
Abstract
The DNA base excision repair (BER) pathway repairs alkylation and oxidative DNA damage caused by endogenous and exogenous agents, including chemotherapeutic agents. Upon removal of the damaged base AP endonuclease 1 (Ape1), a critical component of the pathway cleaves the abasic site to facilitate repair. Ape1 is a multifunctional protein which plays a role not only in DNA repair but it also functions as a reduction–oxidation factor, known as Ref-1 in the literature, to increase the DNA binding ability of several transcription factors involved in different growth signaling pathways. Elevated levels of Ape1 have been linked to resistance to chemotherapy, poor prognosis, and poor survival. Reducing the amount of Ape1 protein in cancer cells and tumors using RNA interference and anti-sense oligonucleotide technology sensitizes mammalian tumor cells to a variety of laboratory and chemotherapeutic agents. Therefore, selective inhibition of Ape1's DNA repair activity is a promising avenue to develop novel cancer therapeutics. Antioxid. Redox Signal. 11, 651–667.
Introduction
Importance of DNA repair pathways and cancer
DNA base excision repair (BER) pathway
The DNA base excision repair (BER) pathway repairs alkylation and oxidative damage caused by endogenous and exogenous agents, including radiation and chemotherapy-induced damage (24, 44). The BER pathway recognizes and repairs single base lesions including N-alkylated purines (N3-methyladenine, N7-methylguanine, and N3-methylguanine), 8-oxo-7,8-dihydroguanine(8-OxoG), thymine glycols, 5-OH and 6-OH dihydrothymine, uracil glycol, 5-hydroxycytosine, and urea residues, in addition to a number of additional adducts (4, 24, 37). Repair of the damaged base is initiated by a DNA glycosylase (Fig. 1) which specifically recognizes and excises the damaged base. Different DNA glycosylases recognize specific and different types of base damage. Glycosylases are of two types—monofunctional and bifunctional glycosylases. Monofunctional glycosylases [e.g., N-methyl purine DNA glycosylase (MPG)] excise the damaged base to generate an apurinic/apyrimidinic (AP) or abasic site. In contrast, bifunctional glycosylases, in addition to exhibiting glycosylase activity, also have an AP lyase function (20, 27). Bifunctional glycosylases such as 8-oxoguanine DNA glycosylase (OGG1), Nei endonuclease VII like, NEIL1, NEIL2, and NTH not only excise the damaged base but also nick the phosphodiester backbone 3′ to the AP site (24, 37). Removal of the damaged base by a DNA glycosylase creates an AP site, and AP sites are also generated by spontaneous base loss in the genome (29, 87).

The second critical component of the pathway is the multifunctional protein apurinic/apyrimidinic endonuclease (Ape1). Following hydrolysis by a DNA glycosylase, Ape1 processes the AP site by making an incision in the phosphodiester backbone immediately 5′ to the AP site. This incision creates 3′OH and 5′ deoxyribose phosphate (5′dRP) termini. At this stage, repair can proceed via one of two pathways (Fig. 1). The short-patch BER (SP-BER) pathway repairs regular AP sites. In the short-patch pathway, DNA polymerase β (Pol β) removes the 5′ dRP moiety via its dRPase activity and uses the 3′OH terminus to insert the correct base. Subsequently, DNA ligase III/XRCC1 (X-ray cross-species complimenting 1) seals the nick and repair is completed. The long-patch BER (LP-BER) pathway preferentially repairs modified (oxidized, reduced) AP sites. In this minor BER pathway. a flap of three to eight nucleotides surrounding the AP site is displaced. The correct nucleotides are inserted by DNA polymerase β, δ or ɛ, along with proliferating cell nuclear antigen (PCNA) and replication factor-C (RF-C). Following resynthesis, flap endonuclease 1 (FEN1) removes the displaced strand and then the nick is sealed by DNA Ligase I or DNA Ligase III/XRCC1 (37). Oxidative DNA lesions can also be excised by the recently identified Neil glycosylases NEIL1 and NEIL2 that show homology to the E. coli endonuclease VIII (5, 53 –55, 69, 122). The AP sites generated are processed by Ape1 and subsequent repair is completed. While there are several different DNA glycosylases to excise the damaged base and generate AP sites, there is only one Ape1 protein, which can process the AP sites generated and facilitate repair, thus emphasizing its significance in the BER pathway.
The Ape1 protein, an essential component of the BER pathway
Based on the method of incision, AP endonucleases can be classified into two classes. The Class I AP endonucleases are also known as AP lyases (or β-lyases) as they process the AP sites by the β-elimination reaction and cleave the phosphodiester backbone 3′ to the AP site, generating a 5′ phosphate and a 3′ α,β-unsaturated aldehyde end. This AP lyase activity is usually associated with complex DNA glycosylases which are responsible for repairing oxidatively damaged DNA (27). The Escherichia coli endonuclease III and endonuclease VIII (52) and the human homologue NTH1 (57, 58, 64) belong to this class of endonucleases. Class II AP endonucleases are the major class of endonucleases and are also known as hydrolytic endonucleases, as they hydrolyze the phosphodiester backbone 5′ to the AP site, creating normal 3′OH and 5′ deoxyribose phosphate termini. Based on homology, Class II AP endonucleases can be further classified into two families, the exonuclease III (xth) and the endonuclease IV (nfo) family. The exonuclease III family consists of human Ape1 in addition to enzymes from various phyla, and these enzymes possess a strong AP endonuclease activity (29, 37, 110, 119, 133). In addition to the endonuclease activity, Ape1 also possesses a 3′-repair diesterase activity. Although this 3′-repair diesterase activity is much weaker (almost 200-fold weaker) (20) than the AP endonuclease activity, it is important in the removal of 3′ blocking lesions such as phosphoglycolate moieties in order to complete repair (20, 37, 42, 104). In the BER pathway, Ape1 is responsible for processing AP sites generated as a result of the action of both types of DNA glycosylases (29, 32, 117). This processing of AP sites by Ape1 then facilitates complete repair of the damaged base. The endonuclease IV family of enzymes is the second major family of Class II AP endonucleases which include the E. coli endonuclease IV and Apn1 from Saccharomyces cerevisiae (yeast) which is responsible for 90% of AP endonuclease activity in S. cerevisiae (37, 73, 108, 128). Apn1 can repair both alkylation and oxidative damage, including oxidized abasic sites and unlike Ape1, Apn1 has a higher 3′ repair-diesterase activity (43). Although the enzymes from both families share the AP endonuclease function, they do not share sequence or structural similarity (29, 99).
