
Abstract
Gap-junction channels connect the cytoplasm of adjacent cells, allowing the diffusion of ions and small metabolites. They are formed at the appositional plasma membranes by a family of related proteins named connexins. Mutations in connexins 26, 31, 30, 32, and 43 have been associated with nonsyndromic or syndromic deafness. The majority of these mutations are inherited in an autosomal recessive manner, but a few of them have been associated with dominantly inherited hearing loss. Mutations in the connexin26 gene (GJB2) are the most common cause of genetic deafness. This review summarizes the most relevant and recent information about different mutations in connexin genes found in human patients, with emphasis on GJB2. The possible effects of the mutations on channel expression and function are discussed, in addition to their possible physiologic consequences for inner ear physiology. Finally, we propose that connexin channels (gap junctions and hemichannels) may be targets for age-related hearing loss induced by oxidative damage. Antioxid. Redox Signal. 11, 309–322.
Introduction
In addition to this traditional view, strong evidence suggests that undocked or unopposed hemichannels can open to allow communication between the cellular interior and the extracellular space under both physiologic and pathologic conditions (5, 83). Connexins are encoded by a family of homologous genes. A screening of the human genomic database identified 20 connexin genes (102). Connexins all have the same topology in the plasma membrane; with the amino and carboxyl termini, and one intracellular loop facing the cytoplasm, four transmembrane domains, and two extracellular loops. Although the homology between connexins is high, important differences between these proteins are found in the intracellular loop and the carboxyl terminus where many regulatory elements act, like kinases and cytoskeletal binding proteins. Additional diversity of GJCh is produced by the formation of heteromeric channels (in which a hemichannel is constituted from more than one connexin type) and/or heterotypic channels (produced by the docking of two hemichannels, each made by a different connexin). These connexin combinations may produce channels with particular functional and regulatory properties (6, 7, 66). The importance of GJCh for human physiology was pointed out by the findings of many genetic diseases associated with mutations in different connexin genes. Nine connexin genes have been implicated in diverse human hereditary disorders, like cataracts, Charcot-Marie-Tooth disease, oculodentodigital dysplasia, and inherited nonsyndromic or syndromic deafness. The latter condition is associated with a variety of mild to profound skin disorders (39). Of all connexin-associated diseases, deafness is the most important in terms of frequency in the human population. Although inherited deafness is genetically heterogeneous, mutations in the gene encoding Cx26 (GJB2) have been shown to account for a large proportion of cases in every population tested, whereas four other connexins, Cx30, Cx31, Cx32, and Cx43, have also been linked to either nonsyndromic or syndromic sensorineural hearing loss (48, 49, 75). The Cx43 mutation in deafness is controversial. Whereas the two mutations found in Cx43, L11F and V24A, are probably located in the Cx43 pseudo gene on chromosome 5, a recent report by Jian-Juo Yang et al. (107) describes a new mutation in the functional Cx43 gene in Taiwanese deaf patients. This review summarizes the most relevant and recent information about different mutations in connexin genes found in human patients, with emphasis on Cx26. The possible effects of the mutations on channel expression and function are discussed, in addition to their possible physiologic consequences for inner ear and skin physiology. Finally, we propose that connexin channels (gap junctions and hemichannels) may be targets for age-related hearing loss induced by oxidative damage.
