore, Fancc−/− hematopoietic stem/ progenitor cells show increased chromosomal aberrations and have a lagging oxidative DNA damage repair. These results point to a potential link between oxidative DNA damage and hematopoietic stem cell defect in FA. The observed inefficient repair of oxidative damage in Fancc−/− hematopoietic stem cells may lead to decrease of stem cell quality (self-renewal capacity), which may ultimately cause premature exhaustion of the hematopoietic stem cell pool leading to BM failure.
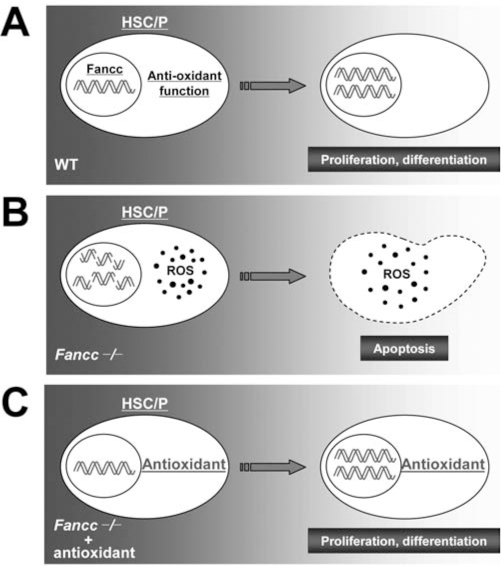
One important issue concerns oxidative stress as a pathological factor in FA disease progression. Numerous reports indicate that FA cells, including HSC/P cells, are hypersensitive to oxidative stress (5, 8, 11, 27, 30, 38, 43, 73, 77, 84, 85, 106, 107). A number of hypotheses regarding the effect of oxidative stress in FA have been suggested, including the proposal that ROS could damage DNA and inability of FA hematopoietic stem/progenitor cells to repair such damage would result in exacerbated genomic instability leading to apoptosis and malignant transformation (Fig. 5).
Redox-Sensitive FA Proteins
Because FA hematopoietic progenitor and stem cells have high rates of stress-induced apoptosis and reduced repopulating ability, FA proteins are subsequently believed to play an important role in the maintenance of normal hematopoiesis during oxidant metabolism. Consistent with the observations that cells derived from FA patients are intolerant of oxidative stress, an extensive body of evidence suggests that FA proteins play crucial role in oxidative stress signaling in variety of cell types including hematopoietic stem/progenitor cells.
Three major FA proteins, FANCA, FANCC, and FANCG, as parts of the FA protein complex, are found to associate with a variety of cellular factors that primarily function in redox-related processes (Table 2). For example, the FANCC protein interacts with NADPH cytochrome P450 reductase and glutathione S-transferase P1-1 (11, 43), two enzymes involved in either triggering or detoxifying reactive intermediates including ROS. Fancc−/− mice with deficiency in the antioxidative enzyme Cu/Zn superoxide dismutase demonstrated a defective hematopoiesis (30). Another FA protein, FANCG, interacts with cytochrome P450 2E1, a member of the P450 superfamily that is associated with the production of reactive oxygen intermediates, and mitochondrial antioxidant enzyme peroxiredoxin-3 (27, 69), suggesting a possible role of FANCG in protection against oxidative DNA damage. Significantly, Saadatzadeh et al. (84) recently showed oxidant hypersensitivity of Fancc−/− cells was due to an altered redox regulation and hyperactivation of ASK1, a serine-threonine kinase that plays an important role in redox apoptotic signaling. Moreover, oxidative stress induces complex formation by two major FA proteins, FANCA and FANCG (77).
