
Abstract
An imbalance in reducing and oxidizing (redox) systems favoring a more oxidative environment is present in asthma and linked to the pathophysiology of the defining symptoms and signs including airflow limitation, hyper-reactivity, and airway remodeling. High levels of hydrogen peroxide, nitric oxide (•NO), and 15-F2t-isoprostane in exhaled breath, and excessive oxidative protein products in lung epithelial lining fluid, peripheral blood, and urine provide abundant evidence for pathologic oxidizing processes in asthma. Parallel studies document loss of reducing potential by nonenzymatic and enzymatic antioxidants. The essential first line antioxidant enzymes superoxide dismutases (SOD) and catalase are reduced in asthma as compared to healthy individuals, with lowest levels in those patients with the most severe asthma. Loss of SOD and catalase activity is related to oxidative modifications of the enzymes, while other antioxidant gene polymorphisms are linked to susceptibility to develop asthma. Monitoring of exhaled •NO has entered clinical practice because it is useful to optimize asthma care, and a wide array of other biochemical oxidative and nitrative biomarkers are currently being evaluated for asthma monitoring and phenotyping. Novel therapeutic strategies that target correction of redox abnormalities show promise for the treatment of asthma. Antioxid. Redox Signal. 12, 93–124.
I. Introduction
II. Redox Reactions Form the Basis for Aerobic Life
Cellular respiration is the quintessential reduction–oxidation (redox) reaction in aerobic organisms. Cellular respiration takes place within the mitochondria and is fundamental for production of the energy that is required to maintain the ordered state of the cell. Hence, redox reactions form the basis for the most important physiologic process that takes place in healthy cells. Simply defined, oxidation is the loss of electrons and reduction is the gain of electrons. However, most oxidation reactions in cells are accomplished by the removal of hydrogen atoms. In cell respiration, glucose loses electrons in H atoms and serves as the electron donor, while oxygen is the terminal electron acceptor.
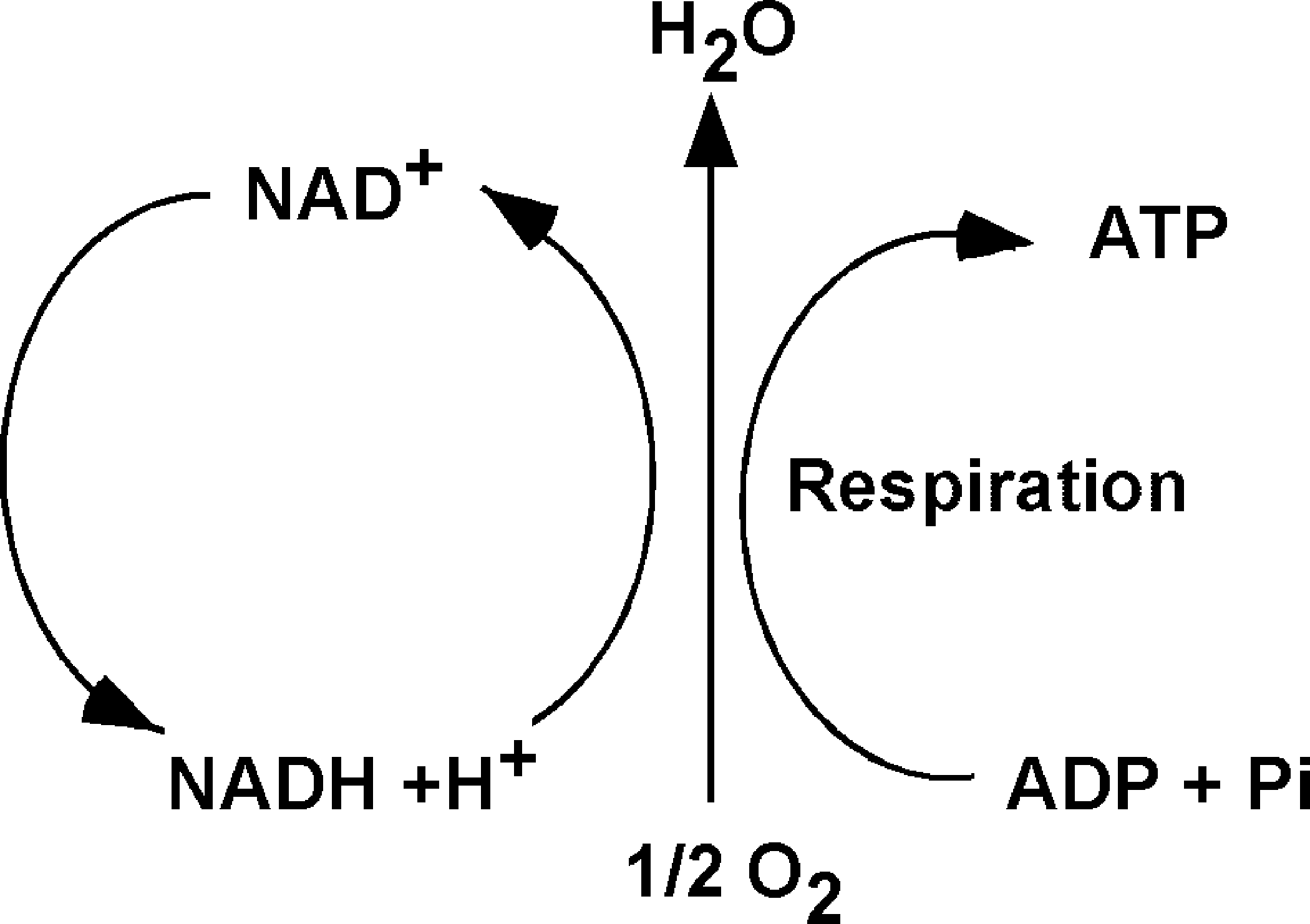
Generally, redox reactions are tightly regulated and occur in multiple steps, in which the electrons are shuttled by carriers, also called redox couples. Common redox couples include NAD+/NADH, NADP+/NADPH, and reduced to oxidized glutathione (GSH/GSSG) (Fig. 1). Chance et al. pioneered the study of oxidation and reduction states of proteins in the respiratory (electron transport) chain of various organs (50). Later, Bucher and co-workers developed experimental approaches to estimate the intracellular reduction potential by determining the ratio of NAD+/NADH and NADP+/NADP (32, 302). Subsequently, Buettner et al. suggested that the redox environment in cells, tissues, or in biological fluids might be defined by the reduction potential and reducing capacity of the redox couples present (33). In general, the ratio of the interconvertible oxidized and reduced form of a specific redox couple is used to define the redox environment in biologic systems (302).
III. Redox Systems in the Lung
Oxygen is one of the most abundant elements in our world, constituting 21% of the air we breathe. The abundant supply of oxygen to aerobic organisms enables it to serve as a high capacitance acceptor for electrons. Furthermore, oxygen can damage cells by production of byproducts of respiration or by production of reactive nitrogen and oxygen species (RNS; ROS) (60). Thus, delivery of oxygen to human tissues is tightly regulated by the allosteric binding of oxygen to hemoglobin in red blood cells (113). However, the lungs are unique in having a vast moist mucosal epithelial surface area that is immediately and directly exposed to inhaled oxygen (and airborne reactive pollutants), which dissolve into the epithelial surface lining fluid. This makes the lungs particularly susceptible to environmental oxidant-mediated injury. Furthermore, the lung is exposed to a multitude of airborne microorganisms, and thus also endogenously generates high levels of RNS and ROS to maintain a remarkably sterile lower airway. Altogether, endogenous production of RNS and ROS by metabolic reactions (respiration, phagocytosis) and environmental exposures (air pollutants, cigarettes smoke. particulates) might be expected to produce an oxidizing lung environment (Fig. 2). However, redox state in the healthy lung is primarily reducing. This is attributed to the multiplicity and abundance of antioxidant systems available to the lung. The vast excess of reduced substances over oxidized ones is maintained by a rich array of antioxidant enzymatic and nonenzymatic effectors on the surface of, and within, the epithelial cells in the airways (176).

A. ROS and RNS production in the lung
Many specific classes of lung cells have recently been compared for their capacity to generate ROS in the context of oxidant-induced lung injury, including tracheal epithelial cells, alveolar epithelial type I and type II cells, Clara cells, and vascular endothelial cells. While inflammatory cells such as neutrophils generate highest levels of ROS, alveolar macrophages and eosinophils are also high level producers of ROS.
1. Endogenous reactive oxygen species
Reactive oxygen species include superoxide, hydrogen peroxide and hydroxyl radicals and can be generated by a number of metabolic pathways and are dangerous byproducts of oxygen consumption (Fig. 3).

a. Superoxide
The tetravalent reduction of oxygen during mitochondrial electron transport is a safe process but also can result in formation of superoxide (O2 •−) (60, 108, 132, 133, 272). Another source for intracellular generation of O2 •− is the NADPH oxidase enzymatic system, which is found in neutrophils, monocytes, and macrophages (15, 51, 66, 81). O2 •− can also be generated by mechanisms such as molybdenum hydroxylase reactions (including the xanthine, sulfite, and aldehyde oxidases) and arachidonic acid metabolism (60, 124). O2 •− is unstable, with a half-life of milliseconds. Because it is charged, it does not easily cross cell membranes (21). O2 •− will react, however, with proteins that contain transition-metal prosthetic groups, such as heme moieties or iron–sulfur clusters. These reactions may damage amino acids or cause protein/enzyme function to be lost (112, 356).
b. Hydrogen peroxide (H2O2)
Under biological conditions, the main reaction of superoxide is to react with itself to produce hydrogen peroxide and oxygen, a reaction known as “dismutation” (Reaction 1) (228). Superoxide dismutation can be spontaneous or can be catalyzed by the enzymes superoxide dismutases (SOD).
