
Abstract
Research on oxidative stress focused primarily on determining how reactive oxygen species (ROS) damage cells by indiscriminate reactions with their macromolecular machinery, particularly lipids, proteins, and DNA. However, many chronic diseases are not always a consequence of tissue necrosis, DNA, or protein damage, but rather to altered gene expression. Gene expression is highly regulated by the coordination of cell signaling systems that maintain tissue homeostasis. Therefore, much research has shifted to the understanding of how ROS reversibly control gene expression through cell signaling mechanisms. However, most research has focused on redox regulation of signal transduction within a cell, but we introduce a more comprehensive-systems biology approach to understanding oxidative signaling that includes gap junctional intercellular communication, which plays a role in coordinating gene expression between cells of a tissue needed to maintain tissue homeostasis. We propose a hypothesis that gap junctions are critical in modulating the levels of second messengers, such as low molecular weight reactive oxygen, needed in the transduction of an external signal to the nucleus in the expression of genes. Thus, any comprehensive-systems biology approach to understanding oxidative signaling must also include gap junctions, in which aberrant gap junctions have been clearly implicated in many human diseases. Antioxid. Redox Signal. 11, 297–307.
Oxidative Damage vs. Oxidative Signaling
Oxygen was first implicated in cancer as far back as the 1920s by Otto Warburg (111). Initially his theory of altered oxidative metabolism fell mostly on deaf ears. However, recently there has been a renewed interest in how cancer cells shift their energy production from oxidative phosphorylation to anaerobic glycolysis, the “Warburg Effect,” that is now considered a fundamental property of cancer cells and not just a consequence of malignant cell transformation (3, 54, 116). In contrast to this role of low oxygen tensions in cancer, the discovery of superoxide dismutase in 1968 by McCord and Fridovich (70) led to an explosion of research on the role of reactive oxygen, a product of high oxygen tensions, in the pathologies of biological organisms, and has been specifically connected with not only cancer but also many other human diseases (1, 41). For many years, research in oxidative stress at high oxygen tensions focused primarily on determining the mechanisms by which ROS damage cells by random reactions with the macromolecular machinery of a cell, particularly lipids, proteins, and DNA.
Over these last 4 decades, extensive research has determined many mechanisms on how ROS react with lipids, leading to the peroxidation of biological membranes resulting in necrotic lesions (31), how ROS react with the nucleotides of DNA leading to genetic instability and mutations (9, 31), and how ROS react with proteins leading to metabolic imbalances. Yet, many chronic diseases affiliated with oxidative stress, such as cancer and cardiovascular diseases, are not always a consequence of tissue necrosis, DNA damage, and genetic instability and mutations, or protein damage, but rather to altered gene expression through epigenetic mechanisms (98, 99, 104). Organic peroxides, for example, act as tumor promoters and not as initiators (34, 53, 73, 86), indicating that these oxidants are not mutagens, but rather epigenetic effectors. Hydrogen peroxide has also been confirmed to be a promoter, but not an initiator using two stage in vivo carcinogenesis model systems and transformation in vitro systems (53, 73, 74, 77). These results on the carcinogenicity of peroxides were only several of many experiments indicating that ROS contributed to chronic diseases at nongenotoxic, nondamaging levels. Consequently, in the past two decades, considerable research has shifted to the understanding of how ROS reversibly control the expression of genes at noncytotoxic doses (1, 28, 49, 89), with at least 127 genes and signal transducing proteins reported to be sensitive to reductive and oxidative (redox) states in the cell (1). This shift in thinking should start to shape both the design and the interpretation of experiments on the role of oxidative stress in human disease.
Oxidative Signaling Mechanisms in Mitogenesis and Cancer
Although many intracellular signaling pathways are known to be redox-sensitive, the mitogen-activated protein kinase (MAPK) and nuclear factor-kB (NF-kB) signal transduction pathways have been extensively studied (1, 30, 40, 89). These pathways, either directly or indirectly, are widely involved in many of the cells redox responses (1). Cancer is a disease that involves aberrant communication between and within cells (22, 99), and MAPK pathways are key signaling networks involved in the regulation of normal cell proliferation, survival, and differentiation (22, 79), thus redox regulation of MAPK pathways is one significant and probable link of oxidative stress and cancer. Numerous articles have been published that clearly link oxidative stress with hyperproliferation of cells within a tissue and has been clearly a focus of cancer research, thus understanding the mechanism of redox regulation of mitogenic signaling pathways such as MAPKs will offer more specific strategies of preventions and cures.