Functions of Ape1
The AP endonuclease activity of Ape1
Ape1 is responsible for 95% of the endonuclease activity in the cell and is a critical part of both the short-patch and the long-patch BER pathway (29, 32, 117). Recognition of the damaged base by a monofunctional DNA glycosylase and its subsequent removal generates an AP site. This AP site is recognized by Ape1 that hydrolyzes the phosphodiester backbone 5′ to the AP site, generating a 3′OH and a 5′deoxyribose phosphate (5′dRP) terminus. Subsequently, regular AP sites are repaired via the SP BER pathway whereby the 5′dRP moiety is removed by the dRPase function of Ape1 or DNA Pol β and repair is completed by insertion of the correct base by DNA Pol β and sealing of the nick by DNA ligase III/XRCC1. Modified (oxidized, reduced) and, to a lower extent, regular AP sites, are repaired via LP-BER where DNA polymerase β, δ, or ɛ, along with PCNA and RF-C, fills in a patch of three to eight nucleotides. FEN1 cleaves the displaced stretch of three to eight nucleotides and repair is completed by DNA ligase I or DNA ligase III/XRCC1 (37). Ape1 is essential to complete the repair of AP sites which are generated by the action of different DNA glycosylases on a variety of DNA lesions, including oxidative DNA lesions that can also be excised by the recently identified NEIL glycosylases (5, 53 –55, 122). Thus, Ape1 is functionally involved in the short-patch and long-patch BER pathways. As discussed above, Ape1 has a strong 5′ AP endonuclease activity, in addition to which it also has a 3′-repair diesterase activity which is important for the removal of 3′ blocking lesions generated as a result of the β-lyase function of DNA glycosylases involved in the repair of oxidative or radiation-induced DNA damage (20, 37, 42, 104). Blocking lesions such as 3′-phosphate groups and 3′-phosphoglycolate moieties generated by the action of oxidative agents such a bleomycin, radiation (IR), and also formed at single-strand breaks are removed by Ape1's 3′-phosdiesterase function so that the subsequent steps of BER can take place and repair can be completed (19, 20, 99, 104, 124). In addition to its hydrolytic and 3′-diesterase functions, Ape1 also has a 3′-5′ exonuclease activity which is important to process 3′ mispaired termini (22) and for the removal of unnatural nucleoside analogs (21, 23).
AP sites formed as a result of the action of DNA glycosylases can also be generated by spontaneous base hydrolysis in the cell (100), and Ape1 is required to further process these AP sites in order to complete repair. If left unrepaired, these AP sites can be cytotoxic and mutagenic as they can block the replicating polymerase (87, 132, 141). Thus, although AP sites can be generated by the action of several different damage specific DNA glycosylases, only Ape1 can process these AP sites and facilitate repair, thus emphasizing its significance in the BER pathway. Furthermore, importance of Ape1 to normal cellular functioning is highlighted by the embryonic lethality of Ape1 knockout mice (80, 138). Elevated levels of Ape1 in cancer cells have been postulated to be a reason for chemotherapeutic resistance (15, 37, 75, 77, 109, 114, 121, 130) and inhibition of Ape1 has been shown to increase cell killing and apoptosis and also sensitize cancer cells to chemotherapeutic agents. Inhibition of Ape1 using DNA antisense and RNA interference technology is known to increase cell killing and apoptosis and also sensitize cancer cells to chemotherapeutic agents (14, 81, 102, 129). These findings demonstrate the uniqueness of Ape1 as a molecular target in therapeutics.
Other repair functions of Ape1
The multifunctional Ape1 protein has several other functions accompanying its DNA repair and redox regulatory activities. As part of its DNA repair activity, Ape1 removes 3′ blocking lesions generated by DNA glycosylases which recognize oxidative damage (Ogg1, Neil) (33, 37, 54) via its 3′-phosphodiesterase and 3′-phosphatase activities to facilitate complete repair. Ape1 also possesses a 3′–5′ exonuclease activity which is important for the removal of deoxyribonucleoside analogs which can impede repair (19, 22, 23, 34). In addition to the afore mentioned functions, Ape1 has been shown to inhibit the activation of PARP1 (poly(ADP-ribose) polymerase 1) during oxidative damage repair, thus preventing the cells from undergoing apoptosis (105). A relationship between Bcl2 and Ape1 resulting in decreased repair has been reported (72) in addition to negatively regulating the parathyroid hormone gene (PTH) (13, 25, 78, 101), being involved in granzyme A (GzmA) aided NK cell-mediated killing (39, 93) and it has been implicated in nucleotide incision repair (NIR) (65, 66). Ape1 has also been suggested to play a role in negatively regulating the Rac1/GTPase to prevent oxidative stress (103) and to regulate vascular tone and endothelial NO production (71) (Fig. 2).