Oxidative Stress Response in FA Hematopoietic Cells: A p53 Connection
The tumor suppressor p53 is a key transcription factor that activates vital damage containment procedures to restrict aberrant cell growth in response to DNA damage, oncogene activation, and loss of normal cell contacts (28, 55). By eliciting cell cycle arrest and DNA damage response to oxidative DNA damage and oncogenic stress, p53 restricts cellular growth by inducing senescence, growth inhibition, or apoptosis that maintain genomic stability (37). Thus, p53 plays a major role in the prevention of cancer. Consistent with this, emerging evidence suggest that p53 deficiency may increase cancer development in patients with FA and FA mice. For example, studies have found a higher proportion of human papillomavirus–positive squamous cell carcinomas (SCC) in patients with FA than in healthy controls. Furthermore, SCC in FA patients is probably associated with the inactivation of p53 by HPV-associated oncoproteins rather than by direct mutagenesis, indicating that loss of functional p53 facilitated the tumor development in these cases (46, 57). Mice deficient for Fancd1 or Fancd2 have accelerated tumor development in Trp53-deficient background (34, 40). In addition, Fancc deficiency accelerates the development of certain blood and solid tumors in mice heterozygous at Trp53 (24). Moreover, these studies demonstrate that FA proteins and p53 cooperate in apoptosis and cell-cycle checkpoint control following DNA damage (25, 34, 54). Thus, p53 may function to prevent the propagation of damaged DNA through apoptosis. In FA patients, this leads to stem cell depletion, which may cause congenital abnormalities and bone marrow failure. Loss of p53 function may predispose to cancer by allowing premalignant cells to survive (41, 93).
Primary cells from FA patients and knockout mice are uniquely hypersensitive to oxidative stress-induced DNA damage and growth arrest. This suggests FA proteins may interplay with p53 in oxidative stress response. Encouraged by recent reports that p53 deficiency increases cancer development in patients with FA and FA knockout mice (24, 34, 40, 46, 57), we formally tested the hypothesis that FA proteins may functionally interact with the p53-activating signals in response to oxidative stress. Our unpublished results suggest that two major FA proteins, Fanca and Fancc, may coordinate with p53 in the regulation of oxidative stress response. This notion is supported by (i) hypersensitive response to oxidative stress in tissues in vivo and in cells in vitro derived from Fanca−/− or Fancc−/− mice is correlated with a persistent p53 overactivation; (ii) manipulation of p53 signaling alters H2O2-induced cell-cycle checkpoint (Fig. 6) and DNA damage response in primary Fanca−/− cells; and (iii) the functional status of p53 dictates the kinetics and persistence of response to oxidative stress in FA cells.