Gap-Junction Networks in the Cochlea
The cochlea is the structure in the inner ear that contains the transduction machinery to sense the vibration transmitted from the middle ear after a sound stimulus. It is formed by three adjacent and paralleled tubular compartments: the scala media, the scala tympani, and the scala vestibule (Fig. 1). The principal cellular components of the cochlea are epithelial cells, fibrocytes, and receptor cells named hair cells, which are located in the wall of the tubular compartments. These compartments are filled with two types of solutions: (a) the perilymph, the ionic composition of which is identical to that of the extracellular solution, fills the scala tympani and scala vestibule; and (b) the endolymph, which possesses a high concentration of K+ (150 mM), fills the scala media (Fig. 1). Another important functional property of the cochlea is the high positive potential of the endolymph (approximately +80 mV), termed the endocochlear potential (Fig. 1). The endocochlear potential is probably a K+ equilibrium potential produced by the stria vascularis, a two-layered epithelium forming the wall of the scala media (Fig. 1) (101). The key elements are the potassium channels Kir 4.1 in the plasma membrane of intermediate cells and K+ transporters in the basal membrane of marginal cells of the stria vascularis (see review in ref. 101). The endocochlear potential is critical during the activation of hair cells. Hair cells in the basilar membrane of the scala media transform the mechanical stimuli produced by sounds into electrical signals transmitted to the brain. They are polarized cells, with the ciliated apical membrane exposed to the endolymph, and the cell body contacting the perilymph (or cortilymph). Sound causes vibration of the basilar membrane, inducing the deflection of hair cell cilia with subsequent opening of mechanosensitive nonselective cation channels (21) that allow endolymphatic K+ to enter into hair cells, resulting in their depolarization. The endocochlear potential directly contributes to the high sensitivity of hair cells to mechanical stimulation because it creates a large driving force for K+ influx of ∼160 mV, the difference between the resting potential of hair cells (−80 mV) and the endolymph potential (+80 mV). After activation of hair cells, K+ is released to the perilymph (Fig. 1). It has then been proposed that K+ can circulate from the perilymph to the endolymph through the “cochlear lateral wall” (reviewed in ref. 41). This is supported by observations that inhibition of K+ flow from the perilymphatic space inhibits endocochlear potential, and perilymphatic perfusion of K+-free solution rapidly and prominently suppresses the endocochlear potential (41). This recirculation pathway may include different cellular components of the cochlear wall. K+ moves first through supporting and epithelial cells in the basilar membrane, to fibrocytes in the spiral ligament (lateral wall connective tissue), and eventually to epithelial cells of the stria vascularis, from which the K+ is released to the endolymph. It has been hypothesized that the K+ circulation may be favored by the formation of two independent syncytia in the cochlea lateral wall: an epithelial gap-junction network (supporting cells and epithelial cells on the basilar membrane), and a connective tissue gap-junction network (fibrocytes of spiral ligament and epithelial cells of stria vascularis (Fig. 1) (51). It is proposed that these two networks mediate the K+ circulation from perilymph to endolymph (41, 100). However, the loss of Cx30 in the mouse ear does result in significant loss of the endocochlear potential generated by the stria vascularis (17) without changes in the K+ concentration and volume of the endolymph. Mutations in Cx26 (V84L) that do not affect ionic conductance, but selectively affect IP3 permeability, have been genetically linked with deafness (4). Thus, the exact role of gap-junction channels in the K+ circulation, and in the generation of the endocochlear potential, has yet to be directly demonstrated.