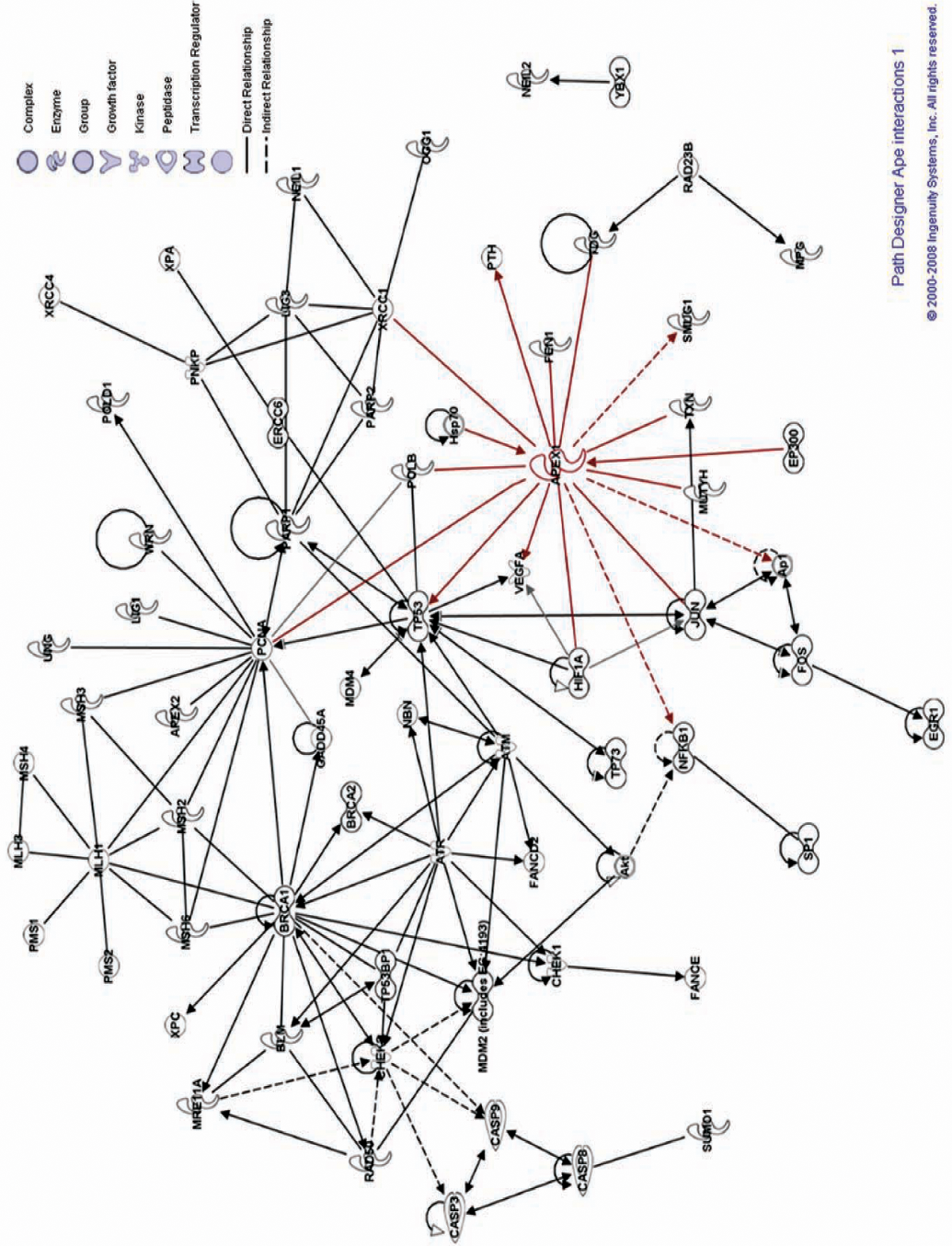
The redox function of Ape1
In addition to the AP endonuclease function of Ape1, it also functions as a reduction/oxidation (redox) signaling factor (Fig. 3) and is therefore also referred to as
The repair and redox functions of Ape1 are distinct from each other
As discussed previously, Ape1 is a multifunctional protein with roles in DNA repair as well as in redox signaling in the cell. These two important functions of Ape1 are functionally distinct from each other and are encoded by distinct regions of the protein (137). The AP endonuclease or DNA repair activity which is a critical component of the BER pathway resides in the C-terminal portion of the protein. The AP endonuclease activity is mediated by the active site residues His 309, Glu 96, Asp 238, and Asp 308, where H309 is the catalytic residue (6 –8, 11, 37, 45, 50, 88, 98). The redox regulatory activity of Ape1 which is important for the control transcription factors resides in the N-terminal sequences of the protein and a conserved Cys 65 residue is crucial for this function of Ape1 (9, 35, 37, 45). These two important functions of Ape1 can be functionally separated from each other, and disruption of one of its activities does not affect the other. Several reports have shown that disruption of Cys 65 by site-directed mutagenesis (21, 35) or by using a redox specific inhibitor, impairs the redox function of Ape1 but does not affect its DNA repair ability (112, 143).