H2O2 can also be produced by oxidase enzymes, including xanthine oxidase, monoamine, and amino acid oxidase (60). Once formed, the oxidizing potential of H2O2 may be amplified by eosinophil and neutrophil derived peroxidases, eosinophil peroxidase (EPO) and myeloperoxidase (MPO), respectively (103, 135, 184, 341) (Reaction 2).
The capacity to generate H2O2 varies among cell types. Kinnula et al. has shown that alveolar macrophages produce high levels of H2O2. Type II cells have the capacity to release an excessive amount of H2O2 whereas endothelial cells produce low amounts of H2O2 (178). Interestingly, the rate of inactivation of catalase via H2O2 production is the highest in Type II cells (178). This confirms that the generation of H2O2 depends upon resident and inflammatory cells in the lung.
c. Hydroxyl radical (•OH)
The hydroxyl radical is an extremely reactive oxidizing radical that will react to most biomolecules at diffusion controlled rates (54), which indicates that reactions occur nearly immediately with biomolecules. The hydroxyl radical is several orders of magnitude more reactive towards cellular constituents than superoxide radicals, and many orders more reactive than hydrogen peroxide. Much of the damage done by superoxide and H2O2 in vivo is due to their production of hydroxyl radicals (•OH) in a series of reactions catalyzed by traces of transition metal ions (60). In these reactions, superoxide acts as the reducing agent. The reduced metal catalyzes the breaking of the oxygen-oxygen bond of hydrogen peroxide to produce a hydroxyl radical (•OH) and a hydroxide ion (HO−). The classic example is the iron-catalyzed Haber–Weiss Reaction in which Fe3+ is reduced to Fe2+, followed by the Fenton Reaction in which the Fe2+ catalyzes the transformation of H2O2 into hydroxyl radical (•OH) (133).
An alternative pathway for •OH formation in vivo may involve myeloperoxidase (MPO) and eosinophil peroxidase (EPO). Under physiological concentrations of halides, MPO produces hypochlorous acid (HOCl) and EPO produces hypobromous acid (HOBr). Studies of •OH with spin-trapping agents and chemical trap (138, 267) have demonstrated that hypohalous acids can generate •OH after reacting with O2
•− (Reaction 3). •OH can react with different molecules such as protein (38), DNA, and lipids (111).
d. Protein modifications by MPO and EPO
Influx of inflammatory cells, which contain, enzymatic systems such as EPO and MPO (Reaction 3) can produce ROS. EPO and MPO are enzymes that accelerate oxidative protein modifications. EPO selectively uses Br− (bromide) to form HOBr (hypobromous acid) (Reaction 2) (226, 341). EPO is the only human enzyme that selectively generates reactive brominating species, thus brominated products serve as fingerprints of atopic/eosinophilic inflammation. MPO is the most abundant protein stored in neutrophil granules, and secreted during cell activation (185). MPO selectively uses Cl− as substrate to generate HOCl (hypochlorous acid) (103, 341) (Reaction 2). These enzymes are secreted by inflammatory cells and produce protein oxidative damage through the production of reactive brominating species (RBS), reactive chlorinating species (RCS), and reactive nitrogen species (RNS). Specific brominated and chlorinated targets in plasma serve as signatures for EPO- and MPO-dependent, i.e. eosinophil- and neutrophil-dependent, oxidative injury (Fig. 4).

2. Reactive nitrogen species
The RNS synthesized in the lung is nitric oxide (•NO) which is produced by nitric oxide synthases [NOS, EC 1.14.13.39] (321). All NOS convert L-arginine to NO and L-citrulline in a reaction that requires dimeric enzyme, oxygen, NADPH, and cofactors flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), tetrahydrobiopterin, calmodulin, and iron protoporphyrin IX. There are three forms of NOS, the inducible NOS (iNOS or NOS2), neuronal NOS (nNOS or NOS1), and endothelial NOS (eNOS or NOS3) (Table 1). Active NOS are dimeric, and each monomer is comprised of an N-terminus oxygenase domain that binds the heme, tetrahydrobiopterin, and substrate L-arginine. The carboxy terminus of NOS monomers bind the flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), and NADPH (320). In general, nNOS and eNOS are constitutively expressed in neuronal and endothelial cells, respectively, dependent on increases in calcium to bind calmodulin that results in enzyme activation and picomolar levels of NO production (91, 318 –321). Immunohistochemical studies reveal the presence of the three isoforms of NOS in the airway. NOS III is primarily localized to pulmonary endothelial cells, and NOS I in nonadrenergic, noncholinergic inhibitory neurons (19, 113). NOS II is continuously expressed in normal human airway epithelium at basal airway conditions (19, 113, 127, 128, 187, 320). NO is also produced by the upper respiratory tract epithelium within the nasopharynx and paranasal sinuses, most likely by NOS II (211). There is evidence that epithelial NOS II activity is a major determinant of NO present in exhaled breath (196). The iNOS is regulated at the level of transcription and mRNA stability, is calcium independent, and produces nanomolar levels of NO (320, 322). Regulation of iNOS expression varies in different cell types, but typically is increased by cytokines and pro-inflammatory factors, interferon gamma, TNF-alpha, and IL1-beta (126, 127). NO synthesis by iNOS is also regulated by availability of substrate arginine and cofactor tetrahydrobiopterin.
The carboxy-reductase domain transfers electrons to the heme iron of the oxygenase domain, which then binds oxygen and oxidizes arginine to generate NO and citrulline (320). Since the early 1990's, NOS have also been shown to generate superoxide by spin trapping/EPR spectroscopy and H2O2, which is presumed to derive from superoxide dismutation (271). This occurs when NADPH is oxidized by the enzyme in the absence of L-arginine (270). Thus, conditions that decrease arginine availability to NOS will lead to greater superoxide formation. Arginase, a critical enzyme in the urea cycle, converts arginine to ornithine and urea. There are two isoforms, arginase 1 and arginase 2, both of which play a regulatory role in •NO and superoxide synthesis by modulating the availability of arginine for NOS (151, 349) (Fig. 5).
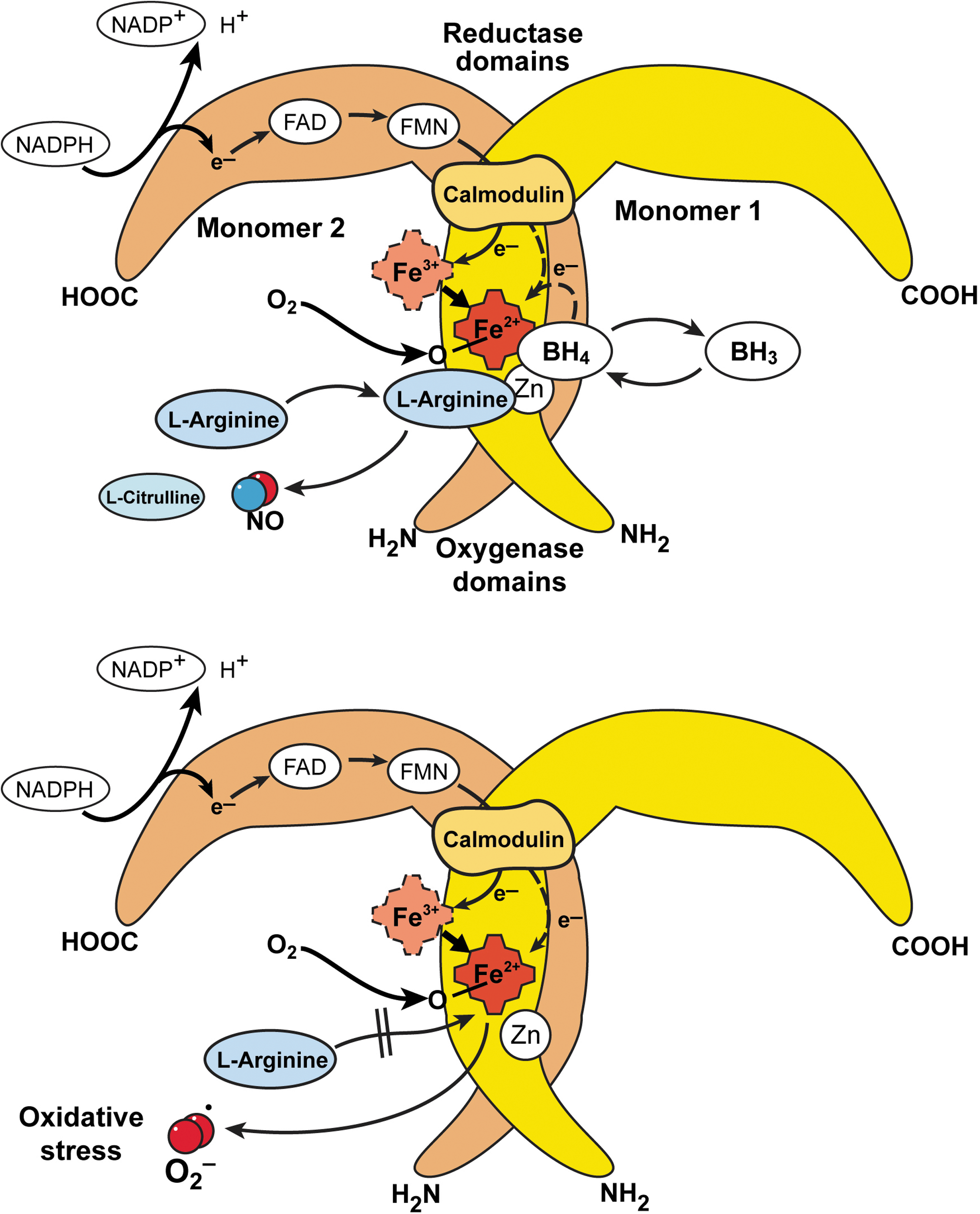
The unpaired electron of •NO makes it highly reactive (311). Since •NO is freely diffusible, consumption of •NO can occur at different sites within the cell, extracellular fluids, and intravascular compartments (195). The diffusion of NO may be most limited by its many possible chemical reactions (87, 342) (Fig. 6). When metabolized, •NO gives rise to a group of compounds collectively known as the reactive nitrogen species (RNS) that possess their own unique characteristics. In biologic systems, up to 40% of the NO synthesized may be consumed by chemical reactions (87, 343). Autooxidation of •NO with O2 results in the formation of nitrite (NO2 −). NO2 − is also a substrate for hemeperoxidases such as MPO and EPO. Peroxidase-catalyzed oxidation of NO2 − results in the formation of nitrogen dioxide radical (NO2 •) or related molecules (1 –3, 29). These substances contribute to the nitration of phenolic compounds, such as tyrosine, to form dimerized (dityrosine) and nitrated (3-nitrotyrosine) products, which are stable.