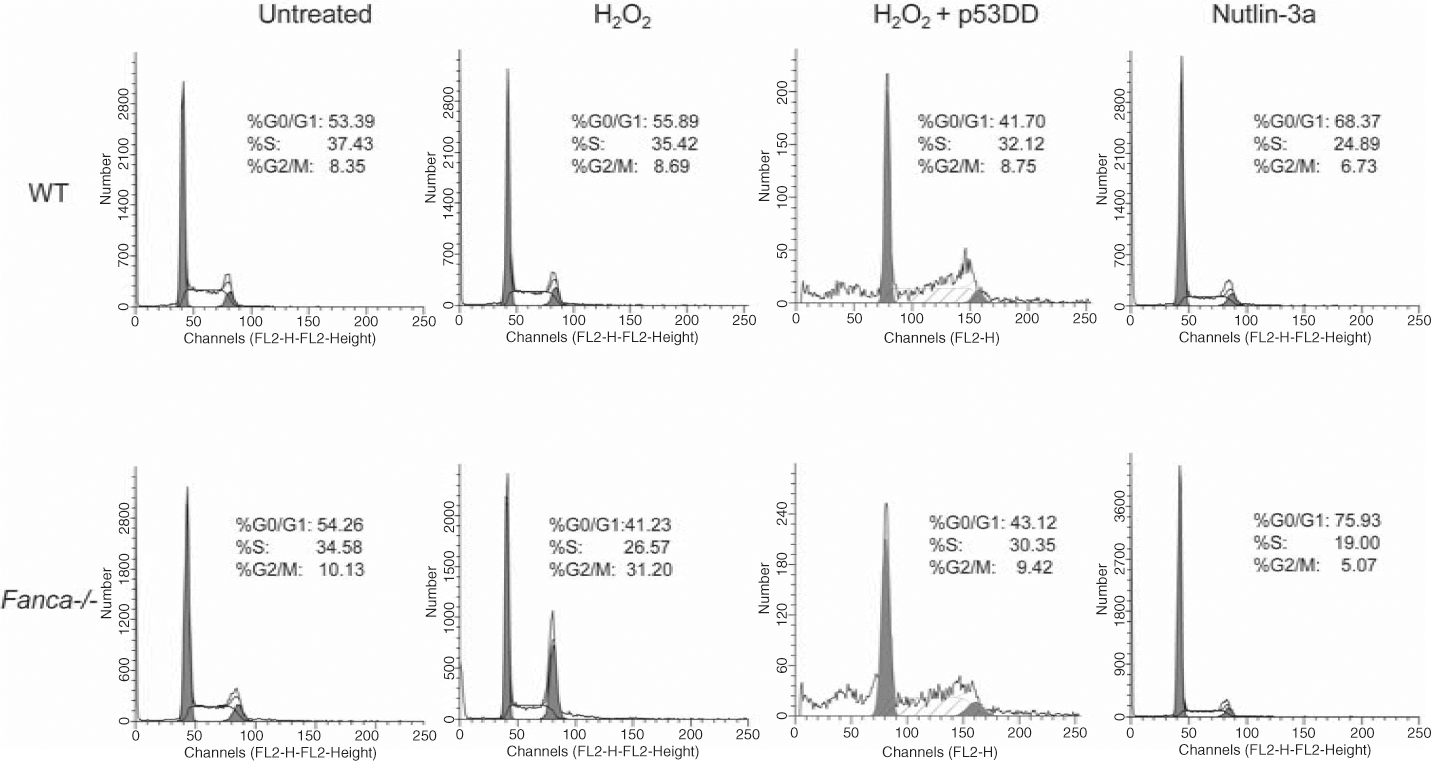
The mechanistic link between p53 signaling and FA has not been well defined. The involvement of p53 in FA pathophysiology has been highlighted by recent studies that show mice deficient for Fancd1, Fancd2, or Fancc have accelerated tumor development in Trp53-deficient background (24, 34, 40). Furthermore, developmental defects and increased apoptosis in Fancd2-deficient zebrafish could be corrected by knockdown of p53, suggesting p53-dependent apoptosis may be an underlying mechanism for developmental defect in the
Fancd2−/− fish (54). Some reports suggested that the activation of p53 leads to an increase in ROS that, perhaps by interfering with mitochondrial function and/or integrity, contributes to cell death. In addition, the higher levels of ROS appear to be part of the feedback loop that stabilizes p53 resulting in more p53 activity (26). Cellular stresses may increase mitochondrial ROS generation and increase p53 protein levels in some cell lines, whereas antioxidant NAC and the Cu/Zn SOD inhibitor can abolish the stress-induced increase in ROS and p53 levels (7). While these studies have not formally conducted in FA cells, the difference in redox status may result in different levels of p53 activation in WT and FA cells. In WT cells, ROS attack chromosomal DNA and generate oxidative DNA damage, mainly in the form of 8-oxo-deoxyguanosine (8-oxo-dG). The damage can induce the activation of p53 by phosphorylation (for example, phospho-p53 at Ser20–p53Ser20), leading to the repair of the oxidative DNA damage (Fig. 7). Loss of FA function leads to increased level of ROS or reduced repair of the oxidative DNA damage. Consequently, FA cells accumulate higher level of oxidative DNA damage, leading higher level of p53 activation. Additional investigations into functional interaction between the p53 and FA pathways in oxidative DNA damage stress response may aid us in better understanding how cells can bypass the normal checkpoints and continue to proliferate in the presence of damaged DNA and oncogenic activation. In the context of FA, new insights on the role of FA proteins in oxidative DNA damage response/repair can suggest new pathways and proteins to target for therapeutic prevention of cancer progression of the disease.