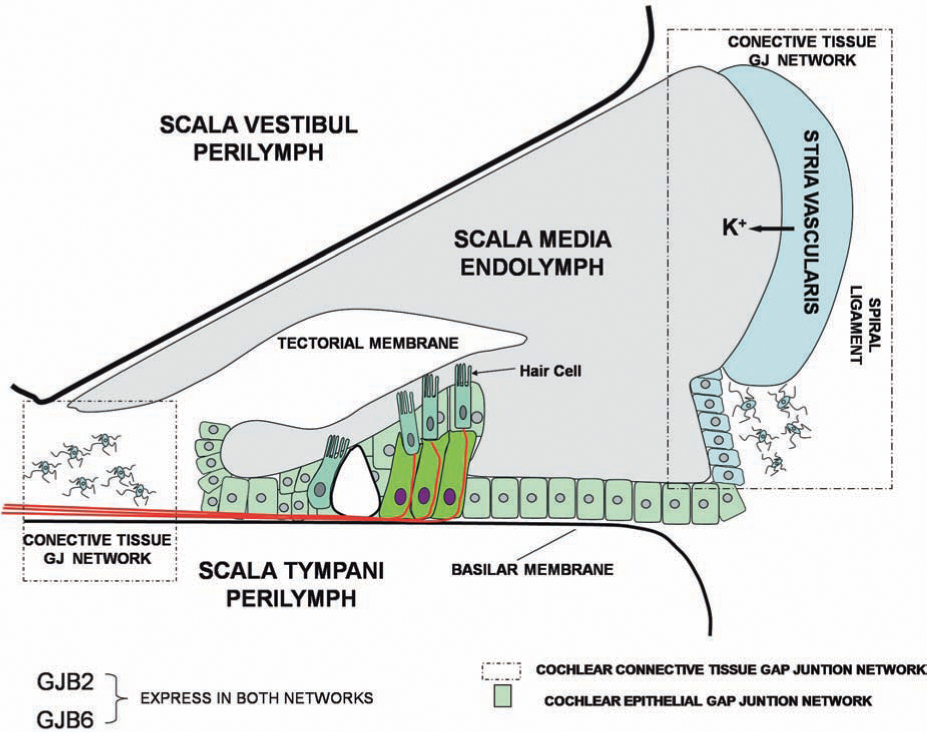
Connexin Expression in Inner Ear and Gap-Junction Function
For obvious reasons, most of the information with respect to connexin distribution in the inner ear has been obtained from animal models, mainly rodents, restricting our understanding of connexin physiology to the supposition that similar distributions would happen in humans. In rodents, several connexins (Cx26, Cx30, Cx31, Cx32, and Cx43) have been detected in the epithelia and connective tissue of the cochlea (15, 32, 36, 47, 55, 56, 60, 104, 112), supporting the formation of a gap-junction network in these tissues. In addition, gap-junction plaques (32 –35, 50, 51) and extensive intercellular coupling (4, 35, 64, 84 –86, 110, 111) were observed in both the cochlear connective tissue and the organ of Corti. Strong antibody co-labeling has been observed for Cx26 and Cx30 in these regions (1, 35, 55, 56, 68, 92), and immunogold studies have shown an equal distribution of these connexins within individual gap-junction plaques (33). Moreover, coimmunoprecipitation experiments under conditions that favor isolation of hemichannels suggested that Cx26 and Cx30 co-oligomerize in cochlear tissue (1, 33, 92). Together, these observations strongly support the existence of heteromeric Cx26/Cx30 gap-junction channels in the cochlea, although heterotypic forms could also exist. Although the GJChs are largely nonselective for ions, they do present selectivity to larger molecules like second-messengers nutrients and fluorescent tracers. The molecular permeability of gap junctions consisting of Cx26 and Cx30 was studied previously in vitro by using dyes of different charge and molecular weight, including Lucifer Yellow (LY) (charge, −2; molecular mass, 443 Da) and neurobiotin (NB) (charge, +1; molecular mass, 287 Da). In heterologous expression systems, gap junctions between cells expressing Cx26 alone are equally permeable to both LY and NB, whereas Cx30 gap junctions are far more permeable to NB than to LY. In addition, human Cx26 and Cx30, coexpressed in HeLa or human kidney cells, form functional heteromeric channels that are permeable to cationic and anionic tracers (92, 108). However, because homotypic Cx30 channels are more permeable to cationic tracers (108), it is possible that heteromeric Cx26/Cx30 channels may have different permeability properties than their respective homomeric counterparts, increasing the functional diversity of GJCh in the cochlea. In addition, cells expressing Cx26/Cx30 heteromeric channels showed faster Ca2+ intercellular signaling than did cells expressing homomeric Cx26 or Cx30 channels (92). These permeability differences may have critical functional consequences. Interestingly, in rat after the onset of hearing (P12–P13), LY transfer was not evident in any recordings from supporting cells immediately adjacent to hair cells (Deiters cells, inner border cells), suggesting a significant decrease in the numbers of Cx26 homomeric channels between these cells in hearing animals (45). The relative contributions of homotypic and heterotypic GJCh to the cochlea physiology remains to be resolved.
In addition to the well-known function of GJCh in intercellular communication, hemichannels present in the non-appositional plasma membrane may open under several physiologic and pathologic conditions (19, 20) to increase plasma-membrane permeability. Functional hemichannels are present in the mature organ of Corti and allow uptake of large anionic molecules under certain conditions (113), as well as the release of ATP.
