
Abstract
Endoplasmic reticulum (ER) stress induces an adaptive program called the unfolded protein response (UPR), which affects activity of an array of kinases and transcription factors. Previous reports provided evidence for activation of nuclear factor-κB (NF-κB), the major transcription factor regulating inflammatory processes, by ER stress. However, recent investigation also suggested that preceding ER stress suppresses activation of NF-κB by subsequent exposure to inflammatory stimuli. Although ER stress induces activation of NF-κB in the early phase, consequent UPR may inhibit NF-κB-dependent cellular activation in the later phase. This article summarizes current knowledge on potential mechanisms underlying the biphasic, bidirectional regulation of NF-κB by ER stress. Antioxid. Redox Signal. 11, 2353–2364.
Introduction
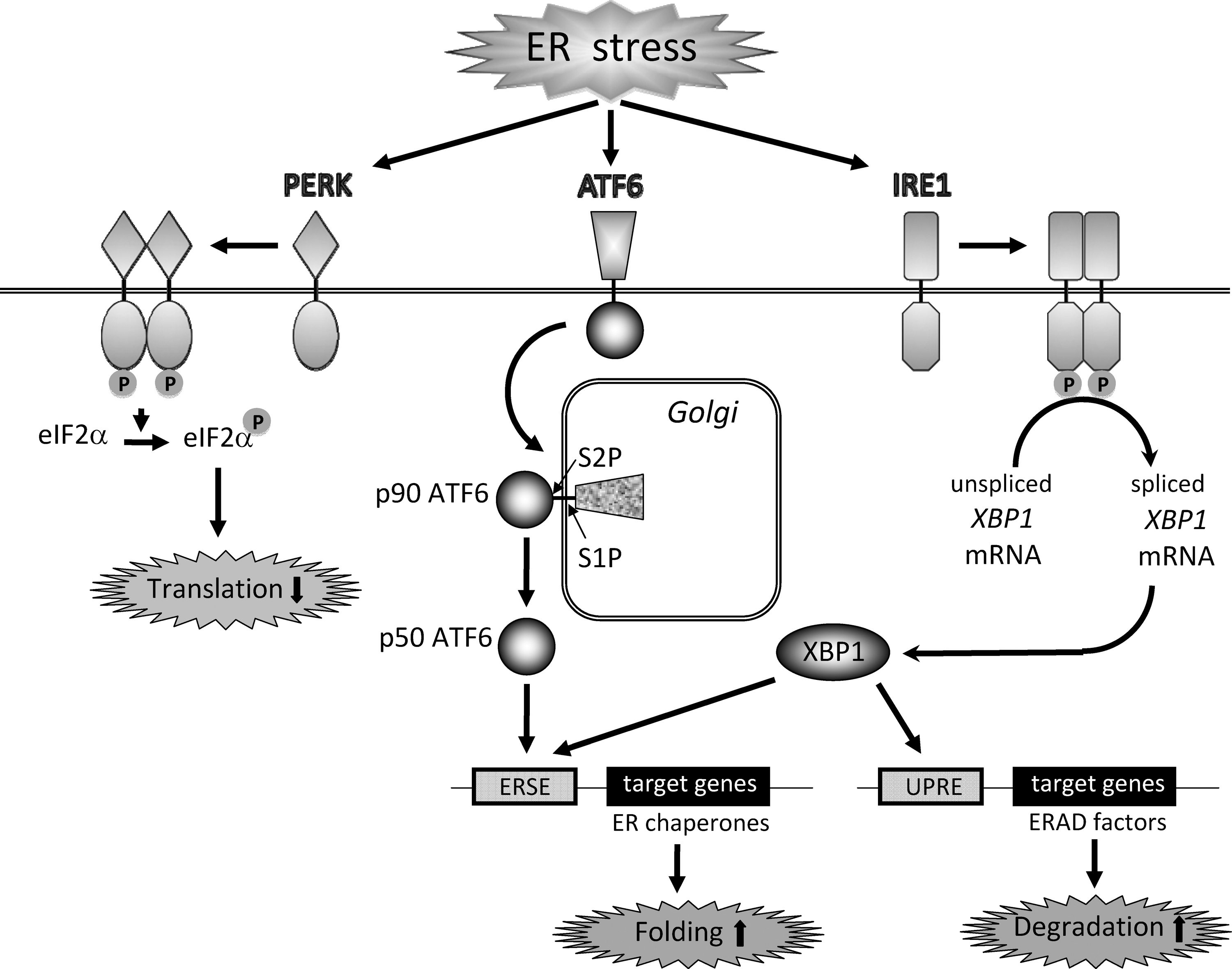
Endoplasmic reticulum (ER) stress is defined as accumulation of unfolded or misfolded proteins in the ER, which triggers an adaptive program called the unfolded protein response (UPR). The UPR alleviates ER stress by suppression of protein synthesis, facilitation of protein folding via induction of ER chaperones, and reinforced degradation of unfolded proteins (71). Three major transmembrane transducers for sensing ER stress have been identified in the ER. Those are RNA-dependent protein kinase-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring ER-to-nucleus signal kinase 1 (IRE1). Activation of PERK leads to phosphorylation of eukaryotic translation initiation factor 2α (eIF2α), which causes general inhibition of protein synthesis. In response to ER stress, p90ATF6 transits to the Golgi where it is cleaved by the proteases S1P and S2P, yielding a free cytoplasmic domain (p50ATF6) which functions as an active transcription factor. Similarly, activated IRE1 catalyzes removal of a small intron from X-box-binding protein 1 (XBP1) mRNA. This splicing event creates a translational frameshift in XBP1 mRNA to produce an active transcription factor. p50ATF6 and XBP1 subsequently bind to the ER stress response element and the UPR element, leading to expression of target genes including an ER chaperone 78 kDa glucose-regulated protein (GRP78) and genes involved in ER stress-related degradation (71) (Fig. 2).
ER stress is implicated in a wide range of pathologies including infection, ischemic injury, neurodegenerative disorders, and metabolic diseases such as diabetes mellitus (3). Several recent reports also suggested that ER stress is induced under inflammatory situations. For example, systemic inflammation caused by lipopolysaccharide (LPS) resulted in ER stress in various organs in mice (30). The UPR is induced in some types of glomerular diseases (38, 44), chronic intestinal inflammation (73), and autoimmune disorders (67, 57, 94). These data indicate a possible link between ER stress and inflammatory events. Indeed, recent investigations suggest the potential of ER stress and UPR for regulating NF-κB activity positively or negatively. This article summarizes current knowledge on molecular mechanisms underlying the bidirectional regulation of NF-κB by ER stress during inflammation.