Ape1 is ubiquitously expressed and though there are several reports showing that Ape1 is localized to the nucleus, cytoplasmic localization of Ape1 has also been reported (34, 74, 97, 113, 134). In addition to exhibiting a heterogeneous and complex pattern of staining, localization of Ape1 is tissue specific and even differs between neighboring cells (113, 134). Localization of Ape1 in the cytoplasm may be associated with its role as a mitochondrial DNA repair protein (37, 97, 124). Noting Ape1's role in redox control of transcription factors, presence of Ape1 in the cytoplasm may be important to maintain these transcription factors in a reduced state prior to their transport to the nucleus (34). Ape1 has also been shown to accumulate in the nucleus and mitochondria in response to DNA damage (97). Thus, it appears that the intracellular localization of Ape1 is regulated; however the significance of its subcellular localization is still not well understood.
Inhibition of DNA Repair as a Target in Cancer
DNA repair pathways are important to maintain the genomic integrity as highlighted by several cancer predisposing syndromes which harbor germline mutations in DNA repair genes (24, 44, 59). Currently, chemotherapy and radiation therapy are the mainstream treatment options available to treat cancers. The cytotoxic effects of most chemotherapeutic agents and radiation are related to their ability to induce DNA damage. The ability of cancer cells to identify and efficiently repair such DNA damage undermines the efficacy of these agents (111, 127). Therefore, inhibiting DNA repair proteins leading to reduced repair of damaged DNA in cancer cells is an attractive approach to combat chemotherapeutic resistance and to increase efficacy of therapy. Although it may sound contradictory to inhibit DNA repair pathway proteins, since cancer promotion and deregulated cellular growth is aided by deficient DNA repair pathways, it actually makes sense to block DNA repair, given the predominance of DNA damage during cancer treatments with chemotherapy and IR, which would allow for increased efficacy of the DNA damaging agent (12, 92). Thus, inhibiting specific proteins from selected DNA repair pathways in cancer cells could provide us with a selective way to sensitize cancer cells to chemotherapeutic agents and also combat their resistance to chemotherapeutic agents (90, 92).
Consequences of inhibiting the BER pathway in cancer
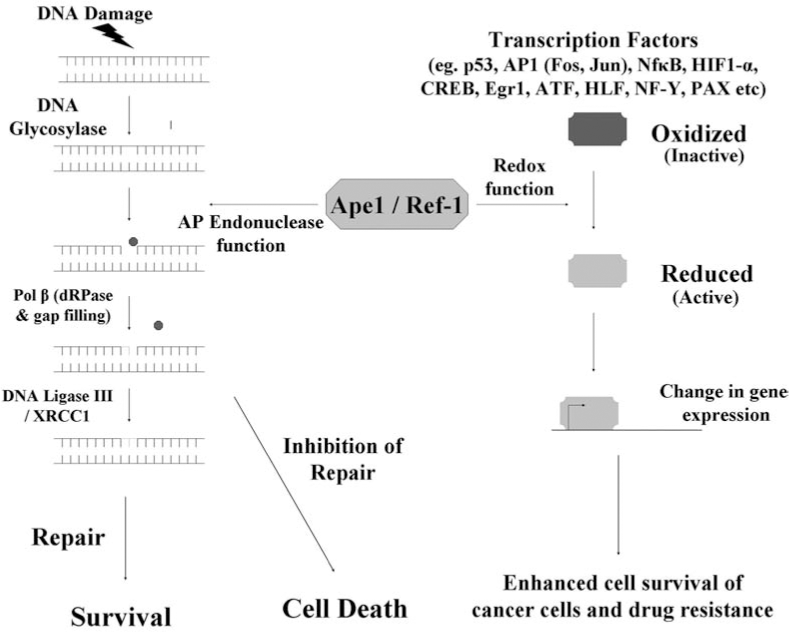
The BER pathway is responsible for repairing DNA damaged by endogenous and exogenous agents including chemotherapeutic agents. In addition to repairing damaged lesions, the proteins of the BER pathway exhibit several important protein–protein and DNA–protein interactions (38) and a delicate balance exists between the levels of all the BER proteins (49). In cancer cells, the upregulation of the BER proteins results in imbalanced repair and can lead to resistance to chemotherapeutic agents; modulating or inhibiting the activities of these BER proteins can lead to deregulated repair, resulting in sensitivity to chemotherapy agents. However, the central idea that presence of robust DNA repair mechanisms leads to resistance to chemotherapeutic agents (12, 26, 31, 56, 82, 92, 120) has been challenged by some studies. Roth et al. (116) showed that absence of Aag (3MeA DNA glycosylase) in the bone marrow (BM) cells of the myeloid lineage from Aag −/− mice are resistant to alkylating agents (MMS), as compared to the wild-type BM cells. They speculated that initiation of repair by Aag and subsequent incomplete repair of the lesions in wild-type BM cells may be more toxic than the inability of Aag null BM cells to initiate repair of these damaged lesions. This effect was specific to the myeloid lineage of the Aag −/− mice and was not observed in embryonic stem cells (ES), primary embryonic fibroblasts (PEF) and cells from the lymphoid lineage in the BM, indicating that this effect is tissue specific, as well as likely lesion specific. In general, it has been shown that presence of DNA repair contributes to resistance to chemotherapeutic agents. DNA glycosylases show quite a bit of functional redundancy (80), and the action of all the DNA glycosylases results in the formation of AP sites. For instance, overexpression of 3MeA DNA glycosylases in S. cerevisiae and E. coli leads to increased sensitivity to alkylating agents and spontaneous mutations, possibly due to an imbalance between the levels of the DNA glycosylase and Ape1 proteins and also due to the build-up of unrepaired AP sites (116). Accumulation of these unrepaired AP sites can lead to (Fig. 4) single-strand breaks, increased apoptosis, and enhanced cytotoxicity (47). Ape1 is required to process these AP sites and along with the rest of the BER proteins can facilitate the ensuing completion of repair. Increased expression of the BER pathway proteins in cancer cells can result in efficient repair of damaged lesions and can reduce the effectiveness of chemotherapeutic agents. Therefore, keeping in mind the importance of the BER pathway in the repair of damage induced by chemotherapy, exploiting the BER pathway and its proteins by inhibiting them would increase the efficacy of chemotherapy, thus making it an attractive target to develop novel means in order to combat chemotherapeutic resistance (120).