The importance of GJCh in the cochlea has been highlighted because of its disruption in several forms of nonsyndromic and syndromic deafness (100). Notably, abnormality of the genes GJB2 and GJB6, encoding Cx26 and Cx30, respectively, are the most frequent genetic causes of deafness (for a complete updated list of mutations, see
Nonsyndromic Deafness
The most common form of genetic deafness, nonsyndromic hearing loss, has been predominantly associated with mutations in the GJB2 gene, encoding Cx26. To date, 90 mutations in the Cx26 gene have been associated with nonsyndromic deafness, which account for half of congenital cases of hearing impairments (a complete and updated list of mutations can be found in:
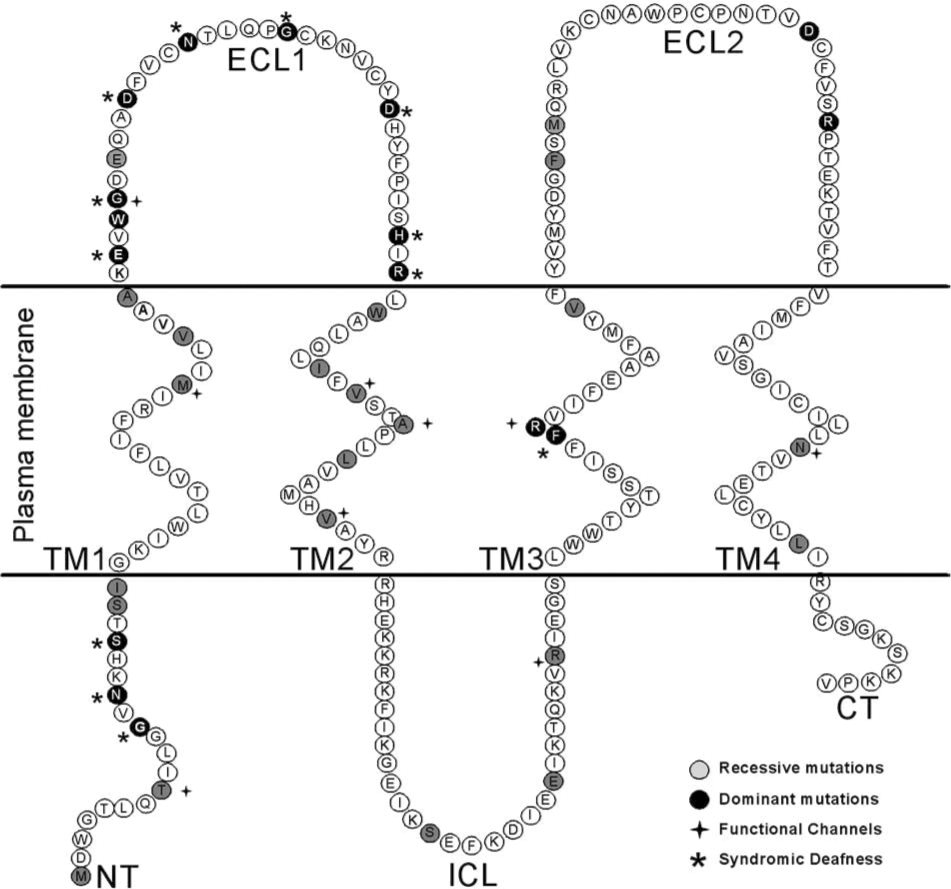
The list includes only mutations that have been study in some detail using exogenous expression systems. For a complete list of mutation see (
By far the most commonly found disease-associated mutations of Cx26 are deletions in two regions of GJB2 (35delG and 235delC (28, 52, 57, 99, 106). 35delG and 235delC are the most common Cx26 mutations found in caucasoid families and east Asian populations, respectively. These mutations result in a frameshift and premature termination of the protein. 35delG is caused by errors in replication of a string of Gs at this location, similar to the high-frequency mutation site in CFTR. Less-common point mutations of Cx26 associated with deafness have been characterized in recombinant expression systems (shown in Table 1). The majority of Cx26 mutations tested are loss-of-function mutations (Table 1), resulting from mistargeting or trafficking of the channels to the plasma membrane, or failure to form normal open channels (Fig. 3). Three loss-of-function mutations have been shown to allow targeting to the plasma membrane but do not support gap-junction plaque formation [G12V, W77R, and R184P (13, 24, 65, 96)]. Mutations S19T, W44C (W44S), E47K, L90P, R127H, and F161S are loss-of-function mutations that form gap-junction plaques in exogenous expression systems but fail to open intercellular channels (13, 24, 65, 68, 74, 91, 96, 98). Mutations V37I, A40G, G59V, I82M, S113R, delE120, V153I, M163V, D179N, R184Q, and L214P were shown to be nonfunctional in Xenopus oocytes, but difficulties with immunofluorescence in these cells did not allow assessment of whether these mutants reach the plasma membrane or are trapped in an intracellular compartment (13, 71, 74, 109).

Several deafness-causing mutations (T8M, M34T, V84L, A87S, V95M, R143W, N206S) form functional channels with similar conductance to wtCx26 channels, but more subtle differences in gating or permeability (4, 8, 13, 71, 90, 96, 98, 109). Interpretation of these differences is complicated by the observation that they can vary between expression systems. For example, whereas Wang and collaborators (98) found that mutant R143W, expressed in N2A cells, forms gap-junction channels with junctional conductance similar to that of the wild type, Mese and colleagues (71) found the opposite when the mutant was expressed in oocytes. A similar situation was found for mutant V95M, which is apparently not functional when expressed in HeLa cells (4) but forms functional channels between HEK293 cells (109). These discrepancies suggest that important regulatory elements for gap-junction function may be missing in some cellular systems, supporting the need to use the most physiologically relevant system to study the pathogenic mechanism of deafness-associated Cx26 mutations.