MAPK is not only activated by ROS (38, 71) but actually require the synthesis of H2O2 (14, 42, 52, 87, 88, 105, 106). Sundaresan et al. (88) were the first to demonstrate, using a vascular smooth muscle cell system, that a transient burst of H2O2 by an extracellular ligand, namely platelet derived growth factor (PDGF), is essential in the activation of extracellular receptor kinase-MAPK (ERK-MAPK). They demonstrated that the transfection of catalase, which dismutates H2O2, into these cells, prevented the activation of ERK-MAPK by PDGF (Fig. 1A and B). They also demonstrated that downstream events of MAPK activation, such as proliferation and increased cell motility, were also inhibited by catalase (Fig. 1E and F). This was one of many studies that demonstrated endogenous growth factors (extracellular ligands) generate ROS, which are then required downstream in intracellular signaling to successfully transmit their signals to the nucleus (58, 90). This seminal paper was one of the first to radically alter our traditional view of oxidative stress from an exclusive concept that reactive oxygen was always detrimental, via oxidative damage of cellular components, to one that reactive oxygen is also part of normal signaling mechanisms that can alter gene expression in an undamaging way. Thus, the pathologies of reactive oxygen can be linked to damage or cell signaling or both, depending on concentrations that range from being too low, to moderate or extremely high conditions.

Gap Junctions, a Major Link in Systems Control of Mitogenesis and Cancer
The successful transmission of an extracellular signal from the membrane to the nucleus via intracellular signal transduction pathways in solid tissue cell types is also dependent upon intercellular signals through gap junctions (98, 99, 104). Gap junctions allow the passive transmission of intercellular signals within a tissue that ultimately modulate signal transduction (99). Gap junctions consist of two hexameric channels, termed connexons, docked to each other between adjacent cells. The family of proteins that form the hexameric connexon is termed connexins. Tissue homeostasis is maintained by opened gap junctional channels and, although the transient closure of channels is a normal response to growth factors, chronic inhibition of gap junctional intercellular communication (GJIC) by oncogenes, tumor promoters, or the chronic exposure to growth factors and cytokines via compensatory hyperplasia and chronic inflammation can lead to increases in proliferation and decreases in apoptosis and differentiation, leading to pathologies such as cancer and cardiovascular diseases (99).
The evidence for the role of GJIC in cancer has been extensively reviewed (25, 36, 50, 51, 55, 64, 72, 75, 81, 92) and the following are highlights of these papers: (a) exogenous and endogenous tumor promoters reversibly inhibit GJIC; (b) structure–activity relationship models showed a high concordance between inhibition of GJIC and the tumorigenicity of chemicals; (c) oncogenes inhibit GJIC; (d) tumor suppressor genes upregulate GJIC; (e) anti-tumor promoters upregulate GJIC; (f) restoration of GJIC in tumorigenic cells via transfection with gap junction genes results in normal growth and morphology of cells; (g) antisense gap junction genes transfected into cancer cells augment foci formation; (h) connexin 32 (Cx32) knockout mice administered with a single dose of an initiator, diethylnitrosamine, had 3.3 to 12.8 times increase of preneoplastic foci as compared to the wild type, while mice not treated with diethylnitrosamine did not exhibit preneoplastic lesions, indicating that the deletion of Cx32 promoted the carcinogenic effect of the initiator; (i) Cx32 knockout mice also exhibited increased levels of radiation and chemical-induced lung and liver tumor formation; and (j) the formation of heterotypic gap junctions between metastatic cells and cells of the target tissue leads to preferential metastasis. Intercellular communication through gap junction channels is a key component of the systems control of cellular events within a tissue that allows for the coordination of intracellular control of the metabolism and expression of genes between contiguous syncytium of cells into an organized hierarchal multicellular system. Interruption of this intercellular communication system allows for a change in the epigenome of the cells that permits aberrant cell growth and organization into tumorigenic tissues independent of the host organism's tissue.