Nitrite was previously considered an end-product of NO, but studies now indicate that nitrite can be recycled to generate bioactive NO. Nitrite reduction to NO occurs in blood and tissues by many mechanisms, such as by xanthine oxidases or reaction with hydrogen ions. Thus, nitrite can serve as a storage pool for NO production at times when NO synthases may be unable to function. •NO is also rapidly oxidized by reaction with oxyhemoglobin (HbO2), resulting in formation of methemoglobin (Hb3+) and NO3 − (8). The rapid reaction of •NO with free radicals (radical–radical reaction) has emerged as one of the major routes to the formation of RNS (8). •NO reacts with superoxide to form peroxynitrite (ONOO−). ONOO− can nitrate tyrosine residues and alter levels or function of enzymes, structural and signaling proteins (13, 120, 224). Tyrosine nitration can cause either gain or loss of protein function (13). On average, proteins are composed of 4% tyrosine residues, but chemical nitration of isolated proteins modifies only a subset of tyrosine residues, and the basis for this selectivity is not fully understood. This suggests that an innate property of the target protein or its location may predispose it toward nitration (13). In acid environments, ONOO− can be protonated to yield peroxynitrous acid (ONOOH), which rapidly decomposes to NO3 − via the intermediate formation of OH• and NO2-like species. ONOOH can also react with thiol residues to form S-nitrosothiols (SNO), which have been proposed as a potential unique signaling mechanism induced by nitrosative stress (149). The exact mechanism by which S-nitrosation occurs in vivo is still unclear, but it involves the formation of •NO-derived intermediates with the redox equivalence of NO+ (the primary candidates are N2O3 and ONOOH) and (di)nitrosyl iron complex (113, 273).
3. Environmental exposures
Because the lung interfaces with the external environment, it is frequently exposed to airborne oxidant gases and particulates, and thus prone to oxidant-mediated cellular damage.
a. Atmospheric ozone (O3) and particulate matter pollution
Ozone, a component of photochemical air pollution, is formed from volatile hydrocarbons, halogenated organics, and oxides of nitrogen in the presence of sunlight (244). Ambient ozone levels usually vary between 20 and 40 parts per billion (ppb); moderate elevations in levels are usually 70–120 ppb (335). There is a great deal of evidence which shows that high concentrations of ozone can be harmful to the lung (73, 165, 190, 239, 242, 244, 261). Ozone can react directly with unsaturated fatty acids and cell membranes to produce lipid ozonation products, which are small, diffusible, and relatively stable (169, 170, 202). Particulate matter pollution is one of the most serious air pollution problems in urban environments (56). The size of the particle is very important since it will determine where the particle will come to rest in the respiratory tract when inhaled (56). One of the most dangerous forms of particulate matter pollution is diesel exhaust particle. Diesel exhaust particles are a polyaromatic hydrocarbon, a hydrophobic molecule that can diffuse easily through cell membranes. Diesel exhaust particles may therefore modify cell growth and differentiation (56).
b. Cigarette smoke and environmental tobacco smoke
Environmental tobacco smoke or secondhand smoke is a complex mixture of gases and particles that include smoke from the burning cigarette (sidestream smoke) and exhaled mainstream smoke. Environmental tobacco smoke contains a large number of components, and many of them are toxic to epithelial cells. Cigarette smoke contains >4,000 chemicals and poisons, including 50 that are known to cause cancer. Some of the chemicals in cigarette smoke are carbon monoxide, cyanide, arsenic, mercury, and NO. Furthermore, cigarette smoke generates or contains ∼1014 oxidative molecules per puff such as hydrogen peroxide and superoxide. Furthermore, environmental tobacco smoke leads to activation of phagocytes augmenting release of free radicals. Because free radicals cause oxidative damage to macromolecules such as DNA, lipids, and protein, they are believed to be involved in the pathogenesis of many diseases (333).
4. Oxidative processes in biology
The formation of ROS and RNS is an essential prerequisite for neutrophils, monocytes, macrophages, and eosinophils to kill certain bacteria. These phagocytic cells use NADPH oxidase enzymatic systems to generate O2 •− directly as part of their armamentarium against invading microorganisms (15, 51, 66, 81). They can also form HOCl through myeloperoxidase-catalyzed oxidation of the Cl− ion by H2O2 (21). •NO is also involved in mononuclear cell-mediated killing of Mycobacterium tuberculosis and other pathogens in rodents and is toxic to tumor cell lines in vitro (252). In the upper respiratory tract of humans, NO appears to be important in maintaining ciliary function and may have a role in sterilizing the mucosa. The heme protein cytochrome P450 catalyzes a series of reactions that detoxify lipid-soluble drugs and toxic metabolic byproducts. This enzyme uses high-energy electrons transferred from NADPH to add hydroxyl groups to potentially harmful hydrophobic hydrocarbons dissolved in the lipid bilayer (88). Such reactions convert water-insoluble drugs or metabolites that would otherwise accumulate in cell membranes into water-soluble compounds, which then diffuse out of the cell and are excreted in the urine. Cytochrome P450 also exploits the reactivity of the iron–oxygen complex to catalyze oxidation of a number of endogenous compounds and xenobiotics (21). These examples show that ROS and RNS play important physiologic functions and yet can also cause extensive damage. Tissue health is maintained under physiologic conditions by antioxidants.
B. Antioxidants in the lung
The balance between physiologic functions and damage is determined by the relative rates of formation and the removal of ROS and RNS, and free radicals. All aerobic organisms use a series of primary antioxidant defenses to protect against oxidative damage. An antioxidant is most simply defined as a molecule capable of slowing down or preventing redox changes in the cell.
The lungs have developed several endogenous antioxidant systems to deal with the production of free radicals. These systems may be divided into enzymatic and nonenzymatic groups. The enzymatic antioxidants include superoxide dismutases (SOD), catalase, glutathione peroxidases, heme oxygenase, glutaredoxin, thioredoxin, and peroxiredoxin. These antioxidant enzymes usually require trace metal cofactors (109). SOD, for example, consists of proteins co-factored with copper, zinc, or manganese (109). Iron is required as a co-factor for catalase (218). The most well-researched nonenzymatic antioxidants include lipid-soluble vitamin E (tocopherol), vitamin A, and carotenoids (including beta-carotene), and water-soluble vitamin C and glutathione (GSH). Glutathione, which is synthesized intracellularly from amino acids cysteine, glycine, and glutamate, is capable of scavenging free radicals either directly or enzymatically via glutathione peroxidase. In addition, GSH is crucial to the maintenance of enzymes and other cellular components in a reduced state (59 –62).
1. Nonenzymatic lung antioxidants
The nonenzymatic antioxidants can be classified depending whether they are hydrophilic or hydrophobic. In general, hydrophilic antioxidants react with oxidants in the cell cytosol and/or bloodstream, whereas the hydrophobic antioxidants protect the cell membranes from lipid peroxidation. Nonenzymatic antioxidants react directly with the oxidants. Such antioxidants are said to be ‘scavengers;’ their roles are unavoidably suicidal.
a. Vitamin E (alpha-tocopherol)
Vitamin E is an important hydrophilic antioxidant. It protects the cell membrane from oxidation by reacting with lipid radicals, such as lipid peroxyl radicals (LOO•) that are produced during lipid peroxidation reactions (233, 336). Alpha-tocopherol is the predominant form of vitamin E in tissues and the primary form in supplements. However, gamma-tocopherol is the major form of vitamin E in plant seeds and in the US diet, yet has drawn little attention compared with alpha-tocopherol. Recent studies indicate that gamma-tocopherol may be important to human health. Gamma-tocopherol appears to be a more effective trap for lipophilic electrophiles than is alpha-tocopherol (162).
b. Vitamin C (ascorbic acid)
Vitamin C is a hydrophilic vitamin that can directly scavenge O2 •− and •OH by forming the semidehydroascorbate free radical that subsequently is reduced by GSH (227). Vitamin C, however, is usually not considered a major antioxidant because it also has pro-oxidant properties. It is probably the only cellular reducing agent other than O2 •−capable of converting Fe3+ to Fe2+, which then reacts with H2O2 to form •OH (291). Whether the pro-oxidant or antioxidant properties of vitamin C prevail in any particular tissue is determined by the extent of available iron stores; iron overload favors excess oxidant generation (21, 291).
c. Glutathione
Glutathione (GSH) is the predominant nonprotein thiol in the cells and is important for maintenance of the cellular redox (302). GSH is a cysteine-containing peptide found in most forms of aerobic life, and is present in high concentration in blood and lung (39 –41, 58, 62). Independent of the GSH system (see later), free GSH can function as a water-soluble antioxidant by interacting directly with radical intermediates in nonenzymatic catalyzed reactions. Lung epithelial lining fluid contains up to 300 micromolar concentration of GSH (290), and >90% of the GSH is maintained in the reduced form. Scavenging of O2 •− by GSH leads via several steps to the formation of thiyl radicals (GS•) and H2O2, which is a radical propagation reaction (21, 113). Increased intracellular GSH is a response to oxidative stress (59, 278), and a critical determinant of cellular tolerance to oxidizing environments (277). Reactive oxygen species increase GSH through induction of γ-glutamyl cysteine synthetase, the rate-limiting enzyme of GSH biosynthesis (281). Uptake of GSH into cells (84, 230), and export of the oxidized form to overcome an accumulation of GSSG within the cytosol occurs rapidly in conditions of oxidative stress (59).