Activation of NF-κB by ER Stress
Pahl and Baeuerle reported that ER stress has the potential to induce activation of NF-κB. They found that various ER stress inducers including tunicamycin, brefeldin A, 2-deoxyglucose, and thapsigargin increased DNA binding activity of NF-κB as well as κB-dependent gene expression (61). Thereafter, several investigators reported molecular mechanisms involved in the activation of NF-κB by ER stress (Fig. 3).
IRE1 pathway and tumor necrosis factor receptor-associated factor 2 (TRAF2)
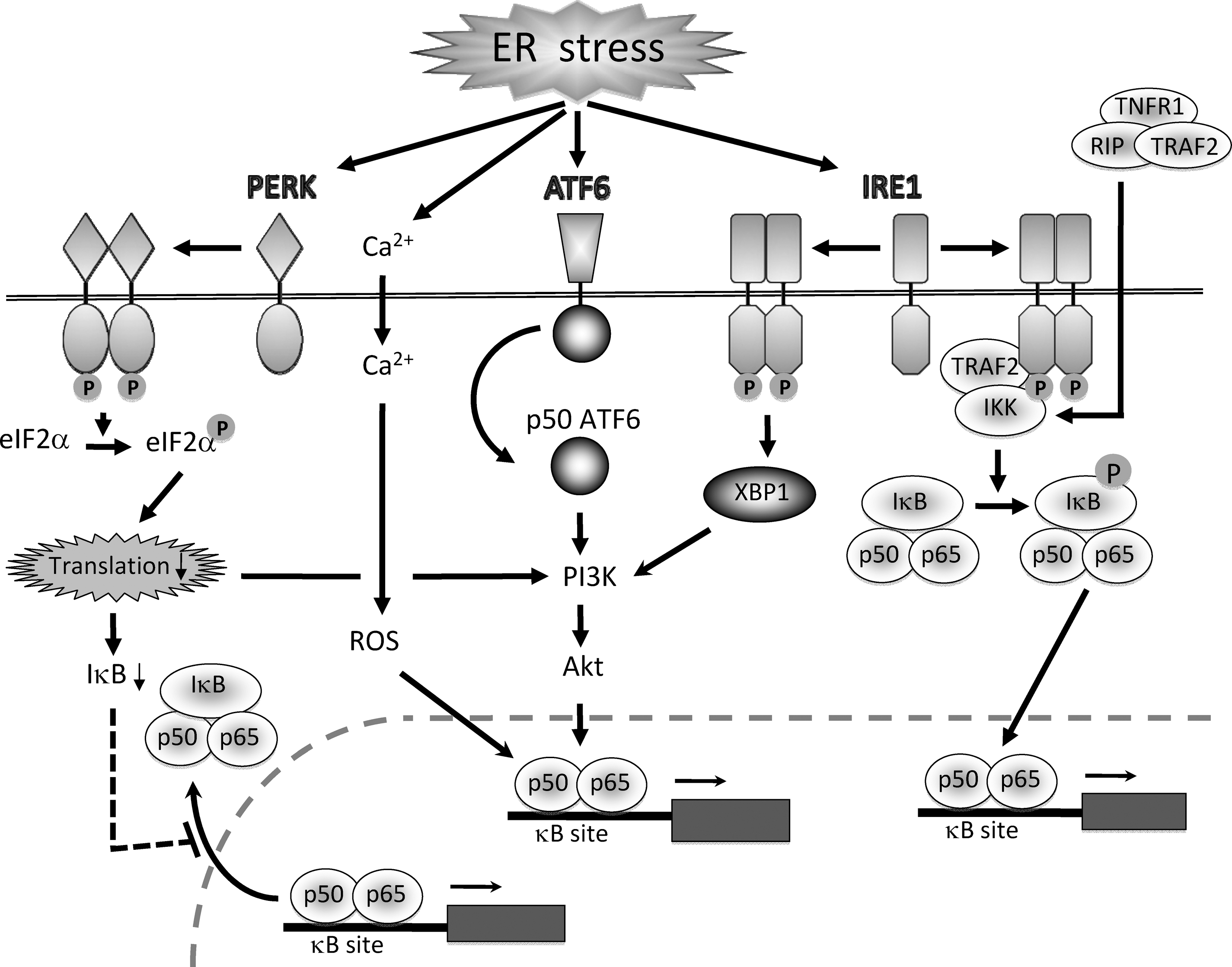
Kaneko et al. investigated involvement of the IRE1 pathway in the activation of NF-κB by ER stress. They found that transfection with a kinase-defective, dominant-negative mutant of IRE1 inhibited thapsigargin-induced, but not tumor necrosis factor-α (TNF-α)-triggered activation of NF-κB (41). Because the cytosolic effector domain of IRE1 interacts with the C-terminus of TRAF2, they also examined involvement of TRAF2 in ER stress-induced NF-κB activation. They found that transfection with a dominant-negative mutant of TRAF2 effectively suppressed thapsigargin-induced activation of NF-κB (41). They concluded that IRE1 and TRAF2 are required for the activation NF-κB by ER stress. The crucial roles of IRE1 and TRAF2 were also confirmed by Hu et al. (33). They found that: a) tunicamycin- or thapsigargin-induced NF-κB activation was impaired in IRE1α knockdown cells and IRE1α–/– cells, b) reconstitution of the IRE1α–/– cells with IRE1α cDNA recovered ER stress-induced activation of NF-κB, c) knockdown of TRAF2 inhibited ER stress-induced IκBα degradation and NF-κB activation, d) under ER stress, IRE1 formed a complex with IKK through TRAF2, and e) kinase activity of IRE1 was essential for activating the IKK complex and consequent phosphorylation and degradation of IκBα.