The Link Between Inflammatory ROS and FA Leukemogenesis
Certain chronic inflammatory conditions have long been known to link to cancer. There is compelling evidence that chronic inflammation increases the risk of human cancers such as hepatocellular carcinoma, colon and bladder cancers, B cell lymphomas, and visceral malignancies (44, 62, 92, 97). Chronic inflammation in the intestinal or bronchial epithelia also promotes carcinomas of the colon and lung (20). Other inflammatory diseases like Barrett's syndrome and Crohn's disease are linked to the development of esophageal cancer and bowel cancer, respectively (3, 9). In these pathological conditions, unresolved inflammation provokes cell turnover coupled with ROS generated at sites of inflammation, leading to chromosomal DNA mutations and malignant transformation of the cells (2, 10).
Oxidative stress is considered to be an important pathogenic factor in leukemia-prone bone marrow diseases like FA (5, 8, 11, 27, 30, 38, 43, 69, 73, 77, 84, 85, 106, 107). The expression of inflammatory mediators, particularly the pro-inflammatory cytokines tumor necrosis factor-alpha (TNF-α), interleukin-1beta (IL-1β), and IL-6 in these patients is often associated with increased production of ROS either as a component of their immune response or as a consequence of increased metabolism (59, 63, 64, 96). Thus, the presence of pro-inflammatory cytokines and increased oxidative stress in these patients may account for profound physiologic changes, including the development of BM failure and progression to leukemia. Many studies (24, 51, 81, 108) have shown a correlation between elevated circulating pro-inflammatory cytokines and anemia in patients with leukemia-related BM diseases but direct evidence for the mechanistic link between inflammation and leukemia is lacking.
The inflammatory cytokine TNF-α is considered as one important pathological factor involved in the abnormal hematopoiesis In FA. Studies from our laboratory and others have suggested excessive apoptosis of FA hematopoietic cells induced by TNF-α, which is overproduced in FA patients,?? may contribute to the pathophysiology of BM failure frequently occurring in FA children. The recent pioneer work from the laboratories of Wade Clapp and Laura Haneline (31 –33, 51, 88) has demonstrated that ex vivo culture of Fancc−/− BM cells leads to an increase in cytogenetic abnormalities and myeloid malignancies that are associated with an acquired resistance to TNF-α, suggesting FA hematopoietic cells are prone to clonal hematopoiesis and malignancy. It is well established that TNF-α-induced ROS production involves the c-JUN NH2-terminal kinase (JNK) and nuclear factor-kappa B (NF-κB) pathways (71, 98). TNF-α-induced ROS activate the JNK kinase, which in turn leads to more ROS production, and sustained JNK activation in NF-κB-deficient