Other nonenzymatic antioxidants include β-carotene (scavenger of superoxide anions and peroxyl radicals), uric acid (hydroxyl radical, superoxide, peroxyl radical scavenger), bilirubin (lipid peroxyl radical scavenger), taurine (hypochlorous acid quencher), albumin (transition metal binding, glutathione precursor and hydrogen peroxide scavenger), and cysteine and cysteamine (donators of sulfhydryl groups).
2. Enzymatic lung antioxidants
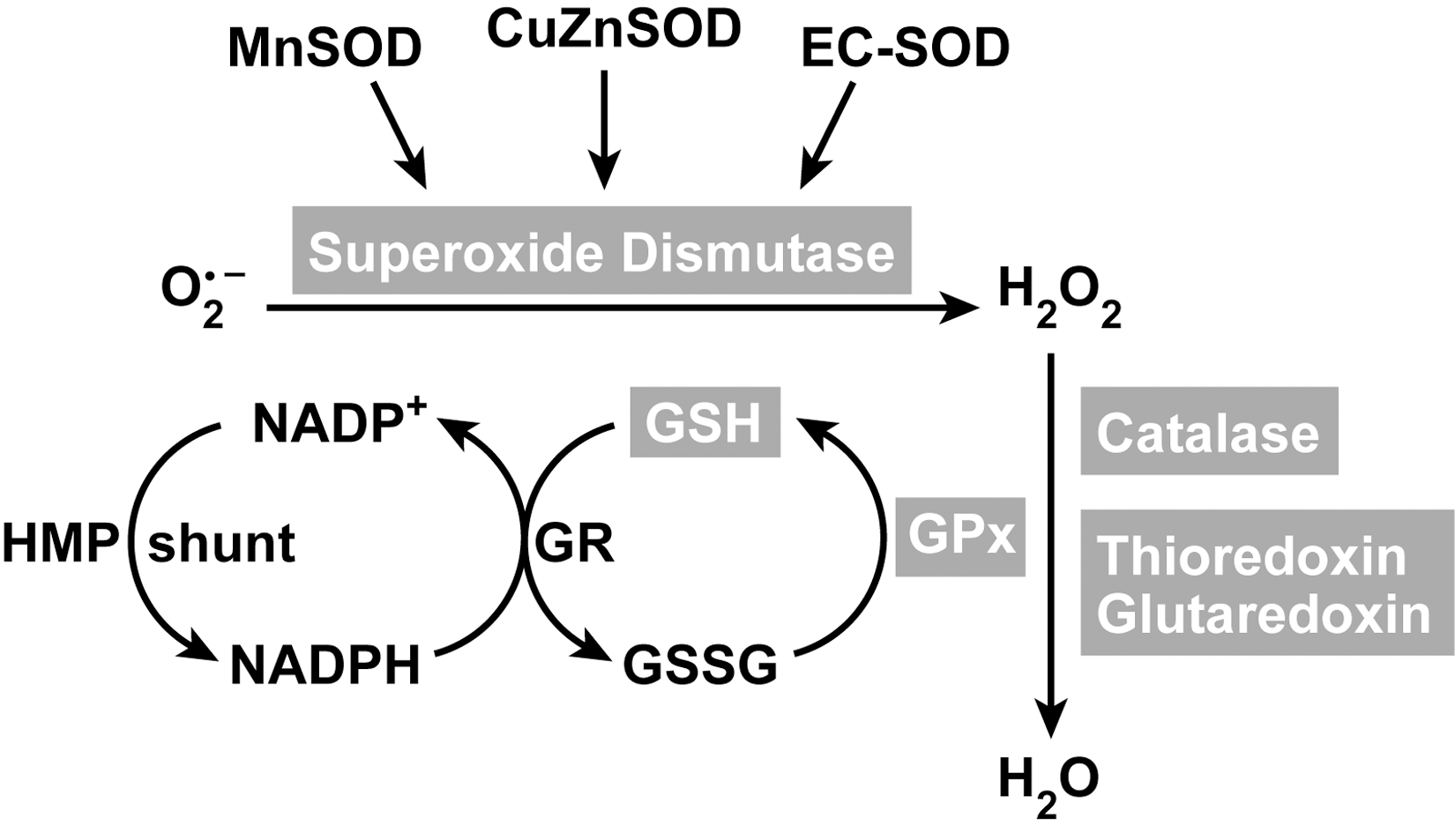
The detoxification pathway of superoxide to water is a result of multiple enzymatic antioxidants (Fig. 7). The major enzymatic antioxidants are discussed below.
a. Superoxide dismutases (SOD)
Superoxide dismutases (EC 1.15.1.11) are ubiquitous enzymes with an essential function in protecting aerobic cells against oxidative stress and are essentially present in every cell in the human body. They catalyze the reaction of superoxide radicals to hydrogen peroxide. Superoxide dismutase enzymes contain metal ion cofactors that, depending on the isozyme, can be copper–zinc, manganese, or iron. Human lung epithelium expresses three forms of eukaryotic SODs that are located on three different chromosomes (Table 2). The distribution of the three SOD isoforms in the lung has been reviewed previously (177), with CuZnSOD expression in bronchial epithelium, alveolar epithelium, mesenchymal cells, fibroblasts, arterioles, and capillary endothelilal cells (98, 193, 266). MnSOD is expressed in the airways, especially in the septal tips of alveolar duct and arterioles near the airways (57). Furthermore, MnSOD is also moderately or highly expressed in respiratory epithelium, alveolar type II epithelial cells, and alveolar macrophages (65, 194). EC-SOD is found in bronchial epithelium, alveolar epithelium, epithelial cells lining intrapulmonary airways, alveolar macrophages, and endothelial cells lining both arteries and veins (256, 257).
The copper–zinc superoxide dismutase (CuZnSOD) protein constitutes up to 80–90% of the intracellular SOD activity and is mainly found in the cytosol, although it also is present at low levels in lysosomes, peroxisomes, nucleus, and intermembrane space of the mitochondria (72). CuZnSOD is expressed in lung cells, such as bronchial epithelial, alveolar macrophages, and capillary endothelium of the lung (53, 63, 82). The gene located on chromosome 21q22.1 gives rise to a 16 kDa protein, each containing a catalytic Cu2+ metal ion which bridges via a histidine residue to a Zn+ ion (20, 109). Active CuZnSOD is a homodimeric protein and accelerates the spontaneous dismutation of superoxide radical by >40-fold through the cyclic oxidation–reduction of its Cu2+ metal ion (109). The reactions are very fast, and do not require reducing equivalents, enabling the reaction to proceed in the absence of any energy input.
In addition to this reaction, CuZnSOD may have peroxidase activity (310). At high levels, hydrogen peroxide reduces the Cu2+ to produce Cu+-O or Cu2+-OH, which either can oxidize the adjacent histidine residue in the monomer, inactivating itself, or oxidize residues in other proteins (7, 125, 355). CuZnSOD may also nitrate tyrosine in proteins via a reaction involving peroxynitrite (24, 74), and it is also reported to catalyze the release of NO from nitrosothiols (166). Over 90 genetic polymorphisms of the CuZnSOD have been described in the causation of the neurodegenerative disease amyotrophic lateral sclerosis (20). However, the lack of abnormalities in genetic deletion of CuZnSOD in mice (283) has led to the belief that pathologic consequences of mutations are due to gain of function of the enzyme's alternate peroxidase or nitration reactions, and are not due to loss of superoxide dismutase activity.
The Mn superoxide dismutase (MnSOD) protein constitutes up to 10% of the intracellular SOD activity and is mainly expressed in the matrix of the mitochondria. The MnSOD gene is on chromosome 6q25.3, and its sequence has no homology to CuZnSOD. The 25 kDa protein is expressed in the cytosol and imported into the mitochondria where the mitochondrial targeting sequence is cleaved to yield a protein of 22 kDa (324, 344). Each monomer contains a Mn and Zn metal ion, and the functional enzyme is a homotetramer (107). The Mn ion is held in place by the nitrogen of three histidines and the oxygen of one aspartate (20). Superoxide dismutation by MnSOD proceeds through the following reactions:
Unlike CuZnSOD, the MnSOD does not have peroxidase or nitration ability. In fact, MnSOD is inactivated by nitration of the tyrosine 34 residue, which is required for enzyme catalytic activity (65, 216, 351). Further differences include that MnSOD is not inactivated by hydrogen peroxide or cyanide, and this allows distinction among the intracellular SODs on native gels (20, 82). Oxidative stress can strongly upregulate MnSOD gene expression (345). A recent report by Yeh et al. demonstrated that CuZnSOD expression can be upregulated via Nrf2 in rats treated with phenolic acids (352). Mitochondria consume large amounts of oxygen in the cell; MnSOD is the primary protection from the superoxide produced as an intermediary of cellular respiration. As might be expected, genetic deletion of this critical enzyme in mice is inconsistent with life, with death occurring due to mitochondrial pathology and oxidative damage to DNA shortly after birth when animals are exposed to ambient oxygen concentrations (206).