PERK–eIF2α pathway
Jiang et al. reported that PERK-induced phosphorylation of eIF2α is essential for activation of NF-κB by ER stress (40). They first showed that PERK–/– cells exhibited lack of NF-κB activation in response to thapsigargin. They also showed that cells harboring a mutant eIF2α showed blunted activation of NF-κB in response to tunicamycin and thapsigargin. Phosphorylation of eIF2α by GCN2, another eIF2α kinase, also triggered activation of NF-κB. Interestingly, unlike cytokine-induced NF-κB activation, PERK/eIF2α-mediated activation of NF-κB occurred independent of IκB degradation. It is different from the IRE1/TRAF2-mediated NF-κB activation that requires phosphorylation of IKK and IκBα. In agreement with this finding, Deng et al. reported: a) phosphorylation of eIF2α was necessary and sufficient to activate NF-κB, and b) eIF2α phosphorylation was correlated with a decrease in the level of IκBα protein without affecting its phosphorylation and stability. They identified that repression of IκBα translation was important in the decreased IκBα level and NF-κB activation triggered by eIF2α (17). The direct role of eIF2α in the translational suppression of IκBα was also reported by Wu et al. (90).
ATF6 and Akt pathway
In the absence of ER stress, PERK, ATF6, and IRE1 associate with GRP78, and this interaction keeps these transducers in a silent state. In contrast, under ER stress conditions, GRP78 preferentially associates with unfolded proteins instead of these stress sensors, resulting in activation of their downstream signaling cascades for UPR (5). AB5 subtilase cytotoxin (SubAB), discovered in Shiga toxigenic Escherichia coli, rapidly and selectively degrades GRP78 by cleavage between two leucine residues at positions 416 and 417 and induces UPR (65, 89). We found that treatment of renal tubular cells with SubAB caused ER stress and transient activation of NF-κB. This activation of NF-κB was preceded by transient phosphorylation of Akt, and treatment of the cells with selective inhibitors of Akt attenuated SubAB-induced NF-κB activation (Yamazaki H. et al., unpublished observations). SubAB triggered three major branches of the UPR, and only the kinetics of ATF6 activation was correlated with the transient activation of Akt and NF-κB. Indeed, genetic and pharmacological inhibition of ATF6 significantly suppressed SubAB-triggered Akt phosphorylation and NF-κB activation (Yamazaki H. et al., unpublished observations). These results indicate a possible role of the ATF6–Akt pathway in the activation of NF-κB by UPR.
Activation of Akt by ER stress was also reported by other investigators. Hosoi et al. reported that ER stress caused by tunicamycin or thapsigargin upregulated phosphorylation of Akt in glial cells (31). They also observed that inhibitors of phosphatidylinositol 3-kinase (PI3K) attenuated ER stress-induced Akt activation. Currently, it is not fully elucidated how Akt is activated by ER stress, but recent reports indicated possible involvement of IRE1–XBP1 and PERK–eIF2α. Using zebrafish embryonic cells, Hu et al. reported that XBP1 located downstream of IRE1 may have the potential to induce phosphorylation of Akt. They showed that Akt phosphorylation was caused in cells stably overexpressing the spliced form of XBP1 (32). Another report by Kazemi et al. also indicated a role of the PERK pathway for activation of Akt. They showed that an active form of PERK acted upstream of PI3K and turned on the Akt pathway through inhibition of protein synthesis by eIF2α. They also showed that induction of the PI3K pathway was impaired in PERK-/- cells in response to various stimuli that activate PERK (43). In contrast to these data, we failed to get supportive evidence for involvement of the IRE1 and PERK pathways in the activation of Akt in SubAB-stimulated renal tubular cells (Yamazaki H. et al., unpublished observations). The reason for the discrepancy is currently unclear, but underlying mechanisms might be different from trigger to trigger, and/or cell type to cell type.