cells was suggested to depend on ROS. Studies have shown that the production of ROS by TNF-α at inflammatory sites causes DNA damage (1, 92, 99). The persistent high levels of oxidative DNA damage observed in HSC/progenitor cells from TNF-α-injected Fancc−/− mice suggest that a deficiency in the FA pathway renders chromosomal DNA susceptible to ROS attack, thereby increasing oxidative DNA damage (87, 108). Our recent studies have also indicated that TNF-α not only is a pro-apoptotic signal suppressing FA hematopoietic progenitor activity, but also promotes leukemic transformation of FA hematopoietic stem/progenitor cells (50). Specifically, we tested the leukemia-promoting effects of TNF-α in Fancc−/− stem cells in vitro. We found that TNF-α exposure initially inhibited the growth of Fancc−/− bone marrow hematopoietic stem/progenitor cells, but longer term exposure of these cells promoted the outgrowth of TNF-α-resistant cytogenetically abnormal clones that upon transplantation into congenic wild-type mice led to acute myelogenous leukemia. TNF-α induced ROS-dependent genetic instability in Fancc−/− but not in WT cells. The leukemic clones were TNF-α-resistant but retained their characteristic hypersensitivity to mitomycin C, and exhibited high levels of chromosomal instability. Expression of FANCC cDNA in Fancc−/− stem/progenitor cells protected them from TNF-α-induced clonal evolution. The molecular etiology of FA leukemogenesis remains unknown. We hypothesize that FA disease progression to leukemia is governed not only by genetic changes intrinsic to the FA cells, but also by epigenetic and environmental factors and that TNF-α-mediated inflammation is one of the most important epigenetic and environmental factors contributing to FA leukemogenesis. Our studies on the role of TNF-α in FA leukemogenesis suggest that TNF-α exposure creates an environment in which somatically mutated preleukemic stem cell clones are generated and selected for. Our results thus provide direct confirmation of the importance of selective pressure in the evolution of leukemic clones in FA. These studies also suggest a model, in which mutations in the FA genes can cause genomic instability and overproduction of TNF-α, which induces apoptosis through upregulation of ROS and JNK/p38 kinases. Patients with excessive apoptosis of BM cells develop bone marrow failure. Chronic exposure of FA BM cells to proinflammatory cytokine TNF-α selects for progenitor cells that are apoptosis-resistant and acquire proliferative advantage. Patients with these TNF-α-resistant BM cells advance to myelodysplasia (MDS) and acute myelogenous leukemia (AML) via a mechanism involving genomic instability, coupled with inflammation driven by high NF-κB transcriptional activity (Fig. 8).
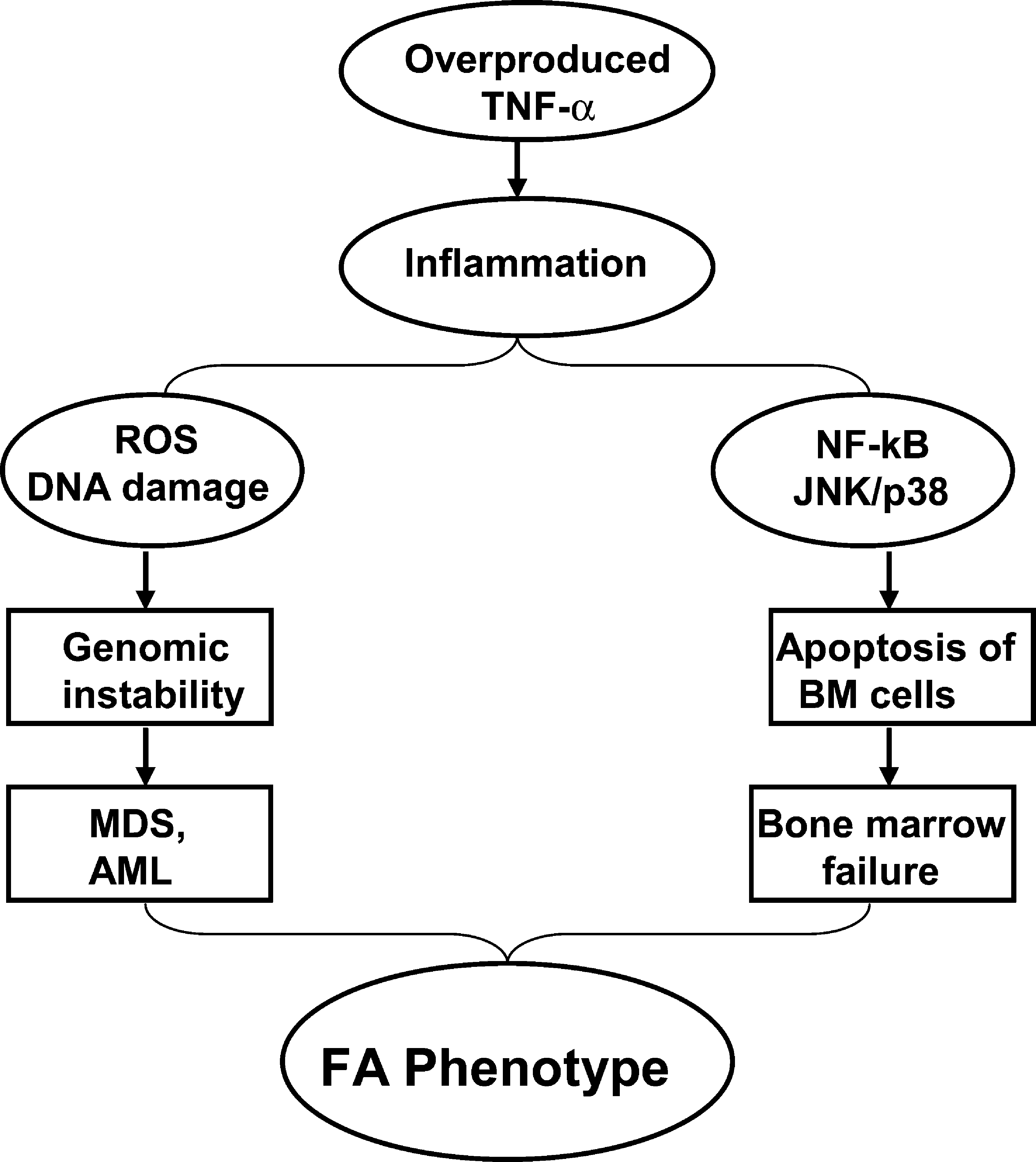