Gap Junctions, Key Cell Structures in the Development of Multicellular Organisms and Tissue Homeostasis
Theodosius Dobzhansky wisely stated, “Nothing in biology makes sense except in the light of evolution” (23). In that context, it might be questioned as to why the gap junction and its associated family of highly conserved genes should be singled out as being more important in the evolution of the metazoan over the mitochondria, spindle fibers, nuclear membrane, tight junction, etc. Clearly, gap junctions are only one of the critical and vital structures/functions of a metazoan. What sets it apart is that it helps create and integrate extracellular phenotypes and functions that the individual cell does not possess, in particular, single cell organisms such as a bacterium. The evolutionary transition from the single cell organism to the first multicellular metazoan required the introduction of several new phenotypes, namely, growth control within this society of grouped cells via “contact-inhibition”; induction of specialized cells or “differentiation” through patterned expression of a large set of genes, not all of which can be expressed at the same time; and controlled cell suicide or apoptosis to allow cells that are useful at one stage of development to be replaced by a new group of cells needed at the next phase of development. The structure/function of specific gap junctions that allows new phenotypes, such as growth control and multiple types of gene patterns to be expressed in cells, all containing the same genome, to allow groups of contiguous, but not gap junction-coupled, cells to differentiate independently of each other, and to allow different phenotypes/functions to emerge when a collection of normal cells aggregates.
In the evolutionary-derived multicellular cellar organisms, three major cell types appeared in the various tissues and organs, namely, the stem cells (totipotent, pluripotent, multipotent, bipolar), the progenitor or transit cells with a finite life span, and the terminally differentiated cells. Homeostatic regulation of cell proliferation, differentiation, and apoptosis of these three cell types was facilitated via extra-(hormones, cytokines, growth factors), intra- (different intracellular signaling mechanisms, such as MAPK; NF-κB), and gap-junctional intercellular communication mechanisms needed to be delicately integrated, with the demonstration that stem cells seem to lack GJIC to restrict differentiation but still maintain growth control via negative secreted growth factors (96, 102). In addition, with experimental evidence linking gap junctions to growth control in progenitor cells (92, 94, 102), and apoptosis in solid tissues (112), the demonstration that cancer cells, characterized by being a disease of homeostasis, lacks growth control, can not terminally differentiate or apoptose properly, seems to support the role of gap junction genes as the “biological Rosetta Stone” that allows one to view them as allowing a systems mechanism to create higher order structures and functions. Without gap junctions, the higher order phenotypes and functions existing during different stages of embryonic/fetal/neonatal, adolescent, adult, and geriatric development could not exist. Endogenous and exogenous chemicals and genetic factors, that influence gap junction function, can cause a wide range of abnormal development and functional processes in many diseases. This illustrates that these gap junction functions are as vital to normal development and function as any other gene. Alteration of the many other non-gap junction genes, that can influence either survival or disease state, is probably affecting gap junctional intercellular communication indirectly.
Last, the connection of some new concepts, namely the role of the quality and quantity of adult stem cells (93) and of the expression and function of gap junctions, must be integrated into any “systems” approach to understand the higher order function of the epigenome that regulates the genomic information. After all, the genomic information is but a “blueprint.” It is the delicate and systematic differential expression of that genetic information that leads to normal development and function. This was beautifully stated by C. Markert (69): “Cells interact and communicate during embryonic development and through inductive stimuli mutually direct the divergent courses of their differentiation. Very little cell differentiation is truly autonomous in vertebrate organisms. The myriad cell phenotypes present in mammals, for example, must reflect a corresponding complexity in the timing, nature, and amount of inductive interactions. Whatever the nature of inductive stimuli may be, they emerge as a consequence of specific sequential interactions of cells during embryonic development. The first embryonic cells, blastomeres, of mice and other mammals are all totipotent. During cleavage and early morphogenesis, these cells come to occupy different positions in the three-dimensional embryo. Some cells are on the outside, some inside. The different environments of these cells cause the cells to express different patterns of metabolism in accordance with their own developing programs of gene function. These patterns of metabolism create new chemical environments for nearby cells and these changed environments induce yet new programs of gene function in responding cells. Thus, a progressive series of reciprocal interactions is established between the cellular environment and the genome of each cell. These interactions drive the cell along a specific path of differentiation until a stable equilibrium is reached in the adult. Thereafter, little change occurs in the specialized cells, and they become remarkably refractory to changes in the environment. They seem stably locked into the terminal patterns of gene function characteristic of adult cells. The genome seems no longer responsible to the signals that were effective earlier in development. Of course, changes can occur in adult cells that lead to renewed cell proliferation and altered differentiation as seen in neoplasms, both benign and malignant, but such changes are very rare indeed when one considers the number of cells potentially available for neoplastic transformation. Possibly mutations in regulatory DNA of dividing adult cells can occasionally lead to new and highly effective programs gene function that we recognize as neoplastic or malignant. However, most genetic changes in adult cells can probably lead to cell death since random changes in patterns of gene activity are not likely to be beneficial.”