Calcium and reactive oxygen species (ROS)
Pahl and Baeuerle reported roles of Ca2+ and ROS in the activation of NF-κB by ER stress (62). Using Ca2+ chelators and antioxidants, they showed that activation of NF-κB by ER stress required an increase in intracellular levels of Ca2+ and ROS. ER-resident Ca2+-ATPase inhibitors that trigger a rapid efflux of Ca2+ from the ER activated NF-κB, and pretreatment with a Ca2+ chelator abrogated this process. The Ca2+ chelator inhibited generation of ROS in response to thapsigargin, suggesting that an increase in Ca2+ precedes ROS formation during NF-κB activation. They also indicated that peroxidase activity of cyclooxygenases or lipoxygenases may be responsible for the ROS production by ER stress (62).
TNF receptor 1 (TNFR1)
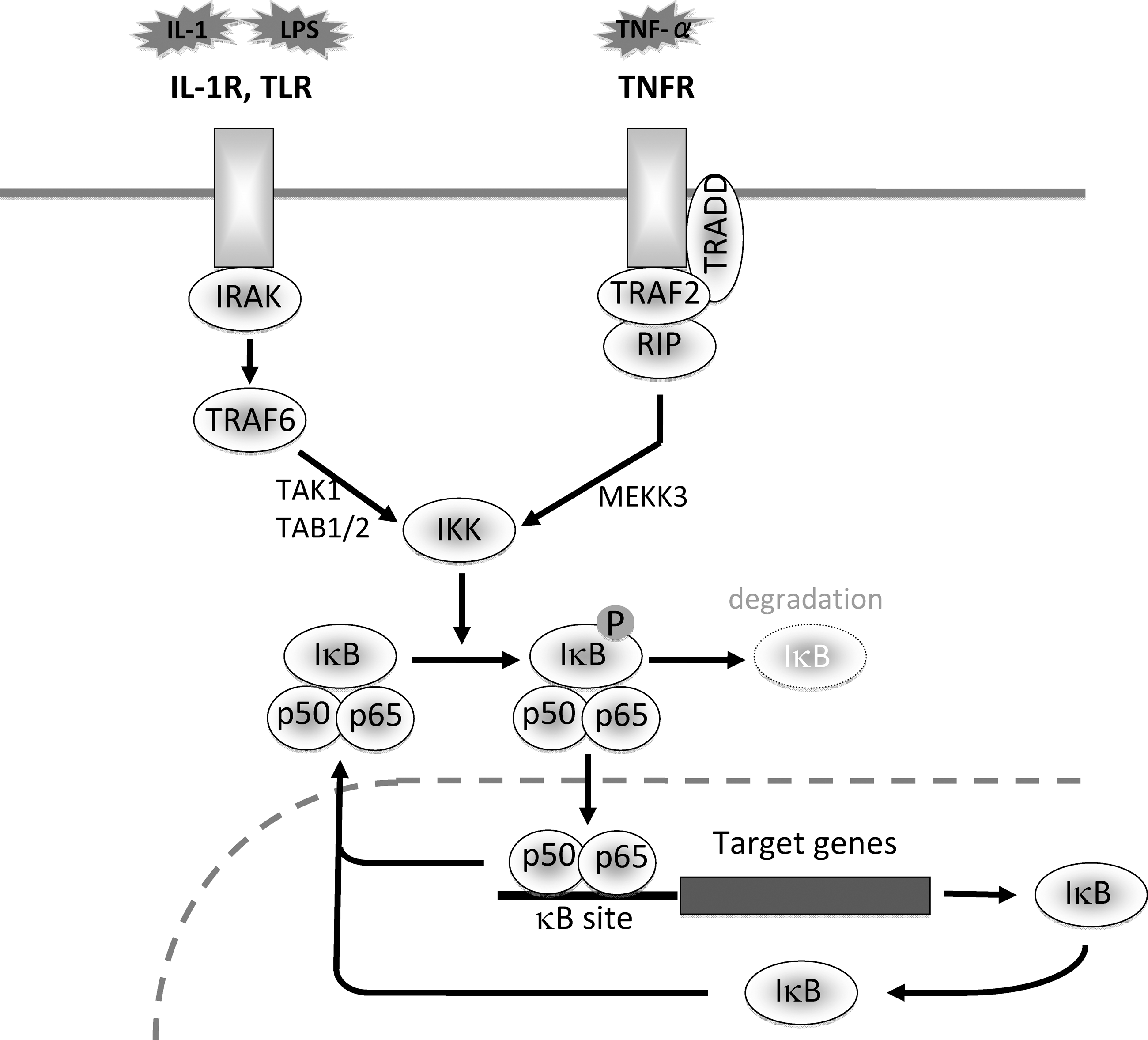
Activation of NF-κB by ER stress may occur independently of the three major branches of UPR. Devin et al. reported that, under ER stress conditions, TNFR1 was accumulated in the ER and formed a complex with TRAF2 and TNF receptor-interacting protein (RIP), a crucial step for activation of NF-κB by TNF-α. This molecular event may lead to activation of IKK even in the absence of TNF-α (18, 95).
Suppression of NF-κB by ER Stress
As described, several investigators reported the potential of ER stress to activate NF-κB. Because of this reason, the proinflammatory aspect of ER stress has been emphasized to date (63). However, we recently reported that preceding ER stress may blunt subsequent activation of NF-κB by inflammatory cytokines. For example, in glomerular podocytes and mesangial cells, expression of NF-κB-dependent genes [monocyte chemoattractant protein 1 (MCP-1) and inducible nitric oxide (NO) synthase (iNOS)] by TNF-α is abrogated by the pretreatment with UPR-inducible agents K-7174 and geranylgeranylacetone (GGA) (25, 78). Induction of UPR by other ER stress inducers also reproduces the suppressive effects of K-7174 and GGA on NF-κB and NF-κB-dependent gene expression (24). These results indicate a possibility that, although ER stress triggers activation of NF-κB in the early phase, consequent UPR has the potential to inhibit activation of NF-κB by inflammatory stimuli in the later phase. This ER stress-induced anergy is reversible and is not due to cellular damage (24). Currently, molecular mechanisms involved in this negative regulation of NF-κB by UPR have not been fully elucidated, but several possibilities can be postulated (Fig. 4).
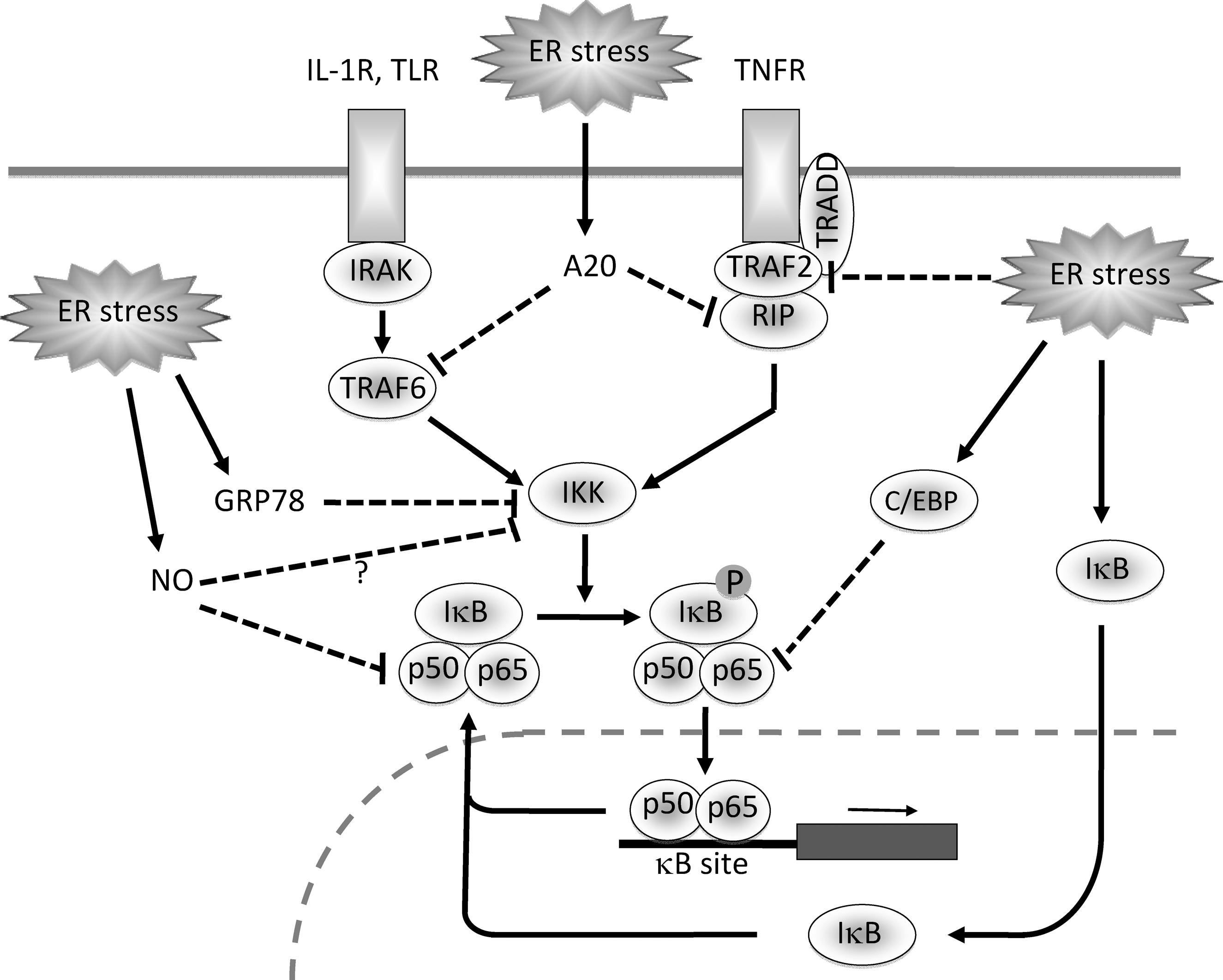
Downregulation of TRAF2
In the TNF signaling pathway, TNFR1, TNFR1-associated death domain (TRADD), RIP, TRAF2, and IKK are essential for NF-κB activation (Fig. 1). First, receptor-bound TRADD recruits TRAF2 and RIP into the TNFR1 signaling complex. TRAF2 then recruits IKK into the TNFR1 complex, and RIP induces kinase activity of IKK (85). Recently, Hu et al. reported that, in thapsigargin- or tunicamycin-treated MCF-7 cells and L929 cells, TNFR1, TRADD, and RIP proteins were maintained at the same levels as those in untreated controls, whereas the level of TRAF2 protein was selectively decreased. The decrease in TRAF2 was not due to transcriptional suppression or increased turnover of mRNA but due to enhanced protein degradation (33). The downregulation of TRAF2 by ER stress was not restricted to the particular tumor cells and may be a more general phenomenon. To our experience, the similar suppression of TRAF2 by ER stress was also observed in glomerular mesangial cells and podocytes (24, 59). Currently, it is not determined whether and how individual branches of UPR contribute to the selective degradation of TRAF2.