Extracellular superoxide dismutase (EC-SOD), a secretory, tetrameric hydrophobic glycoprotein, is the major extracellular SOD in the interstitial spaces of the lungs (100, 219, 220, 258). Each 24 kDa subunit contains a Cu and Zn ion and the active site is similar to the CuZnSOD. The CuZnSOD and EC-SOD have 50% similarity in amino acid sequence. An important characteristic of EC-SOD is that it contains a heparin/matrix binding domain consisting of positively charged arginines and lysines, which is located in the C-terminal region of EC-SOD (171). It is through interaction with heparin and heparan sulfate proteoglycans on cell surfaces and in the extracellular matrix that the extracellular localization of EC-SOD is maintained (100). The heparin/matrix-binding domain is sensitive to proteolysis, which can lead to release of EC-SOD from tissue matrix and sequentially alter oxidant/antioxidant balance. Recent study showed that EC-SOD protects the oxidative fragmentation of heparin/heparan suflate/syndecan-1 (186). The localization of EC-SOD in the lungs is primarily within the smooth muscle region surrounding blood vessels and airways (110). EC-SOD may have an important role in a number of lung diseases, where it modulates oxidant injury, inflammation, hyperoxia-induced lung injury, and pulmonary fibrosis. Polymorphisms are found in EC-SOD; the Arg 213-gly polymorphism (R213G) is frequently found in the human population (4–6%) and is associated with patient outcomes in chronic obstructive pulmonary disease (COPD) and lung injury (10).
b. Catalase
Catalase is a metalloprotein oxidoreductase enzyme (EC 1.11.1.6) and the principal scavenger of hydrogen peroxide when the latter is present at very high concentrations. Catalase is relatively limited in cellular distribution (e.g., peroxisomes and a few other locations). Glutathione peroxidase and peroxiredoxin systems, as classes, are of comparable, if not potentially greater, importance than catalase. The tetrameric hemoprotein undergoes alternate divalent oxidation and reduction at its active site, which contains the porphyrin ring and iron, in the presence of H2O2 (83, 285). The iron is held in place by the four nitrogen atoms of the porphyrin; the fifth valence position is coordinated to tyrosine 358 of catalase, and the sixth valence left free for interaction with substrate. The reaction mechanism proceeds through two steps. First, Fe(3+) reacts with hydrogen peroxide that results in cleavage of the O–O bond in H2O2, and the oxygen remains bound to the sixth valence position of Fe(+5), leading to formation of compound I. Compound I may oxidize a second peroxide molecule to oxygen, while the oxygen bound to the iron is released as water (20). Alternatively, Compound I may undergo inactivation by reduction to Compound II [Fe(4+)] by oxidants, or from itself by formation of a tyrosyl radical (tyrosine 370) under prolonged oxidative stress. Catalase has appreciable reductive activity for small molecules such as H2O2 and methyl or ethyl hydroperoxide (83, 285), but is unable to metabolize large molecular peroxides such as lipid hydroperoxide products of lipid peroxidation. Catalase is effective in the presence of high H2O2 concentrations (43), but under prolonged oxidative stress with oxidation of NADPH, catalase activity drops (181). NADPH binds to the enzyme and stabilizes the structure, and protects catalase from inactivation apparently by reversing accumulation of Compound II (181). The catalase gene located on chromosome11p13 is not generally inducible by oxidant stress (353). Enzyme activity can be regulated by post-translational processes. Under oxidative stress, the Abl family of receptor tyrosine kinases lead to phosphorylation of catalase at tyrosine 231 and tyrosine 386, which results in greater activity and lower cellular H2O2 levels (44). On the other hand, oxidation of tyrosine residues, in particular tyrosine 358, has been linked to loss of catalase activity under oxidative stress, for example, in asthma (119).
c. Glutathione system
The glutathione system consists of reduced (GSH), oxidized (GSSG) and GPx (Fig. 7). It is considered to be the major thiol–disulfide redox buffer of the cell. It is a central mechanism for reducing H2O2. It complements catalase as a reducing system for H2O2 but exceeds catalase in its capacity to eliminate additional varieties of toxic peroxides. Other metabolized substrate species include large molecule lipid peroxides, formed by free radical attack on polyunsaturated lipid membranes and products of lipo-oxygenase-catalyzed reactions (139). The key enzyme in the glutathione system responsible for the reduction of H2O2 are the glutathione peroxidases (GPx, EC 1.11.1.9). The reducing capacity of glutathione peroxidase enzymes are based on high levels of GSH (L-γ-glutamyl-L-cysteinylglycine). Glutathione peroxidases reduce hydrogen peroxide to water by oxidizing glutathione to oxidized/disulfide form (GSSG). The glutathione disulfide (GSSG) that is formed in the course of the reaction is subsequently reduced back to GSH by glutathione reductase, an intracellular enzyme that uses NADPH generated from the hexose monophosphate shunt system as an electron donor (133). Subsequently, GSSG breaks down to its amino acid components for cellular uptake and recycling. The capacity to recycle GSH makes the glutathione system pivotal to the antioxidant defense mechanism of a cell and prevents the depletion of cellular thiols. Four GPx have been described, all selenium enzymes: (a) the classic cytosolic form (cGPx), found in all cells; (b) a membrane-associated glutathione peroxidase phospholipid hydrogen peroxide GPx (90) (PHGPx); (c) another cytoplasmic enzyme, gastrointestinal GPx (giGPx), which was first found in cells of the gastrointestinal tract; and (d) an extracellular glutathione peroxidase (eGPx), first identified as a distinct enzyme in human plasma (354). All members of this family of enzymes can be oxidized by organic hydroperoxides, hydroperoxide, or both, and can subsequently be reduced by glutathione. The existence of multiple forms of GPx is due to the expression of four different gene products (354). All GPx contain a selenium atom in the active site in the form of selenocysteine (SeCys). The alveolar epithelial lining fluid contains a very high amount of both extra and intracellular glutathione peroxidase and micromolar levels of GSH (59 –62). Previous reports have shown that S-nitrosoglutathione (GSNO) is an equivalent effective co-substrate of GPx (106, 146). Glutathione peroxidase use of GSNO leads to release of •NO and reduction of the GSNO storage form (106, 146). GNSO induces eGPx gene expression (59, 64) while overexpression of SOD prevents the induction of eGPx (59).
d. Thioredoxin system
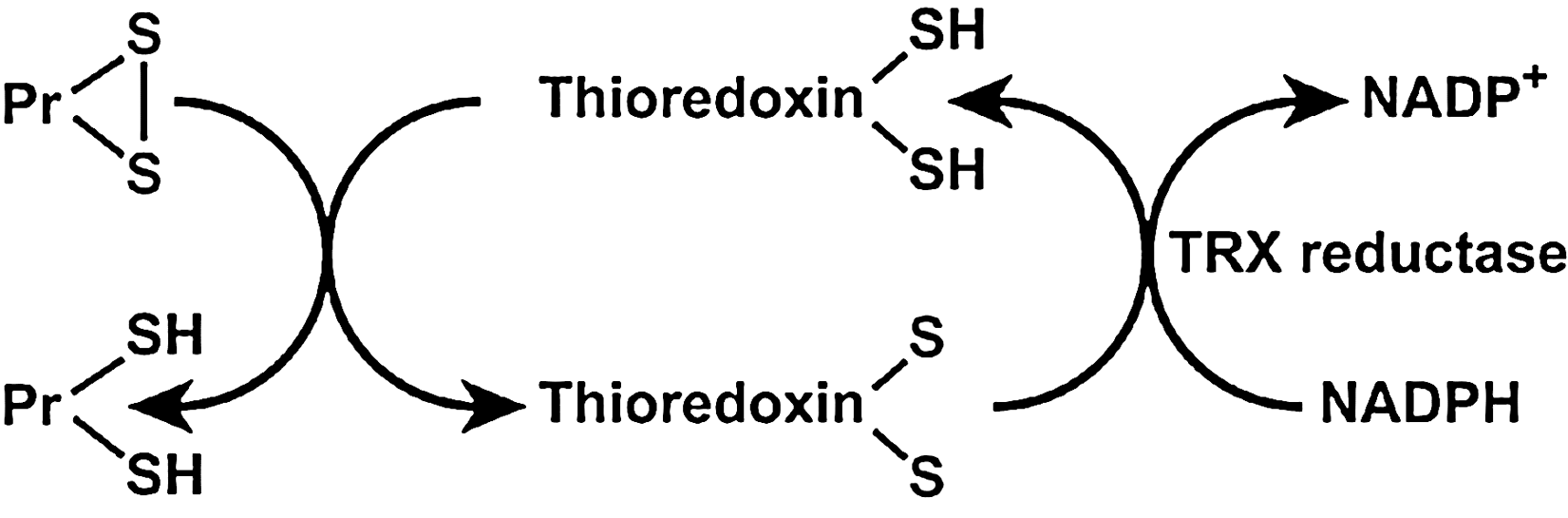
Thioredoxins (TRX) are oxidoreductase enzymes containing a dithiol–disulfide active site (-Cys-Gly-Pro-Cys-) (145). The cysteine residues reverse from a dithiol (-SH HS-) group to a disulfide bridge (-S-S-). The oxidized TRX is a disulfide with one bridge between two cysteines whereas the reduced TRX is a dithiol with two cysteines (145). TRXs are kept in the reduced state by flavoenzyme thioredoxin reductase, via an NADPH-dependent reaction (Fig. 8). Thioredoxin reductases in human are closely related to glutathione reductases. There are two thioredoxins, 1 and 2, with different cellular locations, and there are two thioredoxin reductases, with locations corresponding to the intracellular thioredoxins 1 and 2. The strong reducing activity of the sequence results from the cysteine residues acting as proton donors and cleaving disulfide (S-S) bonds in the target protein (145). Overall, TRXs can reduce protein disulfides (Pr-SH) and protein sulfenic acid (Pr-SO3H) intermediates by cysteine thiol–disulfide exchanges (79). Thioredoxins in human are closely related to glutathione reductase. There are two types of thioredoxins. Thioredoxin 1 is found in the cytoplasm and Thioredoxin 2 in the mitochondria (12). Thioredoxin 1 is a strong scavenger of ROS (142, 245, 246) and inhibits H2O2 in cooperation with the TRX-dependent peroxidase peroxiredoxin (288). Thioredoxin 1 augments gene expression of other antioxidants, such as MnSOD (80). The importance of thioredoxin has been identified in signal transduction, inflammatory response, and other biological functions such as apoptosis, cell growth, and proliferation (153, 247, 250). Specific protein disulfide targets for reduction by thioreoxin are ribonucleotide reductase (284), protein disulfide isomerase (212), and several transcription factors including p53, NF-κB, and AP-1 (102). This small multifunctional protein refolds oxidized proteins and activates transcription factors by reducing cysteine in the DNA binding site (102). Thioredoxins are expressed in bronchial epithelial cells and alveolar macrophages, metaplastic alveolar epithelial cells, and chondrocytes of the bronchus (314).