Inhibition of the DNA Repair Function of Ape1 as a Rational Cancer Target
There are several reasons why Ape1 is a rational target for chemotherapeutic agents: (a) overexpression of Ape1 leads to chemoresistance; (b) cells that lack Ape1 are not viable; (c) knockdown or blockage of Ape1 activity sensitizes cancer cells to chemo agents such as temozolomide (TMZ) and bleomycin. Elevated levels of Ape1 in cancer cells have been postulated to be a reason for chemotherapeutic resistance (14, 37, 75, 77, 109, 114, 121, 130). The importance of Ape1's function in the DNA BER pathway is observed from the lethality of Ape1 knockout mice (80, 138). Specifically knocking down or inhibiting Ape1 using RNA interference and anti-sense oligonucleotide technology hypersensitizes mammalian cancer cells to several laboratory and clinical DNA damaging agents, such as methyl methane sulfonate (MMS), hydrogen peroxide (H2O2), bleomycin, TMZ, and gemcitabine (14, 15, 75, 81, 87, 114, 121, 129, 130). The decrease in cancer cell proliferation and survival after knocking down Ape1 reiterates the importance of Ape1 function. Although these data demonstrate Ape1 is a feasible target for inhibition to sensitize cancer cells, studies involving a reduction in Ape1 mRNA and protein do not allow us to dissect which function, repair or redox, of Ape1 is important for cell growth, cancer promotion and/or progression (14, 15, 75, 81, 87, 114, 121, 129, 130). Fung et al. (47) demonstrated that depletion of Ape1 from cells using siRNA technology causes increased apoptosis and decreased cell growth of cancer cells. They further demonstrated that functional complementation with a yeast homologue (Apn1) of Ape1 deficient in redox activity could restore proliferation potential of the cells. Another report demonstrated that expressing a dominant-negative repair deficient Ape1 protein in cells sensitizes them to chemotherapeutic agents (94). Several studies conducted using a small molecule that binds to AP sites in DNA [methoxyamine (MX)] and blocks Ape1's ability to cut the sugar–phosphate backbone sensitized cancer cells to chemotherapeutic agents (41, 84, 85, 123). Conversely, as we learn more about the redox function of Ape1, we appreciate its critical role in cell growth. Another recent study demonstrated that the redox function of Ape1 is important in hematopoietic differentiation (growth) by using a specific inhibitor of Ape1's redox activity, but did not cause the cells to undergo apoptosis (143). These observations not only suggest a crucial role for both of Ape1's functions in cellular survival and tumor promotion and progression, but also demonstrate differences observed when the redox or repair functions are blocked. Developing specific inhibitors of the two functions of Ape1 would further allow us to discern which of the activities of Ape1 are important for cancer promotion and progression and normal cellular survival. Furthermore, the two functions may play different roles in different kinds of cancer, allowing us to better understand tumor progression. Thus, the consequences of inhibiting of Ape1 in cancer cells point to it being a logical target in cancer therapeutics. Identification of molecules that specifically inhibit Ape1's repair or redox activity should be an effective means to sensitize cancer cells to chemotherapeutic agents and thus impact the development of new and targeted cancer therapies. This review will focus on repair inhibition (see review for information on redox inhibitors) (42).
Existing Ape1 DNA Repair Inhibitors
As discussed above, Ape1's importance in the BER pathway and unique role supports the hypothesis that it is a strong target for cancer therapy; elevated levels in cancer cells and knocking down Ape1 using siRNA leads to increased ability of cancer cells to undergo apoptosis and sensitization to chemotherapeutic agents (14, 37, 75, 77, 109, 114, 121, 130). Thus, combining standard chemotherapeutic strategies with targeted inhibitors of Ape1's DNA repair function would increase the effectiveness of existing chemotherapeutic regimens. Currently three compounds are known to inhibit Ape1's DNA repair activity, methoxyamine (MX) and two compounds, CRT0044876 or 7-nitroindole-2-carboxylic acid (NCA) (91), and an arylstibonic acid compound (118), both identified in high throughput screening (HTS) assays.