The ability of some pathogenic mutations to produce functional channels that conduct ionic current similarly to the wild type, suggests that, in some cases of deafness, potassium recirculation in the inner ear may be normal. Therefore, abnormalities in the exchange of other metabolites through the cochlear gap-junction network may also produce deafness (Fig. 3). For example, mutations V84L, A87S, and V95M produce functional channels that present reduced permeability to the second-messenger IP3 compared with wtCx26 gap-junction channels (4, 109). In organotypic culture of mouse cochlea, injection of IP3 into one supporting cell elicited Ca2+ waves that propagate to the neighboring cells in seconds. The Ca2+ wave is not generated by Ca2+-induced Ca2+ release, but is dependent on the diffusion of IP3 through GJCh (4, 109).
The mechanism by which loss of IP3 intercellular transfer leads to deafness is a matter for speculation. One possibility is that it may indirectly affect K+ spatial buffering through cochlear supporting cells. For example, release of Ca2+ from intracellular stores may gate Ca2+-activated ion currents, especially Cl− currents, increasing anion efflux into the endolymph (Fig. 4). Such a movement of Cl− is likely to promote an equivalent efflux of K+ through membrane channels, preserving the electroneutrality of the cytoplasm and the endolymphatic fluid. This hypothetic mechanism might contribute to the homeostasis of K+, facilitating its re-absorption into the endolymph (54). In summary, loss of gap-junction permeability to IP3 may reduce Ca2+ signalling between supporting cells, thus affecting the KCl balance of cochlear fluids, leading to excitotoxic death of the hair cells. It is important to note that this is a hypothetic model that requires testing, and that the selective loss of gap-junction coupling could affect other metabolites important for cellular viability.

Syndromic Deafness
Connexin-related deafness is sometimes associated with congenital skin disorders, such us Vohwinkel syndrome, keratitis–ichthyosis–deafness syndrome (KID) and palmoplantar keratodermas (Fig. 2 and Table 2). In these syndromes, hearing loss is associated with abnormal epidermal keratinization. As in the sensory epithelium in the inner ear, abundant gap junctions are found in the epidermis, with multiple and overlapping expression of several connexins (Cx43, Cx31, Cx26, Cx30) (10, 14, 29, 31, 63, 103). Thus, to cause skin disease, it has been hypothesized that the mutated Cx26 protein affects the normal gap-junction function of other connexins through a dominant-negative effect over wild-type coexpressed connexins. This hypothesis is supported by: (a) all syndromic mutations are inherited in a dominant way; and (b) in exogenous expression systems, some of these mutants have been shown to act as dominant-negative inhibitors of coexpressed wild-type connexins 26, 30, or 43 (Table 2).
The list includes only mutations that have been study in some detail using exogenous expression systems. For a complete list of mutation see (
Vohwinkel syndrome is characterized by relatively mild sensorineural deafness, hyperkeratosis of the soles, palms, and knuckles, with constriction rings on the digits, sometimes leading to autoamputation. Affected individuals in all Vohwinkel syndrome families analyzed to date are carriers of the D66H mutation (62). In addition, mutation H73R causes Vohwinkel-like syndrome (25). Both mutations are located in extracellular loop 1 (ECL1; Fig. 2), a critical domain for assembly and function of GJCh. Consistently over several experimental systems, these mutations act as dominant-negative effectors of wild type Cx26 or Cx30 gap junction, but not hemichannels (35, 68, 95). Transgenic mice designed to express D66H specifically in the suprabasal epidermal keratinocytes [the cells where Cx26 is upregulated after minor skin trauma (61)] have skin abnormalities similar to those observed in true Vohwinkel syndrome patients (3). In addition, in these transgenic mice, Cx26 accumulates in the cytoplasm of suprabasal keratinocytes, like the observed staining for Cx26 in Vohwinkel patients' skin (3). Interestingly, abundant TUNEL staining in the affected epidermis indicates that either excessive apoptosis, or premature terminal differentiation, contribute to the disease phenotype. In addition, high levels of cell-proliferation markers were observed in the underlying basal epidermis (where the transgene is not expressed), indicating enhanced proliferation of adjacent tissue (3). The authors of this work hypothesized that the mutant protein in the suprabasal keratinocytes disrupts the epidermal gap-junction network, leading to premature terminal differentiation, with retention of cohesion between corneocytes in the stratum corneum. Premature keratinocyte death might produce compensatory basal cell proliferation, leading to massive thickening of the stratum corneum (3).