Induction of CCAAT/enhancer-binding protein (C/EBP)
C/EBP is a family of transcription factors that are required for development, function, and responses to injury of various tissues (52). C/EBP consists of an activation domain, a dimerization bZIP region and a DNA-binding domain. All family members share the highly conserved dimerization domain by which they form homodimers or heterodimers with other family members, and dimerization of C/EBP proteins precisely modulates transcriptional activity of target genes (52). A previous report by Chen et al. showed that ER stress inducers triggered expression and production of C/EBPβ in human hepatoma cells (10). ER stress is known to induce active XBP1 that binds to and activates the UPR element, leading to expression of target genes (71). They found that a putative UPR element was located at the 3' end of the human C/EBPβ gene and that it was responsible for the induction of C/EBPβ by ER stress. They also showed that overexpression of XBP1 caused an increase in transcription mediated by the UPR element in the C/EBPβ gene promoter (10). Consistent with this result, our data showed that, among C/EBP family members, C/EBPβ was preferentially induced by ER stress in several cell types. Although the magnitude was much less, expression of C/EBPα and C/EBPδ was also induced under ER stress conditions (45, and our unpublished data).
It is known that C/EBP family members interact with NF-κB subunits (74) and can inhibit cytokine-induced activation of NF-κB (8, 86). The inhibitory effect of C/EBP on NF-κB may be mediated by protein–protein interactions distinct from the role of C/EBP in gene expression (55). Indeed, Prösch et al. reported that C/EBPα/β interacted with NF-κB p65 subunit and suppressed TNF-α-induced NF-κB activation, even in the absence of direct DNA binding of p65-C/EBP-containing protein complexes to the κB site (66). A recent report by Zwergal et al. (100) also showed that, in monocytic cells, C/EBPβ inhibited activation of NF-κB by TNF-α without affecting its binding to the C/EBP site. They found that, in TNF-α-tolerant cells, TNF-induced phosphorylation of p65 was markedly suppressed, and it was accompanied by formation of C/EBPβ-p65 complexes. In C/EBPβ-/- cells, neither impairment of NF-κB-dependent transcription nor inhibition of p65 phosphorylation was observed. Furthermore, overexpression of C/EBPβ reduced p65 phosphorylation and p65-mediated transactivation. They concluded that C/EBPβ is an essential signaling component for inhibition of NF-κB-mediated transcription in TNF-α-tolerant cells (100).
Taken together, these findings suggest a role of C/EBPβ in the blunted activation of NF-κB in the cells under ER stress conditions. Of note, like C/EBPβ, overexpression of C/EBPα, C/EBPδ, and C/EBPζ may also attenuate activation of NF-κB by TNF-α (45, and our unpublished data). The fact that all of these C/EBP family members had the similar potential indicates that the highly conserved basic region (68) may be required and sufficient for the suppressive effect of C/EBP on NF-κB.
Induction of A20 and IκBα
NF-κB activation triggered by TNF-α is under complex regulation whose different phases lead to distinct gene expression programs. In the regulation of NF-κB, two negative feedback regulators, A20 and IκBα, play crucial but distinct roles (87). Previous reports showed that overexpression of A20 inhibited activation of NF-κB and that, in A20-deficient cells, prolonged activation of NF-κB was observed in response to TNF-α (6, 51). A20-deficient mice developed severe multi-organ inflammation and were susceptible to sublethal doses of TNF-α (51). A20 acts first as a de-ubiquitinating enzyme, which removes Lys63-linked ubiquitin chains from RIP. Next, A20 functions as a ubiquitin ligase by polyubiquitinating RIP with Lys48-linked ubiquitin chains and thereby targets RIP to proteasomal degradation (28, 88). The degradation of RIP results in failure of the TNF signaling. Interestingly, the inhibitory effect of A20 on NF-κB is observed not only in TNF-α-stimulated cells but also in IL-1β-exposed cells, possibly via interfering with TRAF6 that is required for the IL-1 signaling (27).