Methoxyamine (MX), an indirect inhibitor of Ape1's repair activity
Methoxyamine (MX) is an inhibitor of Ape1 which interacts with the aldehydic C1 atom left at the DNA abasic site after removal of the damaged base by a DNA glycosylase (86). MX is considered to be an indirect inhibitor of Ape1 in that it does not directly bind to Ape1 and inhibit its activity. Rather, MX binds to AP sites and prevents Ape1 and Pol β from processing them further and completing repair. As a single agent, MX is not significantly cytotoxic, however MX can potentiate the cytotoxicity of alkylating agents such as TMZ and 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) (41, 84, 123). TMZ predominantly alkylates guanine at the N7 and O6 positions and adenine at the N3 position. The BER pathway repairs N7 and N3 alkylation damage and the action of the DNA glycosylases generate AP sites. MX binds to these AP sites, thus preventing Ape1 from completing the repair and stabilizing the AP site intermediate (41, 84, 123). Clinical trials with MX in combination with TMZ are currently being pursued. A direct repair inhibitor with the ability to directly block Ape1's repair activity should prove to be more potent in sensitizing cells to chemotherapeutic alkylating agents while reducing nonspecific effects. As discussed previously, MX is currently in use to block Ape1's ability to repair AP sites. However, this reagent targets the DNA (AP sites) and not Ape1 directly (86, 115). MX stably interacts with the C1′ aldehyde atom resulting from the removal of the damaged base, producing a stable covalent adduct (86); Ape1 is unable to readily cleave the resulting MX–AP site (46). MX is a simple compound, H3CONH2, with no obvious potential for improvement in efficacy through derivatization and high concentrations of MX are required in cell-based assays (20–50 mM) in order to potentiate cell killing in combination with other agents (85, 94, 123, 139). In addition, since MX binds AP sites and not Ape1 directly, it will also affect the ability of other mammalian enzymes to bind DNA substrates including DNA Pol β (61).
Lucanthone, a direct inhibitor of Ape1's repair activity
Lucanthone, originally identified as a topoisomerase II poison (10), is considered to be a direct inhibitor of Ape1's DNA repair activity. Its extensive use to treat schistosomiasis has shown it to be safe and nearly nontoxic from a clinical standpoint (28). Cancer cells treated with lucanthone exhibited a dose-dependent increase in AP sites, seemingly due to inhibition of Ape1's repair activity and blocking an early step in the BER pathway (95). Patients with brain metastasis treated in combination with radiation and lucanthone showed increased regression of the tumors with the combination, as compared to radiation alone (28). Additionally, lucanthone enhances the cell killing effect of MMS and TMZ in breast cancer cells by the inhibition of Ape1's DNA repair activity (89). However, the evidence of lucanthone, also being a topoisomerase II inhibitor, raises the concern that the tumor cell killing observed could be partially attributed to the off-target effects of lucanthone, which again points to the need of a robust direct inhibitor of Ape1's repair function.
7-Nitroindole-2-carboxylic acid (NCA), a direct inhibitor of Ape1's repair activity
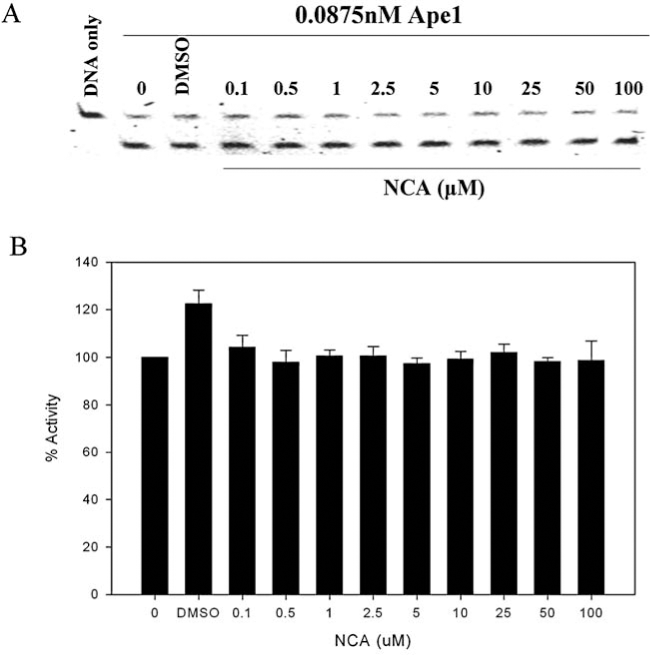
Madhusudhan et al. (91) identified CRT0044876 or 7-nitroindole-2-carboxylic acid (NCA) in a high-throughput screen (HTS) of a library of 5,000 drug-like compounds to be a direct inhibitor of Ape1's repair activity with an IC50 value of ∼3μM. NCA is negatively charged and is reported to inhibit all the DNA repair activities of Ape1 such as the AP endonuclease (repair activity), 3′-phosphodiesterase, 3′-5′exonuclease, and 3′-phosphatase activities of Ape1. Survival analyses in HT1080 human fibrosarcoma cells showed that NCA potentiates the cytotoxicity of MMS, TMZ, H2O2, and zeocin (91). However, efforts to reproduce this repair inhibition have not been realized by our laboratory and by others (51) (Fig. 5A and B).
Arylstibonic acid compounds as inhibitors of Ape1's repair activity
Recently, Seiple et al. (118) screened an NCI Diversity Set library of 2,000 compounds to identify specific inhibitors of Ape1. The authors identified an arylstibonic acid compound 13755 which has a negative charge, as an inhibitor of Ape1's DNA repair activity. This compound shows partial mixed type inhibition in that it binds both to the enzyme and the enzyme substrate complex. Even though these compounds have been reported to inhibit Ape1's DNA repair activity in vitro, they have virtually no cellular uptake and are less attractive as translational agents. Treatment of HOS osteosarcoma cells with 5 μM of the compound in the presence of MMS did not show decreased survival in cytotoxicity assays (118).