e. Glutaredoxin system
Glutaredoxins (GRX) are thiol–disulfide oxidoreductases that use glutathione as a cofactor and catalyze the reversible exchange of GSH with protein thiol groups (P-SH) (Fig. 9). There are two groups of glutaredoxins (Grxs), dithiol GRXs, which contain the Cys-Pro-Tyr-Cys active site motive and the monothiol GRXs lacking the C-terminal active site thiol in its Cys-Gly-Phe-Ser active site (207). Glutaredoxins uniquely also reduce mixed disulfides (-S-S-) with glutathione via a monothiol mechanism (deglutathionylation) where only an N-terminal low pKa Cys residue is required (79, 207) (Fig. 9). It is of note that GRX are dependent on GSH/GSSG concentrations. The human cell contains four GRXs, two dithiol (GRX 1 and GRX 2), one multiple monothiol (GRX 3), and one monothiol (GRX 4) (207). Glutaredoxin also catalyzes the formation of protein disulfide of certain proteins in the presence of a GS-radical generating system (79, 316). The formation of protein–SG mixed disulfide (glutathionylation) by glutaredoxin through a monothiol mechanism may play an important role in protecting against more drastic irreversible modifications of protein thiols, particularly when the redox state of the cytoplasm becomes more oxidizing, as under conditions of oxidative stress (79, 101).
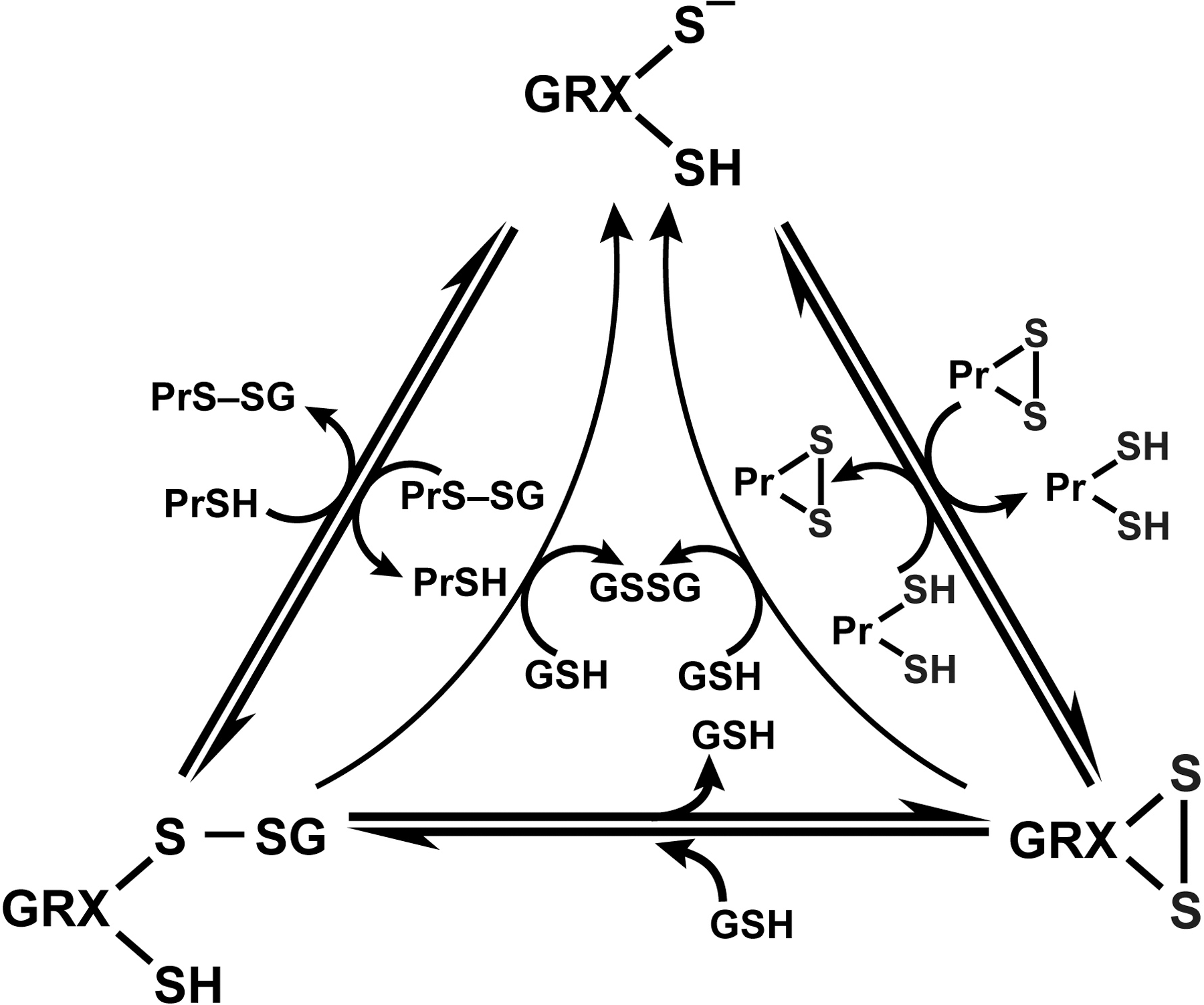
f. The role of protein thiolation (Pr-SH); S-glutathionylation in redox signaling
Maintaining the optimal GSH/GSSG ratio in the cell is critical to cell survival and is important in regulating the redox state of protein thiols. Changes in the cellular redox status, mainly due to decrease in GSH/GSSG ratio, initiates a series of redox-dependent modifications of proteins, lipids, and nucleic acids. With respect to proteins, cysteinyl residues are of particular interest, because their thiol group (Pr-SH) is susceptible to a number of oxidative modifications (30, 96, 122). Dominic et al. showed that cells may resist oxidative stress by protein thiolation (86). Proteins containing cysteine (Cys-SH) residues in the thiolate form (S-) are very likely to undergo oxidative modifications, which can interfere with biological functions. Protein sulfhydryl groups can be present as reduced thiols (Pr-SH), or oxidized to sulfenic (Pr-SOH), sulfinic (Pr-SO2H), or sulfonic acid (Pr-SO3H). Mild sulfhydryl oxidation produces disulfides and sulfenic acids, which are easily converted to disulfides by reaction with an adjacent sulfhydryl reside. Sulfenic acid may also be progressively oxidized to sulfinic acid and then to sulfonic acid. Disulfides and sulfenic acids may be reduced back to the sulfhydryl stage by TRX or GRX or other thiol reductases under high reducing potential. Recent reports have shown that sulfinic acid can also be reduced to the sulfhydryl stage although the reaction requires ATP and, hence, is not a simple reduction reaction (27). Sulfonic acid is not reversibly reduced to sulfhydryl under physiological conditions. It is difficult to accurately evaluate generation of oxidation of sulfhydryls because they are highly reactive and in a dynamic equilibrium. In general, they can be found as intra-or intermolecular disulfides (Pr-S-S-Pr) or mixed disulfides (Pr-S-S-X) with X as a low molecular mass thiol, such as cysteine or glutathione [i.e., S-thiolated proteins (150)]. Since GSH is widely distributed in cell compartments such as in cytoplasm, (1–10 mM of GSH) and mitochondria (5–10 mM GSH) as well as in the extracellular compartments such as epithelial lining fluid of the lung (100 μM GSH), S-glutathionyated (Pr-S-S-G) proteins are likely the main mixed disulfides in the lung (328).
Protein S-glutathionylation is a post-translational modification resulting in the formation of mixed disulfides between glutathione and protein sulfhydryl groups (78, 79). Protein S-glutathionylation can occur by several mechanisms [see recent review by Dall–Donne, (79)]. S-glutathionylation can occur not only during oxidative stress, but also under basal conditions (49, 208, 286). S-glutathionylation is involved in numerous physiological processes such as growth, differentiation, cell cycle progression, transcriptional activity, and metabolism. This suggests that S-glutathionylation is a widespread mechanism of redox regulation and important to basic cell function. The small amount of proteins that are S-glutathionylated in the cell under basal conditions can increase up to 50% under oxidative stress, and is accompanied by decrease of GSH (78, 79). The role for S-glutathionylation of proteins might be storage for GSH or as a protection of protein sulfhydryl integrity against more irreversible modifications and protein damage in response to higher levels of oxidative stress (78, 79). The reaction of GSH with protein thiols occurs by thiol–disulphide exchange and is catalyzed by GRX, enabling protein thiols to respond to a wide range of redox changes (i.e., GSH/GSSG ratio) during oxidative stress and redox signaling [see recent review by Dall–Donne, (79, 150)]. The main feature that makes S-glutahtionylation an attractive mechanism in the cell is its easy reversibility. Deglutathionylation is the process for removal of GSH from the protein mixed disulfides. This occurs when the redox environment becomes more reduced and can happen in an enzyme-dependent or -independent manner (79) (Fig. 9). Thus, S-glutathionylation serves the dual purposes of redox signaling in physiological conditions and protecting proteins from irreversible oxidative modifications during mild oxidative stress (78).