Recently, we found that A20 is induced in cells under ER stress conditions. In thapsigargin-treated mesangial cells, the induction of A20 was observed within 3 h, which was in parallel with the induction of GRP78. The induction of A20 via ER stress was triggered not only by thapsigargin but also by other ER stress inducers. The similar effect was also observed in tubular epithelial cells, suggesting that the induction of A20 by ER stress is a general phenomenon (24). Using a reporter assay, we also showed that transfection with A20 markedly suppressed activation of NF-κB in TNF-α-stimulated mesangial cells, and that knockdown of A20 by A20 microRNA modestly but significantly attenuated the induction of anergy by ER stress in IL-1β- or TNF-α-stimulated cells (24). These results indicated partial involvement of A20 in the blunted activation of NF-κB under ER stress conditions. The fact that A20 interferes with the cytokine signaling at RIP and TRAF6 levels is consistent with our finding that the targets of ER stress in the NF-κB pathway are located upstream of IKK (24). Currently, it is unclear how A20 is induced by ER stress, but a previous report showed that expression of A20 is regulated by NF-κB (47). Because ER stress induces activation of NF-κB in the acute phase, the induction of A20 by ER stress may be caused by the early, transient activation of NF-κB through the IRE1/TRAF2, PERK–eIF2α and/or ATF6–Akt pathways, as described in the section of Activation of NF-κB by ER Stress.
Active export of NF-κB from the nucleus by IκBα is another major mechanism for the suppression of NF-κB (1). After NF-κB activation, IκBα is induced at the transcriptional level. The synthesized IκBα protein enters the nucleus and shuttles NF-κB back to the cytoplasm, leading to subsidence of NF-κB activation (2) (Fig. 1). However, currently, there are no reports on the role of IκBα in the negative regulation of NF-κB by ER stress, and further investigation will be required to clarify this issue.
Other possibilities: Roles of GRP78 and NO
GRP78, also referred to as BiP, is an ER chaperone and a central regulator of ER function via protein folding and assembly, targeting unfolded protein for degradation, ER Ca2+-binding and controlling ER stress transducers including PERK, ATF6, and IRE1 (50). GRP78 is the most well-known multifunctional molecule induced by ER stress. In some cell types, GRP78 might be important in the suppression of NF-κB. We recently found that, in murine podocytes, acute ablation of GRP78 by SubAB caused transient activation of NF-κB. Furthermore, transient transfection with GRP78 significantly inhibited activation of NF-κB by TNF-α (59). This result indicates possible involvement of GRP78 in the induction of anergy by ER stress in certain cell types, although some exceptions may exist (24, 45). In some situations, GRP78 could attenuate cytokine responses through different mechanisms (e.g., via secretion of GRP78). For example, previous reports showed that GRP78 may be secreted, stimulate expression of anti-inflammatory molecules including soluble TNFR2 (p75-TNFR) and IL-1 receptor antagonist and thereby attenuate cytokine responses. This mechanism is possibly involved in the attenuation of inflammation including experimental collagen arthritis (11, 16, 64). Secretion of GRP78 caused by ER stress could contribute to blunted responses to inflammatory stimuli. Using mesangial cells, we also found that GRP78 is secreted into the extracellular space under ER stress conditions. However, we failed to get supportive evidence for involvement of extracellular GRP78 in the blunted response to TNF-α (24). Furthermore, a previous report also indicated a possibility that, under inflammatory situations, GRP78 might be involved in the activation of NF-κB by TNF-α via binding to IKK (73). Regulatory roles of GRP78 in the cytokine responses seem to be complicated, and further investigation will be required to determine exact roles of GRP78 during individual inflammatory contexts.
A previous report suggested that NO is induced in pancreatic β cells under ER stress conditions (46). A number of reports demonstrated the potential of NO for negative regulation of NF-κB, as reviewed by Janssen–Heininger et al. (39). It is believed that NO is involved in a negative feedback loop to block prolonged activation of NF-κB. The inhibitory effect of NO on NF-κB may occur through nitrosylation at cysteine 62 of p50 subunit, leading to prevention of NF-κB from binding to the κB site (15, 54). IKK may also be a potential target of NO, because multiple cysteines are present in the kinase domain of IKK (69). Based on these, NO generated under ER stress conditions could be involved in the acquisition of anergy to inflammatory stimuli.
Regulation of Inflammation by ER Stress
Evidence for UPR during inflammation
ER stress is induced under various inflammatory situations, as summarized in Table 1. We previously reported that systemic inflammation caused by LPS in mice resulted in UPR in several organs including lung, liver, kidney, and spleen (30). During the process of inflammation caused by a wide range of viruses (hepatitis B virus, hepatitis C virus, hepatitis D virus, flavivirus, Borna disease virus, murine leukemia virus, and Moloney murine leukemia virus), ER stress may also be induced because of production of viral proteins (98). Shkoda et al. reported that expression of GRP78 was induced in intestinal epithelial cells of mice suffered from chronic inflammation. They also showed that, in intestinal cells from patients with inflammatory bowel diseases (ulcerative colitis and Crohn's disease), the level of GRP78 elevated. Epithelial cells from patients with sigmoid diverticulitis similarly exhibited an increase in GRP78 protein, suggesting that ER stress responses may be a part of the inflammatory process (73). Suyama et al. reported that ER stress–C/EBP homologous protein (CHOP) pathway had a pivotal role in the acceleration of experimental pancreatitis via induction of caspases and IL-1β (75). UPR may also be involved in the pathogenesis of glomerulonephritis. Cybulsky et al. showed that glomerular GRP78 and GRP94 proteins were upregulated in proteinuric rats with passive Heymann nephritis and that ER stress preconditioning of the rats reduced proteinuria (13). The same group also showed that phosphorylation of PERK and eIF2α was enhanced in glomeruli of rats with Heymann nephritis (12). Inagi et al. reported that, in the anti-Thy1 model of mesangioproliferative glomerulonephritis in rats, expression of ER chaperones GRP78 and 150 kDa oxygen-related protein was upregulated in glomeruli, especially in podocytes and mesangial cells. They also showed that preconditioning with subnephritogenic doses of ER stress inducers ameliorated the pathological manifestations of the disease (38). Several investigators also reported that GRP78 was highly expressed in synovial tissues from rheumatoid arthritis patients and in chondrocytes from osteoarthritis patients, suggesting involvement of ER stress in articular inflammation (7, 67, 70, 94). ER stress and UPR may also be involved in autoimmunity, including autoimmune myositis. Nagaraju et al. reported increased expression of GRP78, CHOP, PERK, and ATF3 in muscles from myositis patients and in a mouse model of myositis. Activation of caspase 12, a critical mediator of ER stress-induced apoptosis, was also observed in this myositis model (57).