Disruption of Gap Junction, a Central Function in Diseases
As noted in the “Oxidative signaling mechanisms in mitogenesis and cancer” section, the dysregulation of gap junctions is a common phenotype of cancer cells. However, altered gap junction functions are also linked to many other human diseases. Chronic exposure to epigenetic toxicants have been implicated in teratogenesis (95), reproductive dysfunction (32, 59, 114), altered muscle contractions in the heart and uterus (16, 17, 19 –21, 46), and implicated in neurotoxic effects (97). More recently, the development of gap junction knockout mouse model systems and the identification of mutated gap junction genes in human diseases have greatly expanded our view of the role gap junctions play in various pathological conditions.
The first human disease specifically linked to a mutation of a gap junction gene, connexin32 (Cx32), was the X-linked form of Charcot–Marie–Tooth syndrome, which is a neuropathy that results from demyelination and axonal degeneration of peripheral nerves (7). The mutations of Cx32 have been implicated in the interruption of normal diffusion of metabolites between the Schwann cell body and its distal processes (27, 84). Since this discovery, many other mutations of various connexins have been linked to human diseases as reviewed by Kelsell et al. (47), such as hearing loss associated with mutations in Cx32, Cx26, Cx31, and Cx30; dominant epidermal diseases with Cx26; dominant skin diseases (erthrokeratoderma variablis) with Cx 31, and Cx 30.3; Clouston's hidrotic ectodermal dysplasia with Cx30, association with visceroatrial heterotaxy with Cx43; and dominant zonular pulverant cataract with Cx46 and Cx50.
Various knockout mouse model systems have produced surprising phenotypic results with some discrepancies between mouse knockout model systems and the human genetic connexin disorders (47). However, many of these studies indicated there is tissue-specific compensation between multiple connexins. The fact that not all connexin knockout model systems result in embryonic lethality and that some tissues that express a predominant form of connexin exhibited no tissue abnormalities while others did, lend credence to multiple connexins compensating for each other. As summarized by Kelsell et al. (47), knockouts for Cx32 resulted in hepatic abnormalities and susceptibility to hepatic tumors and mild peripheral neuropathies; for Cx 26 resulted in embryonic lethality at day 11; for Cx31 resulted in transient placental dysmorphogenesis; for Cx43 resulted in abnormal cardiac development and subsequent embryonic death; for Cx46 resulted in cataract formation; and for Cx 50 resulted in cataract formation and microphthalmia.
Oxidative Control of Gap Junctions
Considering that gap junctions play a critical role in maintaining the balance between proliferation, differentiation, and apoptosis of cells within a tissue, and many of the signal transduction pathways involved in cell proliferation is redox regulated, it is not surprising that gap junctions can also be regulated through redox mechanisms (44, 60 –63, 67, 80, 103, 109). Some of the earliest evidence of oxidative stress in gap junction function are (a) oxidative-dependent hepatotoxic levels of carbon tetrachloride reversibly block GJIC between rat hepatocytes (81a); (b) antioxidants prevent tumor promoter-induced inhibition of GJIC in mouse hepatocytes (80a); (c) paraquat-generated ROS inhibit GJIC in mouse hepatocytes. More recently, we reported that hydrogen peroxide inhibited GJIC in a dose-dependent fashion (103). Free radical scavengers such as propylgallate and Trolox®, which do not affect H2O2 but scavenge its downstream products such as hydroxyl, alkoxyl, or peroxyl radicals, do not reverse the inhibitory effect of H2O2 (101, 103), which implicates that the redox regulation of gap junctions is by H2O2 and not its metabolites. Inhibition of GJIC by H2O2 is reversible (103) indicating that redox regulation of GJIC is not a consequent of irreversible protein damage.