g. Peroxiredoxins
Peroxidredoxins (Prxs, EC 1.11.1.15) have received considerable attention in recent years as a new family of nonseleno peroxidases. Prxs exert their protective antioxidant effects through their broad spectrum of peroxidase activity, whereby hydrogen peroxide, peroxynitrite, and a wide range of organic hydroperoxides (ROOH) are reduced and detoxified. The antioxidant function of Prxs is dependent on redox-active cysteines. Prxs also modulate cytokine-induced hydrogen peroxide levels, which have been shown to mediate signaling cascades leading to cell proliferation, differentiation, and apoptosis. There are at least four different peroxiredoxins, with varying hydrogen peroxide-, lipid hydroperoxide-, and/or phospholipid hydroperoxide-substrate specificities and intracellular locations. Six different types of Prxs have been characterized in human lung (179). The bronchial epithelium showed moderate to high expression of Prxs I, III, V, and VI, the alveolar epithelium expressed mainly Prxs V and VI, and alveolar macrophages expressed mainly Prxs I and III (179).
h. Heme oxygenase
Heme oxygenases are members of the heat-shock family of proteins that play a protective role in inflammation and oxidative stress. These enzymes catalyze the degradation of heme molecules into biliverdin, bile pigments, and generate carbon monoxide and iron. Carbon monoxide and biliverdin have been attributed antioxidant properties (55). Consistent with this role, heme oxygenase-1 knockout mice are more susceptible to oxidative stress (268). Furthermore, induction of heme oxygenase by the repeated administration of hemin suppresses inflammation in the airway in ovalbumin-challenged guinea pigs, a model of asthma (46). Heme oxygenases are expressed in lung inflammatory cells of rats exposed to hypoxia. Recently, heme oxygenase-1 has been reported in human airways during asthma (46); levels in sputum of asthma patients are higher than in controls. Carbon monoxide concentrations are higher in exhaled breath of asthmatics as compared to healthy controls, which also suggests heme oxygenases are increased in human asthma. There are three forms of heme oxygenases. Heme oxygenase-1 is inducible, whereas heme oxygenase-2 and - 3 are constitutive (279). Heme oxygenase is expressed in airway epithelial cells, alveolar macrophages, bronchial epithelial cells, and inflammatory cells of the lungs (279).
IV. The Role of Redox in Asthma
A. Pathophysiology of asthma
Asthma is a chronic inflammatory disease of the lower airways, characterized clinically by reversible airway obstruction and airway hyperresponsiveness. The characteristic feature of asthma is airway inflammation that results in epithelial cell desquamation, mucus production, and airway remodeling. Inflammatory cells in the airway include mast cells, eosinophils, lymphocytes, and activated monocytes, macrophages, and neutrophils (Fig. 10). Research has revealed that a complex interaction of cells and numerous biological active proinflammatory mediators are responsible for the pathogenesis of asthma. Among these mediators, there is overwhelming evidence that endogenous reactive oxygen and nitrogen species are responsible for the airway inflammation of asthma, and that the disequilibrium of the airway reducing state is a determinant of asthma severity (13, 21, 37, 58, 59, 65, 92, 119, 269, 294, 305, 357).

B. Production of ROS in asthma
Enhanced levels of oxidant production are abundantly documented in asthma. Inflammatory cells are increased in asthmatics (Fig. 11) and produce more ROS as compared to control subjects. (21, 35 –37, 145, 159, 160, 269, 305). Airway antigen (Ag) challenge in atopic individuals has been used as an experimental model to study mechanisms/mediators that lead to asthmatic responses and airway inflammation (37, 58, 92). Exposure of asthmatic individuals to appropriate Ag results in both an immediate asthmatic response occurring within minutes and a similar but prolonged late response after many hours. Asthma attacks and experimental Ag challenge are both associated with immediate formation of O2 •− that persists throughout the late asthmatic response (300). As early as 10 min following local instillation of antigen into airways of atopic individuals, over twofold increase in O2 •− generation is noted (37). Reports of O2 •− generation by airspace cells range from 4 × 106 nmol/5 × 105 cells/h (37), with production of superoxide being high at sites of Ag challenge (300). Spontaneous and experimental allergen-induced asthma attacks lead to leukocyte (eosinophil, neutrophil) activation, during which NADPH oxidase is activated and ROS such as O2 •− and its dismutation product, H2O2 are rapidly formed (16, 183).
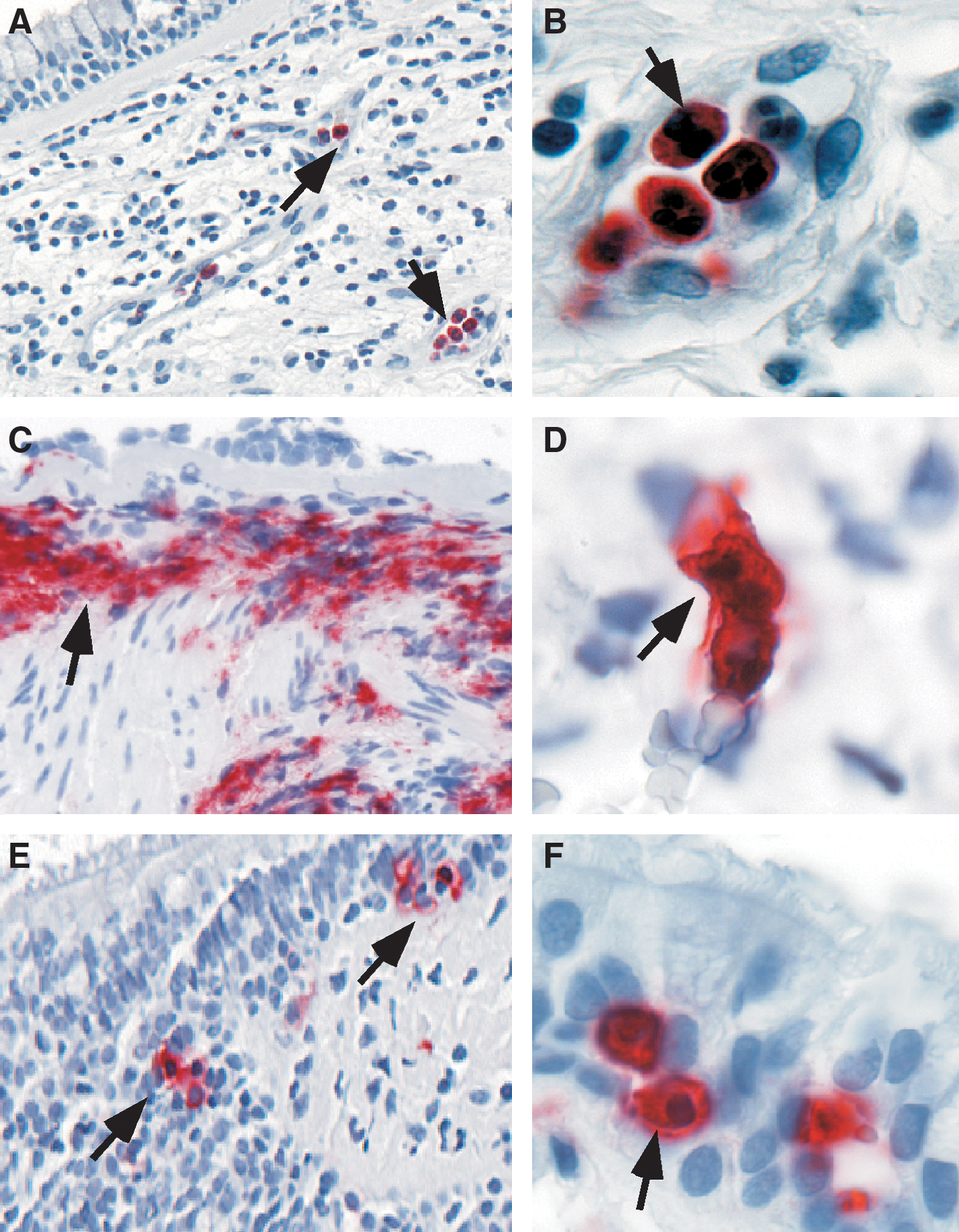
It is widely agreed that a link exists between increase of reactive species and asthma severity. For example, ROS production by asthmatics' neutrophils correlates with severity of reactivity of airways (37, 269, 299). Significant increase in neutrophils have been observed in the late-phase reaction after antigen challenge, in many cases of fatal asthma (189, 238), nocturnal asthma (222), in long-standing asthma even during periods of remission (104), and in patients with steroid responsive intractable asthma (326).
Oxidative modifications are characteristics of asthma (171, 301). Increased levels of eosinophil peroxidase and myeloperoxidase parallel numbers of eosinophils and neutrophils, respectively, and are found at higher than normal levels in asthmatic peripheral blood, induced sputum, and bronchoalveolar lavage fluid. Biomarkers of eosinophil activation include release of granule proteins including EPO (28, 45, 71, 143, 234, 254, 255, 293, 327) and major basic protein (MBP) (123, 134, 144, 340), which are readily found at high levels in blood, sputum, bronchoalveolar lavage and bronchial tissues of asthmatics (Fig. 11). Eosinophils, or MBP, in bronchial biopsies or induced sputa have been traditionally used to judge inflammation and the response, or lack of response, to therapies (8). However, activation of eosinophils and EPO generation of brominating oxidants is more accurately detected by oxidatively modified amino acids, among which 3-bromotyrosine is a unique product of EPO and eosinophils. Increased levels of 3-bromotyrosine are found in asthmatics bronchoalveolar lavage as compared to controls subjects (347). The levels of 3-bromotyrosine are increased further when asthmatics are exposed to experimental segmental antigen challenge (347). Consistent with a pathogenic link of free radicals and asthma severity, 3-bromotyrosine in airways of individuals with severe asthma admitted to the Intensive Care Unit with respiratory failure are elevated ∼100-fold over individuals in the Intensive Care Unit for nonasthma causes (217). Recent studies indicate that urinary bromotyrosine is elevated in asthmatics as compared to healthy controls, and may further increase during exacerbations, highlighting a potential role as a systemic noninvasive biomarker (141, 236).