Potential triggers of ER stress during inflammation
A number of pathophysiological factors may lead to accumulation of unfolded proteins in the ER. Those include hypoxia, nutrient (glucose) deprivation, alterations in the redox balance, changes in calcium homeostasis, failure of post-translational modifications and increases in protein synthesis (50). Currently, inflammatory stimuli or mediators responsible for the induction of ER stress are unclear, but several putative factors have been proposed (Table 2).
Cytokines
Zhang et al. (99) previously reported that, after in vivo injection of IL-6 or IL-1β, expression of GRP78 and CHOP was induced in the liver of mice. Moreover, levels of spliced XBP1 mRNA and mRNA encoding EDEM, an ER degradation-enhancing α-mannosidase-like protein that is regulated by the UPR, were also upregulated in the liver upon IL-6 or IL-1β stimulation (99). Xue et al.(93) showed that, in fibrosarcoma cells, TNF-α induced a) phosphorylation of eIF2α and consequent expression of growth-arrest and DNA-damage-inducible protein 34 mRNA, b) induction of p50ATF6, and c) accumulation of the spliced form of XBP1 mRNA and XBP1 protein, indicating that TNF-α activated the PERK, ATF6, and IRE1 signaling pathways (93). These results suggested that major inflammatory cytokines have the potential to induce ER stress and UPR. Currently, molecular mechanisms involved are not well understood, but cytokines, especially TNF-α, may induce ER stress via generation of ROS (93).
ROS
ROS, abundantly produced by activated leukocytes, serve as signaling molecules for inflammation and determine the pattern of gene expression and inflammation phenotypes through ROS-sensitive transcription factors (49). Recent reports suggested that ER stress and oxidative stress are closely linked with each other (53). We found that cadmium and cigarette smoke induced ER stress and that oxidative stress, especially generation of O2•-, was located upstream of ER stress in cadmium-exposed renal cells and cigarette smoke-exposed pulmonary cells (77, 96, 97). We also reported that ROS, including O2•- and ONOO-, caused ER stress in renal tubular cells (97). Consistent with this result, some previous reports indicated that ER stress may be involved downstream of oxidative stress. For example, the anticancer agent geldanamycin caused expression of GRP78 via a ROS-dependent mechanism (48). In vascular endothelial cells, ONOO- caused increases in the levels of GRP78 and GRP94 proteins (19). In the ischemic brain, induction of ATF4 and CHOP was attenuated in mice and rats overexpressing superoxide dismutase (26). These results suggest that oxidative stress is located upstream of ER stress under various pathophysiological contexts. Currently, it is not fully elucidated how ROS induce ER stress. Previous reports showed that oxidative stress caused inhibition of Ca2+-ATPase (42, 56), a known trigger of ER stress. One possibility is, therefore, that ROS may trigger depletion of calcium store in the ER via inhibition of Ca2+-ATPase (83). Another possibility is that ROS might cause ER stress through generation and accumulation of oxidatively modified, abnormal proteins. Unfolded proteins might also be accumulated in the ER through ROS-induced functional perturbation of ER foldases and/or chaperones (9). Interestingly, Hung et al. reported that ER stress-preconditioning in renal tubular cells conferred resistance to H2O2-induced injury (34).
NO
An excess amount of NO is produced by iNOS, and induction of iNOS is observed in inflammatory diseases (35). It is well known that NO has the potential to induce ER stress, as reviewed by Gotoh and Mori (22). For example, treatment of pancreatic β cells with a NO donor, S-nitroso-N-acetyl-d,l-penicillamine (SNAP), causes expression of CHOP (60). In macrophages treated with LPS plus interferon-γ, generated NO triggers ER stress responses including induction of ATF6 and CHOP (23). Oyadomari et al. reported a) SNAP increased cytosolic Ca2+, b) agents depleting ER Ca2+ induced CHOP expression, and c) calreticulin, a Ca2+-binding ER chaperone, increased Ca2+ content in the ER and protected cells against NO-induced apoptosis (60). NO may induce ER stress via depletion of Ca2+ in the ER. NO inhibits activity of sarcoendoplasmic reticulum Ca2+-ATP-ase (SERCA) by tyrosine nitration within the channel-like domain (84, 91). NO also increases activity of RyR1 and RyR2, the calcium release channel ryanodine receptors, through S-nitrosylation (92). It is therefore speculated that NO depletes ER Ca2+ either by inhibiting Ca2+ uptake from cytosol through SERCA or by facilitating Ca2+ release to cytosol through RyRs.
Complement
C5b and the other late-acting complement components can assemble the two terminal complexes C5b-9 and SC5b-9 and play important roles in several pathological conditions such as immune-mediated inflammation. In addition to the lytic effects of C5b-9, sublytic amounts of C5b-8 or C5b-9 can stimulate cellular activities under pathological conditions (14). Passive Heymann nephritis is a model of membranous nephropathy in humans. In this model, complement C5b-9 induces injury of glomerular podocytes, resulting in proteinuria. Using cultured podocytes, Cybulsky et al. reported that exposure of the cells to C5b-9 directly increased GRP78 and GRP94 mRNAs and proteins. Knockdown of GRP78 via antisense GRP78 enhanced complement-dependent injury of podocytes (13). They also showed that complement induced phosphorylation of PERK and eIF2α in podocytes, and that fibroblasts from PERK-deficient mice were more susceptible to complement-triggered, ER stress-dependent cytotoxicity (12).
Biphasic, bidirectional regulation of NF-κB by ER stress
Implication for pathophysiology
As described, previous reports showed that ER stress triggers activation of NF-κB.
The mechanisms underlying this phenomenon involve TRAF2 and the IRE1 pathway, the PERK - eIF2α pathway, the ATF6 - Akt pathway, and Ca2+ and ROS, as was described in the section of Activation of NF-κB by ER Stress. On the other hand, ER stress and consequent UPR also have the potential to inhibit NF-κB via downregulation of TRAF2 and/or induction of C/EBP, A20, IκBα, GRP78, and NO. Using a selective inducer of ER stress, SubAB, we have evidence that treatment of cells with SubAB causes ER stress and acute activation of NF-κB, whereas this activation is transient and subsided within 24 h (Yamazaki H. et al., unpublished observations). Thereafter, both basal and cytokine-inducible activation of NF-κB are suppressed without cellular damage (59, and our unpublished results). These data evidence biphasic, bidirectional regulation of NF-κB by ER stress. That is, although ER stress activates NF-κB in the early phase, consequent UPR suppresses cellular responses to subsequent inflammatory stimuli in the later phase. The proinflammatory and anti-inflammatory aspects of ER stress may be important both in the initiation of inflammation and in its spontaneous subsidence.