KID syndrome is a rare ectodermal dysplasia characterized by vascularizing keratitis, profound sensorineural hearing loss, and progressive erythrokeratoderma, a clinical triad that indicates a failure in development and differentiation of multiple stratifying epithelia. All Cx26 dominant mutations linked to KID syndromic deafness are located in the N terminal (NT) or in the ECL1 of the protein (Fig. 2). Previous studies suggest that the NT region of connexins is involved in the voltage gating of gap-junction channels and in the recognition between different connexin subunits during oligomerization. Cx26 mutations, G12R, N14Y, and S17F, produced gap-junction plaques, indicating that they do not affect trafficking and gap-junction formation (2, 79). However, these mutations either do not form open channels or significantly affect channel permeability. Previous studies suggest that charged amino acid residues in the amino terminus of connexins form part of the transjunctional voltage sensor of GJCh and play a fundamental role in ion selectivity (76, 77). Results from studies of the voltage dependence of NT mutants predict that residues 1–10 lie within the channel pore. The NT is proposed to contribute to the channel vestibule by folding back toward the pore mouth via the conserved G12 residue (76). In support of this interpretation, the three-dimensional structure of M34A shows that the channel vestibule is blocked by a physical structure that looks like a plug (73). In this work, it is proposed that the plug most likely corresponds to the NT, because the pore region is an ideal location to detect the transjunctional voltage field (73). More recently, these authors showed that partial deletion of the NT results in a marked decrease in this plug mass (72a).
The dynamic properties of the synthetic NT peptide containing the KID mutation N14Y, as revealed by two-dimensional nuclear magnetic resonance and circular dichroism, suggest that this mutation induces profound changes in the local structural flexibility of the NT (2). Therefore, a possibility is that some mutations in the NT domain may produce channels that open with low probability because the plug is stabilized within the pore vestibule. Other Cx26 mutations associated with KID syndrome are clustered in the first half of ECL1 (Fig. 2). Belonging to this group are mutations A40V, G45E, and D50N or D50Y, which have been studied in exogenous expression systems (29, 38, 79, 91). In two different functional expression systems [i.e., Xenopus oocytes and HEK (human embryonic kidney)-293 cells], G45E led to increased hemichannel activity and cell lysis (38, 91) (Fig. 3). Similar results were observed in oocytes expressing A40V (38). In either case, this severe phenotype was rescued by increasing extracellular Ca2+, which closes the hemichannels. These studies support the proposal that treatment strategies should include the development of pharmacologic agents that modulate extracellular Ca2+ concentration, or specifically block Cx26 hemichannels in the epidermis and cochlea.
The other KID mutation located in the ECL1, D50N, did not produce functional channels when expressed in NEB1 cells (an immortalized keratinocyte cell line) but displayed impaired trafficking to the plasma membrane (29). However, plasma-membrane localization of Cx26 is observed in the sweat gland of a KID patient heterozygous for the D50N mutation (29).
These differences between in vivo and in vitro studies suggest that connexin–connexin interactions may change mutant protein behavior with respect to localization and possibly functionality. In support of this idea, D50N is found at gap-junction plaques in cells co-transfected with fluorescently tagged D50N and wtCx26 or wtCx30 (29). FRET analysis demonstrates a proximity between mutant and wt subunits consistent with co-assembly into heteromeric channels that could explain the expression of the mutant at the cell surface.
Finally, the missense mutations G59A and R75W (or R75Q) cause autosomal-dominant, profound hearing loss that has been associated with a mild skin disorder, palmoplantar keratoderma. Expression of Cx26 R75W in transgenic mice causes deafness, which is associated with death of both supporting cells and hair cells (53). Cx26 mutants with different amino acid substitutions of residue R75, including R75W, are unable to form functional gap-junction channels between oocyte pairs (27). However, they do insert normally into the plasma membrane (27) and form functional hemichannels (27), albeit with slightly modified gating. This mutant has a dominant-negative effect on gap-junctional communication mediated by wtCx26 (23, 27, 35, 53, 68, 80), consistent with the dominant nature of deafness cause by the R75W mutation (80). The possible mechanism of gap-junctional communication dysfunction for mutant R75W could be a defective docking, in which interaction of apposing hemichannels does not result in channel opening. However, some form of stable interaction is likely to occur, as at the immunofluorescent and ultrastructural level, R75W gap-junction plaques have been observed in different expression systems (72, 94). Connexons purified from the Sf9 insect cells expressing R75W have very low stability in the detergent dodecyl maltoside compared with those formed by wtCx26, suggesting that R75 is important for intersubunit interactions (72). Because functional hemichannels can be detected in cells transfected with wtCx26, we can speculate that the supposed connexon instability has no biologic relevance, but these studies in exogenous systems are hard to quantitate. The other palmoplantar keratoderma mutation, G59A, is also a loss-of-function mutation (35, 68, 95). Some discrepancies with respect to the effects of this mutation on Cx26 trafficking have been reported, with both complete intracellular localization (35, 68), and effective assembly into gap-junction plaques (95) being reported. Co-expression experiments showed that the plasma-membrane localization of Cx26 G59A is rescued by wtCx26 or wtCx30, but that this mutant has a dominant-negative effect on wild-type connexins (35, 68, 95). The mechanism by which this mutant affects gap-junction function is unknown.