While the role of FA proteins in the regulation of TNF-α-induced ROS production remains to be elucidated, it is likely FA proteins can disrupt downstream ROS signaling by protecting chromosomal DNA from ROS attack or facilitating the repair of oxidative DNA damage. Recently, the Grompe group reported that treatment of Fancd2−/−;Trp53 +/− mice with the antioxidant tempol delayed solid tumor development, possibly through a mechanism involving the enhanced repair of oxidative DNA damage by the antioxidant (105). However, it is also possible FA proteins can influence the expression of antioxidant enzymes (such as glutathione S-transferases and catalase) or the biosynthesis of ROS metabolic molecules such as glutathione. So far, there is no direct evidence for any of these assumptions. Another potential target is the redox-sensitive transcription factor NF-κB whose activation is known to enhance inflammation and promote cancer (10, 23, 60). Indeed, TNF-α-resistant BM hematopoietic stem/progenitor cells advance to acute myelogenous leukemia via a mechanism involving genomic instability coupled with inflammation driven by high NF-κB transcriptional activity (50).
Conclusion
FA is now considered the only human genomic instability syndrome that is uniquely sensitive to oxidative stress. As a bona-fide hematopoietic stem cell disease, FA represents an excellent disease model for studying oxidative stress response in hematopoietic stem/progenitor cells. Since BM failure and leukemia are rarely found in other known genomic instability syndromes such as ataxia telangiectasia, Nijmegen breakage syndrome, xeroderma pigmentosum, and Werner syndrome, further investigation into the function of FA proteins in oxidative damage response and repair will provide information on whether oxidative stress is a common signal that drives FA disease progression to leukemia. Therefore, understanding the relationship between oxidative stress and FA disease progression provides a unique opportunity to mechanistically comprehend and potentially intervene in these physiologically important processes.
Footnotes
Acknowledgments
The work of the authors is supported by NIH Grants R01 CA109641, R01 HL076712, and a Leukemia and Lymphoma Scholar award. We thank Dr. Keqin Ren for graphic support.
Abbreviations
ASK1, apoptosis signal-regulating kinase 1; AML, acute myeloblastic leukemia; BMF, bone marrow failure; FA, Fanconi anemia; GM-CSF, granulocyte-macrophage colony-stimulating factor; H2O2, hydrogen peroxide; HSC/P, hematopoietic stem cells and progenitors; IFN-γ, interferon-gamma; IL-6, interleukin-6; JNK; c-JUN NH2-terminal kinase; MEFs, murine embryonic fibroblasts; MDS, myelodysplasia; NAC, N-acetyl-L-cysteine; NF-κB, nuclear factor-kappa B; redox, reduction/oxidation reaction; ROS, reactive oxygen species; SCC, squamous cell carcinoma; SODs, superoxide dismutases; TNF-α, Tumor necrosis factor-alpha.