Implication for pharmacological effects
Recent reports suggested that some anti-inflammatory agents have the potential to induce ER stress.
Such pharmacological effects are generally considered as a mechanism underlying adverse effects of those agents. However, induction of subtoxic levels of UPR may also be involved in the anti-inflammatory potential of those agents via inhibition of NF-κB.
Non-steroidal anti-inflammatory drugs (NSAIDs)
NSAIDs have been widely used for the relief of inflammation. The anti-inflammatory potential of NSAIDs is primarily due to their ability to inhibit cyclooxygenase enzymes involved in the production of proinflammatory prostaglandins (20). However, several studies demonstrated that certain NSAIDs may exert anti-inflammatory effects independent of cyclooxygenase activity and prostaglandin synthesis (79). Recently, we found that, in murine podocytes, expression of MCP-1 in response to TNF-α was suppressed by indomethacin. This anti-inflammatory potential was correlated with induction of GRP78 and CHOP. The induction of UPR by indomethacin was observed similarly in other cells including mesangial cells and tubular epithelial cells (59). In TNF-α-treated cells, suppression of MCP-1 by indomethacin was inversely correlated with induction of UPR, and inducers of UPR reproduced the suppressive effect. Reporter assays showed that indomethacin as well as thapsigargin attenuated activation of NF-κB by TNF-α, and it was associated with enhanced degradation of TRAF2 (59). Previous reports showed that certain NSAIDs other than indomethacin may also cause UPR including induction of GRP78 (80, 81). Induction of UPR and consequent inhibition of NF-κB may be a more general phenomenon observed in NSAIDs.
Calcineurin inhibitors
Immunosuppressive agents cyclosporine A (CsA) and tacrolimus (FK506) inhibit cytokine production by activated lymphocytes through interfering with calcineurin and are widely used to avoid transplant rejection and to treat autoimmune diseases (72). However, little is known about their effects on the function of nonlymphoid cells. We found that, in renal tubular cells, induction of MCP-1 by inflammatory cytokines was blunted by CsA and FK506. This suppression was correlated with induction of UPR, which was reversible and observed generally in nonimmune cells. Administration with CsA in mice caused rapid, systemic induction of UPR in vivo (45). In TNF-α-treated cells, suppression of MCP-1 by CsA or FK506 was associated with blunted responses of NF-κB. The suppression of NF-κB was reproduced by other inducers of UPR. CsA and FK506, as well as other UPR inducers, caused upregulation of C/EBP family members, especially C/EBPβ and CHOP, and overexpression of either C/EBPβ or CHOP significantly attenuated TNF-α-triggered NF-κB activation. Furthermore, downregulation of C/EBPβ by small interfering RNA partially reversed the suppressive effect of CsA on TNF-α-induced expression of MCP-1 (45). These results suggest that calcineurin inhibitors confer anergy to inflammatory stimuli on resident cells through UPR-dependent induction of the C/EBP family members.
Other anti-inflammatory agents
GGA, also known as teprenone, has been used in clinics for the treatment of gastric ulcer and gastritis. However, recent investigation revealed the potential of this compound as a general cytoprotective agent. It is currently believed that GGA induces 70 kDa heat shock protein (HSP70) and thereby protects various cells from apoptosis (29, 36). GGA also possesses the anti-inflammatory potential. For example, GGA attenuates experimental colitis and ischemia-triggered renal inflammation (58, 76). However, it is currently unclear whether these therapeutic effects are ascribed only to upregulation of HSP70. Recently, we reported novel potential of GGA to induce UPR. In mesangial cells, GGA triggered selective branches of UPR including the ATF6 pathway and the IRE1–XBP1 pathway (21). We also found that GGA blocked activation of NF-κB and consequent induction of MCP-1 by inflammatory cytokines. It was inversely correlated with induction of UPR, and inducers of ER stress reproduced the suppressive effects of GGA. Furthermore, attenuation of UPR by stable transfection with GRP78 diminished the anti-inflammatory effect of GGA (25). These results indicate a UPR-dependent mechanism underlying the anti-inflammatory potential of GGA.
K-7174, a GATA-specific inhibitor, is a putative anti-inflammatory agent that attenuates effects of inflammatory cytokines in certain cell types. A previous report showed that K-7174 has the potential to reduce expression of vascular cell adhesion molecule 1 in cytokine-stimulated endothelial cells (82). Another report also showed that, in human hepatoma cells, suppression of erythropoietin expression by IL-1β or TNF-α was reversed by K-7174 (37). We found that, in glomerular podocytes, induction of MCP-1 and iNOS by TNF-α was abrogated by K-7174. It was correlated with unexpected induction of UPR. In podocytes, induction of UPR reproduced the suppressive effect of K-7174. Furthermore, K-7174-elicited UPR abrogated induction of MCP-1 and iNOS not only by TNF-α but also by activated macrophages (78). These results suggest that a UPR-dependent mechanism may also be involved in the anti-inflammatory potential of K-7174.
Concluding Remarks
Accumulating evidence suggests roles of ER stress and UPR in a wide range of pathophysiologies. In particular, cellular activation and induction of cell injury by ER stress have been extensively investigated under various cellular contexts. However, ER stress/UPR has Janus faces. It is essential to disclose not only its dark side (pathological significance) but also its light side (physiological significance). In particular, opposite, bidirectional roles of ER stress in inflammatory processes should be further investigated in the future. In addition, the extent of anergy caused by ER stress is different from trigger to trigger. For example, in many cell types, TNF-induced activation of NF-κB is more susceptible to ER stress than IL-1-induced NF-κB activation. The mechanisms underlying such differences should also be elucidated in the future.
In this review, I focused only the regulation of NF-κB by ER stress. However, other signaling molecules involved in inflammatory processes may also be regulated by ER stress in a biphasic, bidirectional fashion. Recently, Hosoi et al. (31) investigated an effect of ER stress on the activation of Akt in glial cells. They found that Akt was phosphorylated by ER stress upon short-term exposure to tunicamycin or thapsigargin. However, they also observed that Akt phosphorylation was rather downregulated upon longer-term exposure to the ER stress inducers (31). The Janus faces of ER stress may be important in the dynamic, coordinated regulation of not only NF-κB but also other inflammation-related signaling molecules.