Glutathione (GSH) in its reduced form is the major reductant of H2O2 in mammalian cells, and the depletion of this antioxidant commonly results in greater oxidative damage of macromolecules. This two-electron reduction of H2O2 by GSH to H2O is catalyzed by glutathione peroxidase, and clearly serves as a protective role against peroxide-dependent oxidative injury. A previous report determined the effect of depleting GSH on H2O2-dependent regulation of GJIC in a rat liver epithelial cell line (103). GSH depletion in this study was achieved by either the conjugation of GSH with diethylmaleate (DEM) or by inhibiting γ-glutamylcysteine synthetase, which catalyzes the rate-limiting step of the biosynthesis of GSH, with buthionine sulfoximine (BSO). When cells were treated with BSO for 24 h and DEM for 1 h, GSH level decreased by 72% and 95%, respectively (Fig. 2A and B, (103)). The depletion of GSH alone did not alter GJIC [Fig. 2A, (103)], but the addition of H2O2 to these GSH-depleted cells completely reversed the GJIC-inhibitory properties of H2O2, [Fig. 2B, (103)]. When cells were treated with BSO for only 1 h, no depletion of GSH was observed, and the addition of H2O2 to these GSH-sufficient cells containing BSO resulted in the inhibition of GJIC (Fig. 2B, (103)), which indicates that GSH depletion, and not the BSO, prevented H2O2 from inhibiting GJIC. This paradoxical reversal of the biological effect of H2O2 in the inhibition of GJIC contrasts with the traditional paradigm of oxidative damage where the removal of the primary antioxidative defense system results in increased damage.
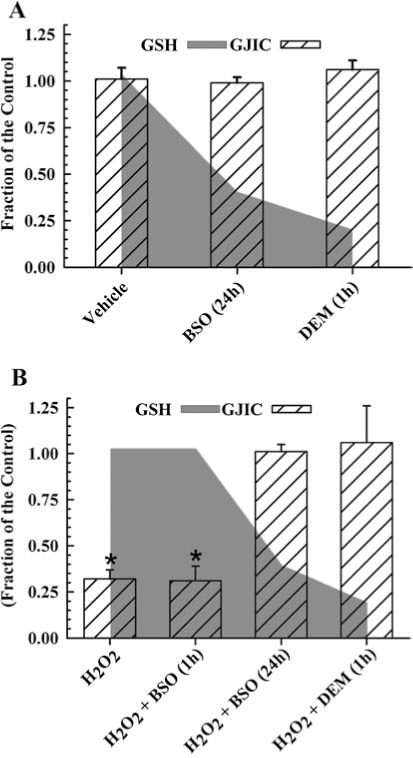
Other examples of this phenomenon where depletion of GSH prevents an oxidative chemical-induced cellular event is in: (a) lindane-dependent inhibition of GJIC in myometrial smooth muscle cells (67), (b) peroxide induction of c-jun (56), and (c) the activation of NF-κB by H2O2 and O2 ·− (35). These results indicate that signaling pathways not only require H2O2 for activation but also GSH. The implications are that reductive scavenging systems, which typically protect against damage from high levels of oxidative stress, also are potentially involved in the normal oxidative signaling systems at lower levels of oxidative tensions.