Need for Specific Inhibitors of Ape1's DNA Repair Activity
There is a clear need for a specific repair inhibitor of Ape1 in order to effectively determine the role of Ape1's repair activity in potentiating the effects of alkylating chemotherapeutic agents. This is required given the importance of inhibiting Ape1 using siRNA leading to sensitization of cancer cells to chemotherapeutic agents (14, 15, 75, 81, 87, 114, 121, 129, 130). However, these studies remove all of Ape1's functions (repair and redox) as well as Ape1's protein–protein interactions (38), making the data difficult to interpret. Thus, identifying specific and potent Ape1 repair inhibitors would facilitate the understanding of Ape1 not only in cancer, but also in dividing normal cells (bone marrow, gut, etc), non-dividing normal cells (neurons), and other diseases where Ape1 has been implicated (17, 47, 60, 67, 106, 124). In addition, these small molecule inhibitors will allow the specific inhibition of Ape1's repair function while keeping its post-translational modifications (18, 68, 70, 97) and subcellular location of Ape1 intact and in determining the effect of blocking Ape1's function on these subcellular events (30, 107, 124). In summary, identification of specific inhibitors of Ape1's repair activity will further our ability to determine the role it plays in cancer promotion and progression, thus making a productive target of chemoprevention.
High Throughput Screening (HTS) Methodology to Identify Specific Inhibitors of Ape1's DNA Repair Activity
High throughput screening (HTS) is an efficient scientific method that allows the assay of large numbers of varied chemical compounds against biological targets in a relatively short period of time. HTS assays are either entirely or partially automated and can be carried out in the 96-well or 384-well format. Robotic automation in HTS helps speed up the process of drug discovery and facilitates the generation of a large amount of scientific data in a short time-frame. Several different libraries of synthetic and drug-like compounds are available for HTS. Typically, the first round of HTS is carried out with a fixed concentration (1–10 μM) of the chemical compound. The positive ‘hits' identified in the primary assay can be re-screened in the same assay and these hits are then followed up in secondary assays to validate them, determine an IC50 concentration, and perform functional cellular assays. Thus, HTS is a promising and rapid methodology to identify potential modulators of the biological activity of the target from a large number of compounds.
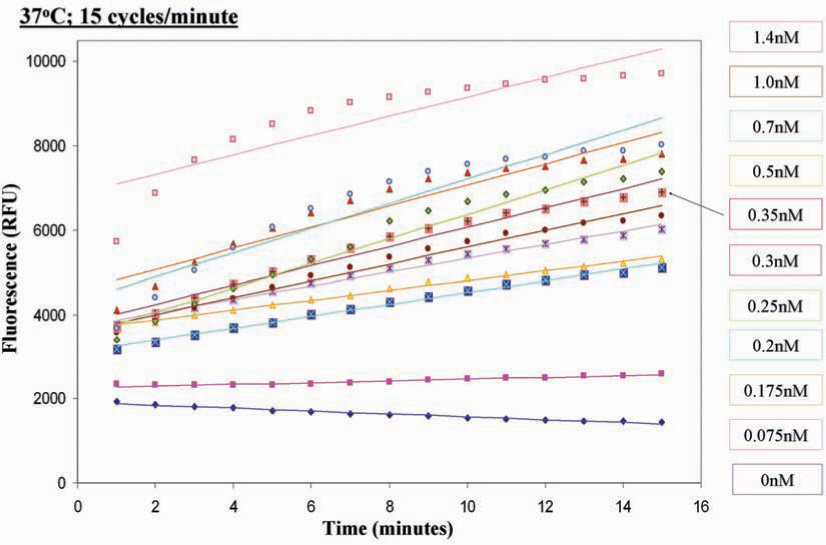
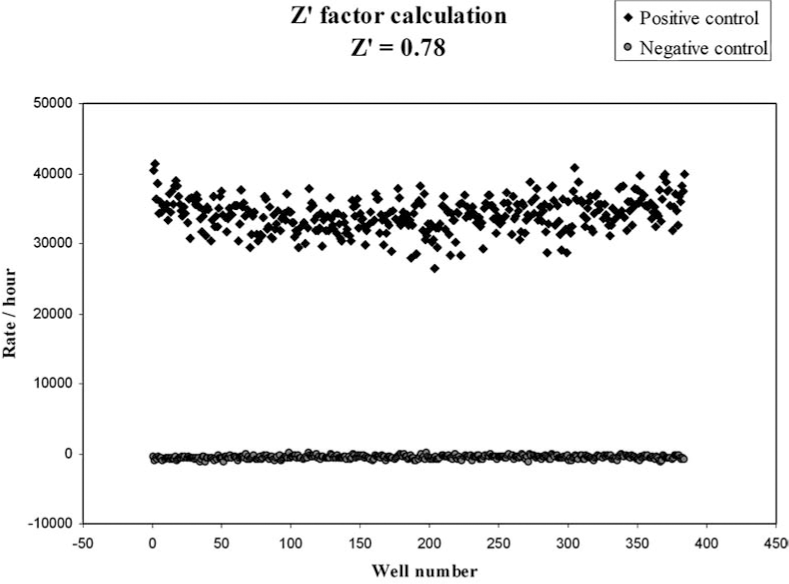
Efforts are under way in our laboratory to identify and develop a novel inhibitor of Ape1's DNA repair activity using the HTS methodology. We have modified the fluorescence-based high throughput screening assay described by Madhusudhan et al. (91) to screen a library of 60,000 synthetic drug-like compounds which follow Lipinsky's rule of five (83) from Chemical Diversity Ltd Inc. (San Diego, CA). We used purified Ape1 protein at a concentration that showed a linear range for the assay over a period of time (Fig. 6). In order to determine the reliability of the assay, we performed control experiments and calculated the Z′ -factor score of 0.78 (Fig. 7). The Z′ -factor reflects the dynamic range between the positive and negative controls and is also a reflection of the reproducibility of the data and reliability of the assay. The Z′-factor values range from 0.5 to 1.0, and a score above 0.5 is an indication of a good assay, with 1 being a perfect assay (142). After an initial screen of the 60,000 compound library, we identified 190 compounds showing ≤50% inhibition of Ape1's DNA repair activity. Of the 190 compounds, 174 were available and re-screened in the same HTS assay. Forty-five compounds showing ≤40% inhibition of Ape1's repair activity were identified after two rounds of HTS screening. These 45 compounds are now being further validated in a secondary gel-based AP endonuclease assay to further assess inhibition of the compounds. On completion of the validation of the 45 compounds in the secondary assays, promising compounds will then be tested for their ability to specifically inhibit Ape1's DNA repair ability in in vitro cellular assays, including cell growth, cytotoxicity, and apoptosis assays.