Other Connexin Mutations Involved in Deafness
Mutations in other connexins have been detected in deaf patients. Deletion in GJB6 (Cx30), “del(GJB6-D13S1830),” is the second most frequent mutation causing prelingual hearing impairment in Spain and is also common in other European countries and in Israel (26). Dominant mutations in the Cx30 gene have been identified, T5M (40) and 63delG (9). Normal trafficking and gap-junction plaques were observed in HeLa cells or keratinocyte cells transfected with TM5/EGFP, but dye-coupling experiments fail to showed functionality (18), suggesting that T5M is a loss-of-function mutation. Interestingly, other Cx30 mutations associated with skin disease (G11R, V37E, A88V) showed impaired trafficking of the protein to the plasma membrane (18).
Mutations in the gene for human Cx31 (GJB3) are associated with disorders of the skin and auditory system. Mutations in Cx31 gene (GJB3) produce dominant or recessive inherited deafness (58, 105). Recessive mutations in Cx31 (141delI, I141V) (58) are located in the third transmembrane domain, whereas dominant mutations (R180X, E183K) (105) are clustered in the ECL2. The pathogenic mechanism of these mutants is unknown. However, mutation 66delD, associated with a dominant syndrome of hearing loss and peripheral neuropathy, has defective trafficking to the plasma membrane, and the functionality assessed with dye transfer is impaired compared with wtCx31 (30).
Gap-Junction Channel and Environmental and Age-Related Hearing Loss
Although age-related hearing loss is polygenic and multifactorial in etiology, a consensus indicates that the cochlea is the main affected auditory organ (59). The low-frequency pattern of hearing loss is interpreted as possibly representing a disorder of the stria vascularis, whereas the high-frequency loss is probably due to changes in hair cell function (59). The effects of aging on connexin expression and gap-junction function in the cochlea have yet to be investigated. However, in other systems, aging induces changes in connexin expression. For example, in the aging heart, Cx43 is drastically reduced in the sinoatrial node, contributing to decreases in the conduction velocity observed in older hearts (46). Similar studies should be done in the cochlea to determine the effect of aging on Cx26 or Cx30 expression and its potential phenotypic consequences.
Noise is the most-studied environmental factor causing hearing loss, with long-term noise exposure leading to loss of the outer hair cells and ultimately loss of the inner hair cells (59). In rats after acoustic trauma induced by noise exposure (54.2 dB), Cx26 expression was upregulated in the cochlea lateral wall (43), suggesting that noise may regulate connexin expression. The cochlea is also a very metabolically active tissue that produces significant levels of reactive oxygen species (ROS), which increase to detrimental levels with reduced production or function of the endogenous enzymes that protect the cell from ROS damage. Loss of antioxidant defense or increase in ROS production appears to be consistently associated with the aging process in many tissues and has also been associated with changes in gap-junction expression or function (37, 67, 87). Loss of antioxidant enzymes has also been associated with noise-induced damage in the ear (44). Two classes of antioxidant enzymes are active in the cochlea: enzymes involved in glutathione (GSH) metabolism (glutathione S-transferase, GST; glutathione peroxidase, GPX1; and glutathione reductase, GSR) and enzymes involved in the breakdown of superoxide anions and hydrogen peroxide (e.g., catalase, CAT; and Cu/Zn superoxide dismutase, SOD1) (44, 59, 69, 70). Studies of knockout models of Gpx1 and Sod1 have shown that deletion in these two antioxidant genes can lead to both age-related and noise-induced hearing loss (59, 70). Oxidative stress affects connexin expression and GJCh function in many systems. For example, in astrocytes, reoxygenation after hypoxia disrupts gap-junctional communications (67). In addition, treatments with antioxidant agents modify connexin expression. Cx26 was induced in a transgenic mouse line that expresses the gene CrtB, encoding phytoene synthase, which could produce the potent antioxidant phytoene (a type of carotenoid) endogenously (87). This was consistent with previous findings that carotenoids enhance gap-junctional communications by inducing the expression of connexins genes and resistance to oxidative stress. Taurine in rat hepatocytes prevents the reduction in Cx32 induced by oxidative stress (37), thus protecting against H2O2-induced reduction in gap-junctional communication in the liver. Melatonin, the potent free radical scavenger hormone, protects astrocyte gap junctions from oxidative damage induced by reoxygenation (67). Thus, in addition to its role in familial deafness, Cx26 may be affected by oxidative stress in the cochlea, contributing to age-related hearing loss or damage cause by sound (Fig. 4). The latter hypothesis should be tested experimentally. However, we can speculate that treatment with antioxidants (like carotens, taurine, or melatonin) may protect gap-junction function from oxidative damage induced by age or sound, contributing to protection from hearing loss.