Oxidative Integration of GJIC and Mitogenic Signal Transduction
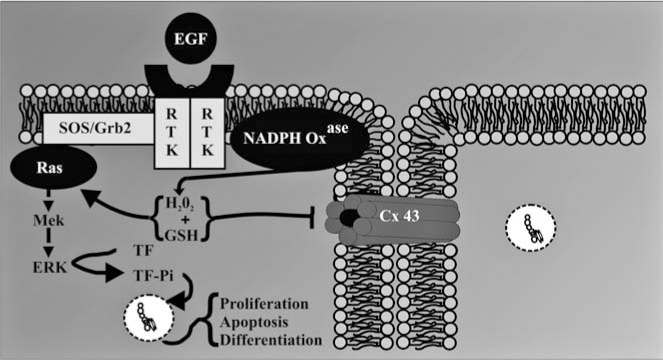
A common response of a cell to growth factors is a transient production of H2O2 that is an essential coactivator of mitogen-activated protein kinases (MAPKs) (24, 90). NADPH oxidase, an enzyme that synthesizes H2O2, is a well-characterized enzyme known to play an antimicrobial role in phagocytic cell types (6, 57). More recently, various NADPH oxidases in nonphagocytic cells have been implicated in oxidative signaling and have been specifically linked to the activation of various MAPKs in response to different growth factors and cytokines (2, 5, 8, 11 –14, 48, 65, 66, 68, 78, 82, 83, 85, 91, 107, 110, 115).
Diphenyleneiodonium (DPI) is a selective inhibitor of NADPH oxidase that has been extensively used to link the activity of this enzyme with specific cell functions and signaling mechanisms (90). Preincubation of DPI with the F344-WB cells prevented epidermal growth factor (EGF)-induced inhibition of GJIC (Fig. 3) (101), thus implicating NADPH oxidase in EGF-induced inhibition of GJIC. Inhibition of GJIC by EGF was also prevented by blocking MAPK/ERK kinase (MEK) activation with PD98054 (Fig. 3) (101), which agrees with previously published results (18, 43). These results are not unexpected considering that the MEK–ERK pathways are redox regulated, as cited above.

A major implication of these results is that gap junctions serve as a modulator of a signal transduction pathway, specifically MAPK (Fig. 4). Hypothetically, if gap junctions were not closed, then the generation of H2O2, an essential signaling cofactor of MAPK, in response to extracellular ligands could result in the dilution of H2O2 from the target cell into the neighboring cells, thereby decreasing the concentration of H2O2 to a nonthreshold level that would be insufficient for the activation of MAPK (Fig. 5). Superoxide and hydroxyl radicals are transient and unstable ROS that are unlikely to travel significant distances from the site of synthesis. However, H2O2 is a fairly stable intermediate capable of migrating from the site of production. Although there are no reports of H2O2 traversing the gap junction channels, this molecule, like H2O, is a very small polar compound and could conceivably traverse the channels similar to H2O. This dilution effect of a low molecular weight signaling cofactor would be quite prominent in an asymmetric environment that is typical of real tissues. Thus one significant role of gap junctions would be to maintain above threshold concentrations of positive signaling cofactors, which is consistent with closure of gap junction channels during mitogenesis. This hypothesis also allows for the prediction that the opened channel state, typically seen for differentiation and apoptosis, is needed to prevent threshold concentrations of negative signaling cofactors. These examples demonstrate how extra-, intra-, and intercellular signaling pathways might interact to coordinate the epigenetic expression of genes in response to ROS (Figs. 4 and 5).

Signaling Pathway-Specificity of Antioxidants: Implications in Prevention and Therapy of Diseases
Traditionally, oxidative stress was viewed as a consequence of too much reactive oxygen produced for the innate reductive scavenging system of a cell to handle, resulting in cellular damage. Certainly this is true for many acute oxygen toxicities that can lead to tissue necrosis, such as that seen in ischemia and reperfusion. But this is an overly simplistic view, considering that reactive oxygen as well as the innate scavenging systems is also involved in redox sensitive signaling. Considerable progress has been achieved in defining specific redox-dependent targets of intracellular oxidants (26) and the role of antioxidants similarly play specific roles, both redox sensitive and redox insensitive, in cell signaling and consequent gene expression profiles (4). Thus, understanding the mechanisms by which oxidative compounds promote and antioxidants prevent the development of tumors must begin integrating the different forms of cell signaling (102). To continue using the simple assumption that ROS are always detrimental to the organism and that high levels of antioxidants must be beneficial due to their scavenging properties of ROS is no longer acceptable. For example, previous intervention studies with antioxidant supplementation to human populations proved either ineffective, such as the N-acetylcysteine EUROSCAN studies in Europe (108, 113), or actually detrimental to human health, such as increased lung cancer of smokers in the β-carotene studies (CARET trials in USA and ABTC trials in Finland) (37, 113).