In the secondary gel-based AP endonuclease assay, a concentration of Ape1 protein in the linear range was titrated against a range of concentrations of the compounds. As a control, all the compounds tested in the gel-based AP endonuclease assay are compared to NCA, a known inhibitor of Ape1's DNA repair activity, at a concentration of 10 μM (91). One of the compounds identified in the HTS screen, AB-01, demonstrated consistent inhibition of Ape1's DNA repair activity both in the HTS and the secondary gel-based AP endonuclease assay. As seen in Fig. 8A and B, compound AB-01 is able to inhibit Ape1's DNA repair activity. At a concentration of 10 μM, inhibition of Ape1's DNA repair activity by AB-01 is better than that demonstrated by NCA at the same concentration.

Conclusions
Ability of cancer cells to recognize and repair chemotherapy-induced damage is an important factor in resistance to chemotherapy (90). Therefore, inhibiting DNA damage repair pathways and using inhibitors against specific proteins of these pathways is an excellent strategy to develop targeted therapies for cancer treatment. (12, 31, 59, 90, 92). Ape1 is a an essential protein functioning in the BER pathway which repairs damage caused by endogenous as well as exogenous DNA base damage including chemotherapy-induced DNA damage (24, 37, 44). Ape1 is unique such that it is the only cellular protein that can process the AP sites generated as a result of the action of the DNA glycosylases. It is also the only DNA repair and redox protein in the cells and there is no backup for the critically important repair function of Ape1 in cancer cells. This makes it a unique target, particularly since it has two important functions that work independently of one another. In addition to its important role in normal cellular functioning, altered or elevated levels of Ape1 have been observed in a variety of cancers, including breast cancer, gliomas, sarcomas (osteosarcomas, rhabdomyosarcomas), ovarian and multiple myeloma, among others (37, 75, 77, 109, 114, 121, 130) which have been speculated to be a cause of resistance to chemotherapy. In addition, these heightened levels of Ape1 have been linked to tumor promotion, progression, and poor prognosis associated with shorter relapse-free survival and poor outcome from chemotherapy (77). There is a vast amount of data showing that downregulating or inhibiting Ape1 in cancer cells using RNA interference and DNA antisense oligonucleotide techniques can sensitize them to laboratory and clinical chemotherapeutic agents (14, 15, 75, 81, 114, 121, 129, 131). Since Ape1 is involved in the repair of DNA damaged by chemotherapeutic agents, identifying selective inhibitors of the DNA repair or redox activities of Ape1 would make it an excellent target, both from a single agent approach and in combination with chemotherapy and IR. Additionally, it may be coupled with other specific molecular targets in cell cycle or other pathways to thwart the cancer cell from skirting cell death and prevent drug resistance from occurring.
Footnotes
Acknowledgments
Financial support for this work was provided by the National Institutes of Health, National Cancer Institute CA94025, CA106298, CA114571, and CA121168 to MRK, IU Simon Cancer Center Translational initiative pilot funding (ITRAC) to MRK, and the Riley Children's Foundation (MRK).
Abbreviations
AAG, 3Me A DNA glycosylase; BCNU, 1,3-bis(2-chloroethyl)-1-nitrosourea; 5′ dRP, 8-oxoG, 8-oxo-7,8-dihydroguanine; OGG1, 8-oxoguanine DNA glycosylase; Ape1, apurinic/apyrimidinic endonuclease 1; AP sites, apurinic/apyrimidinic sites; BER, base excision repair, NCA, CRT0044876 7-nitroindole, 2-carboxylic acid; DNA Pol β, DNA polymerase β; ES, embryonic stem; Fen-1, flap endonuclease-1; Gzm A, granzyme A; HTs, high throughput screen; HR, homologous recombination; H2O2, hydrogen peroxide; IR, ionizing radiation; ITRAC, IU Simon Cancer Center Translational Research Acceleration Collaboration; LP-BER, long patch-BER; MX, methoxyamine; MMS, methyl methane sulfonate; MMR, mismatch repair; MPG, N-methyl purine DNA glycosylase; NK, natural killer, NEIL, Nei endonuclease VIII like; NO, nitric oxide; NHEJ, non-homologous end joining; NER, nucleotide excision repair; NIR, nucleotide incision repair; PTH, parathyroid hormone; PEF, primary embryonic fibroblasts; PARP, poly (ADP ribose) polymerase 1; Ref-1, redox effector factor-1; SP-BER, short patch-BER; TMZ, temozolomide; XRCC1, x-ray cross complementing factor 1.