Conclusions
In summary, Cx26 mutations produce deafness and skin disease through multiple pathogenic mechanisms. In terms of disease, deafness-associated Cx26 mutants may be broadly classified into two categories: (a) mutations that produce nonsyndromic deafness, and (b) mutations that produce syndromic deafness, in which deafness is associated with skin disorders. Nonsyndromic mutations are very diverse and located in most Cx26 protein domains. They are generally recessive mutations, but a few dominant mutations cluster mainly in the third transmembrane domain and second extracellular loop [important protein domains for constitution of the channel, as defined by mutagenic mapping of the pore lining (89) (Fig. 2)]. All Cx26 syndromic mutations are dominant and are located in the NT domain or in the first extracellular loop (critical domains for voltage sensing, channel gating, and hemichannel permeability) (Fig. 2). In terms of GJCh formation and function, deafness-associated Cx26 mutations could be classified into four types: (a) mutations that affect hemichannel trafficking to the plasma membrane or GJCh assembly; (b) mutations that produce gap junctions, but the channels are non-functional; (c) mutations that produce functional GJCh that have aberrant gating or permeability properties, like reduced IP3 permeability; and (d) mutations that produce functional hemichannels at the plasma membrane that may open under physiologic conditions, affecting ionic balance or the homeostasis of vital metabolites that diminish cellular viability (Fig. 3).
Most functional mutations are located in the transmembrane domains, especially clustering in the second trans-membrane domain, which has also been implicated in lining the pore (89) (Fig. 2). However, nonfunctional mutations are located in any part of the protein, suggesting that the structure of Cx26 gap-junction channels is very sensitive to minor changes in the amino acid sequence, independent of the protein domain where they are present. Deafness associated with Cx26 mutations can be inherited in recessive or dominant forms, depending on whether the mutant connexin can act as dominant-negative subunits for the function of coexpressed wild-type connexins, like Cxs 26, 30, or 43. The mechanism by which some dominant mutations are syndromic and others are nonsyndromic is a matter for speculation. However, it is reasonable to think that constitution of aberrant heteromeric channels is behind the mechanism. This serves to emphasize the fact that, although exogenous expression of connexins in model systems has focused on homomeric connexon channels, in situ, heteromeric channels likely exist or even predominate. Each unique heteromeric connexin assembly may have its own unique properties and physiologic role.
Although it is clear that gap-junction intercellular communication is vital for cochlea and skin physiology, its exact function is unknown. The hypothesis that gap junctions participate in K+ recirculation in the cochlea is both logical and consistent with the physiology of the ear, but remains to be directly tested experimentally. Gap junctions may also be important for other reasons. The permeability properties of some pathogenic Cx26 mutations indicates that circulation of metabolites, like IP3, is critical for cochlea-supporting cell function or survival and that K+ flow cannot be the only function of Cx26 (Fig. 4). Finally, connexin genes are potential candidates for susceptibility to age-related hearing loss or noise-induced hearing damage.
Footnotes
Acknowledgments
This work was supported by Anillo de Ciencia y Tecnología, ACT-46 for ADM and NIH-NCI (CA48049) and NIH-NIGMS (GM55437) for BJN.
Abbreviations
CT, carboxyl-terminus domain; Cx, connexin protein; ECL1, ECL2, extracellular loop 1 and 2, respectively; GJB2 and GJB6, Cx26 and Cx30 genes, respectively; GJ, gap junction; GJCh, gap-junction channels; GPX1 glutathione peroxidase 1; GST, glutathione S-transferase; ICL, intracellular loop; KID, keratitis–ichthyosis–deafness syndrome; NT, protein amino-terminus domain; SOD, superoxide dismutase; TM, transmembrane domain; wtCx, wild-type connexin.