The complexity of oxidant and antioxidant interactions is apparent in a study of the effects of organic peroxides and antioxidants on gap junctions (102). As predicted, the tumor-promoting organic peroxides (33, 39), benzoylperoxide and dicumyl peroxide, inhibited GJIC and activated extracellular kinase (ERK)-MAPK, and the nonpromoting peroxide (33, 39), t-butylperoxide, had no effect on these two endpoints (102), yet t-butylperoxide caused considerably more oxidative DNA damage than BzOOH (45). Resveratrol, an antioxidant found in the products of grapes, such as red wine, and peanuts, prevented the inhibition of GJIC by dicumyl peroxide, which closed gap junction channel through a phosphatidylcholine-specific phospholipase C (PC-PLC) but not benzoylperoxide, which closed channels in a PC-PLC-independent way (102). The effect of resveratrol on ERK-MAPK was less specific where resveratrol blocked the activation of ERK by both benzoylperoxide and dicumylperoxide (102). The specificity of the antioxidant effect of resveratrol is further demonstrated by the lack of a protective effect of N-acetylcysteine (NAC) on preventing activation of ERK and the inhibition of GJIC by either tumor promoting organic peroxides. However, NAC did prevent the general oxidative events of BzOOH, such as the depletion of reduced glutathione (GSH) and the induction of cytotoxicity (102). These results clearly indicate diverse roles that antioxidants can play in cell signaling and the notion that by simply preventing general oxidative damaging events is not sufficient in protecting against oxidative promoters. The closing statement presented in the minireview of Azzi et al. (4) “Finally, the discrepancies in the outcome of the intervention studies may be understood if, instead of considering the simple paradigm of bad oxidants and good antioxidants, scientists will start to talk about the real molecular function of such compounds in each particular situation.” eloquently reflects the importance of studies focusing on the redox and nonredox mechanisms of antioxidants on signaling network systems.
Conclusions
Although oxidative damage is clearly a factor in many pathologic conditions affiliated with oxidative stress, oxidative signaling probably plays a greater role in chronic diseases. Thus, it will be crucial to understand how various oxidants and antioxidants specifically interact with cell signaling systems, and determine what levels are needed for gene expression controlling normal cellular tissue phenotypes. Considering that reactive oxygen is an essential participant in many normal cell signaling systems, then pathologic levels may not always result with an excess generation of reactive oxygen but also from deficient levels. Understanding the specific roles of antioxidants in cell signaling will better enable us to develop more effective and safer intervention strategies. Exposure to high levels of a single antioxidant could easily overwhelm a specific effect of a precise signaling protein or small group of signaling proteins, thus resulting in a toxic rather than beneficial effect. Cell signaling mechanisms affected by redox chemistries are not limited to only the signal transduction pathways of intracellular signaling but also involve intercellular signaling through gap junctions. Thus, any comprehensive-systems biology approach to understanding oxidative signaling must also include gap junctions, in which aberrant gap junctions have been clearly implicated in many human diseases. This is not surprising, considering that gene expression must be coordinated between cells of a tissue in order to maintain tissue homeostasis. We propose a hypothesis that one potential function of gap junctions is to modulate levels of second messengers that are either positive or negative cofactors needed in signal transduction. Several reactive oxygen species, such as superoxide, hydrogen peroxide, nitrous oxide, all have very low molecular weights not much different from that of water, thus can be predicted to readily traverse the gap junction channels, and can consequently serve as ideal second messengers in a network of signaling pathways that include gap junctions. Taking a more comprehensive-systems approach in understanding the redox mechanisms of cell signaling networks will greatly aid efforts in the prevention and treatment of diseases affected by oxidative stress.
Footnotes
Acknowledgments
This research was supported by NIEHS grants #R01 ES013268-01A2 to Upham. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIEHS.
Abbreviations
BSO, buthionine sulfoximine; DEM, diethylmaleate; DPI, diphenyleneiodonium; EGF, epidermal growth factor; ERK, extracellular receptor kinase; GJIC, gap junctional intercellular communication; MEK, MAPK/ERK kinase; MAPK, mitogen activated protein kinase; ROS, reactive oxygen species.