MPO-mediated oxidant modifications also contribute to the pathophysiology of severe asthma (161). Significant (two- to threefold) elevations in chlorotyrosine are recovered from allergen challenged subsegments from asthmatic subjects undergoing segmental allergen challenge (347). Malondialdehyde and thiobarbituric acid reactive products have also been detected in urine, plasma, sputum, and bronchoalveolar lavage fluid that relate to the severity of asthma. Furthermore, 8-isoprostane, a biomarker of lipid peroxidation, is also elevated in exhaled breath condensate in adults and children with asthma (94, 237, 240, 346).
Perhaps most impressive is the striking increase of numbers and amounts of specific proteins that undergo nitration modifications in vivo in the experimental allergen-induced murine model of asthma (119). In murine and human allergen challenge studies, tyrosine nitration increases following allergen exposure of sensitized mice or atopic asthmatic humans (8, 92, 137, 152). The temporal sequence of events and airway localization of nitrotyrosine (13, 92), clearly support a link between eosinophilic infiltration and oxidation events and suggest that eosinophils may contribute to the generation of large number of oxidant products in asthma (29, 119).
C. Inhalation of exogenous ROS or RNS: Contribution to asthma severity
Recent studies have suggested that ozone and diesel exhaust particles have an additive effect on airway hyperreactivity and inflammation in asthma. Ozone increases hyperreactivity, induces IL-5 and granulocyte-macrophage-colony stimulating factor (GM-CSF) in bronchoalveolar lavage, which recruits and enhances the longevity of eosinophils in a mouse model of allergic asthma (175). Ozone also leads to oxidative modification of surfactant proteins, such as SP-A, which causes the lung to be more susceptible to lipid peroxidation and inflammation, and results in reduction of phagocytosis (235). Exposure of human airway epithelial cells to lipid ozonation products in vitro leads to activation of eicosanoid metabolism, phospholipases A2, C, and D, as well as induction inflammatory mediators such as IL-6, IL-8, and prostaglandin E2 (169, 170, 202). This provides evidence of a direct link between lipid ozonation products produced by ozone exposure and ozone-induced inflammation and cell damage (56).
Diesel exhaust particles and their components have been demonstrated to enhance airway hyperreactivity in a murine model of asthma. A recent study by McCreanor et al. demonstrated that adult asthmatics, walking for 2 h in a street with only diesel-powered vehicles, had significant reduction in lung function. These changes were accompanied by increased myeloperoxidase and 8-isoprostane in sputum and exhaled breath condensate, suggesting endogenous production of oxidants in response to the inhaled particulate materials (229, 276).
Tobacco smoke, a mixture of gases and particles that include smoke from the burning cigarette and exhaled mainstream smoke (333), contains >1014 oxidative molecules per puff of smoke, including superoxide and hydrogen peroxide. Active cigarette smoking has been associated in some studies with the development of asthma. Smoking asthmatics have an increased in morbidity and mortality as compared to nonsmoking asthmatics. Furthermore, smoking has a marked detrimental effect on lung function in asthmatic subjects and it increases the risk of severe asthma exacerbation. Cigarette smoke also influences the efficiency of inhaled corticiosteroid treatment in asthma.
Environmental tobacco smoke or second-hand smoke is also related to asthma [i.e., the association between environmental tobacco smoke exposure and pulmonary function is well documented (6, 97, 157, 198, 232)]. A recent report shows that lung function in bartenders improved after legislative ban of smoking in public places (5), and the cohort with preexisting asthma or rhinitis had the greatest increase of lung function after ban of smoking (232). This indicates that those individuals with airway inflammation have the greatest effect from inhalation of ambient free radical species. Etiological studies of the effect of environmental tobacco smoke on adults have found an increased risk of asthma, dose-dependent relationship to wheezing, and a greater risk for more severe airflow obstruction (147, 156, 163, 164, 191, 204, 329). The importance of environmental tobacco smoke in the etiology of asthma in children has been established. Environmental tobacco smoke exposure of children related to parental smoking is associated with poorer lung functions in asthmatic children, and the relative risk of asthma is greater in children exposed to cigarette smoking by both parents compared with smoking of neither parent (67, 147, 156, 158, 337).
Taken together, these data indicate that exogenous oxidant species contribute to asthma severity and asthma pathogenesis.
D. Nitric oxide in the lungs: Relation to oxidative modifications
Evidence supporting increased •NO in asthma is substantial (92, 113, 126, 172, 264). •NO is increased in the lower airway and in the exhaled breath of asthmatics (92, 113, 126, 172, 264) (Fig. 12). Exhaled NO is clinically used as a noninvasive biomarker of asthma and therapeutic responsiveness (113, 308) but some studies suggest limitations of its value (243).
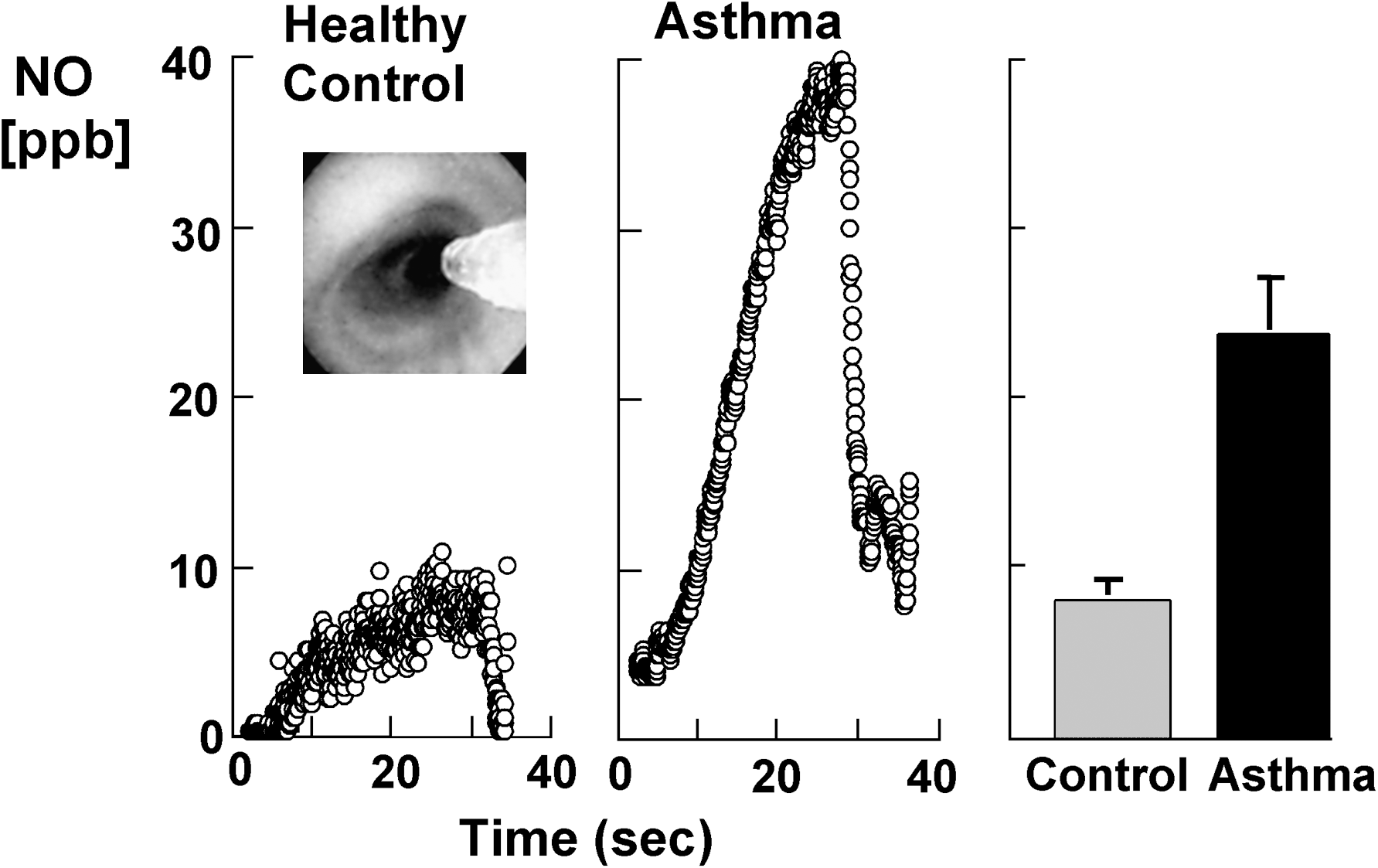
Exhaled •NO in asthmatics increases after allergen challenge during the late asthmatic response (92, 174). It is increasingly suggested that high-output synthesis of •NO is a marker of, and/or contributes to, the airway inflammation that defines asthma. Multiple mechanisms function together to support high level •NO synthesis in the asthmatic airway. Individuals with asthma have 3-fold higher than normal •NO concentrations, and increased NOS2 mRNA and protein in airway epithelial cells (126, 127). This is principally observed in steroid-naïve patients with atopic asthma, and the inter-individual variation in exhaled nitric oxide concentrations can be significant. The increase in •NO concentrations is due to increased transcriptional activation of the NOS2 gene and a greater catabolic breakdown of storage pools of GSNO in the lung related to alterations in the redox state (113, 115, 273).
The biological effects of •NO have been attributed to its binding to guanyl cyclase, but its byproducts also have a biologic role. The biochemistry of NO oxidation products is critical in the balance of beneficial and adverse effects associated with •NO. For example, NO synthesis under oxidative and acidic conditions causes injury, in part because •NO oxidation in weak acid yields ONOOH and HONO (92, 113, 115). The dynamics of •NO metabolism in the asthmatic airway during an experimentally provoked asthmatic response to Ag reveal multiple and sequential reactions, and suggest a multifunctional role for •NO in the airway. In comparison to healthy controls, mild well-controlled atopic asthmatics tend to have increased •NO, NO3 −, and nitrotyrosine but undetectable S-nitrosothiols (SNO) in the lower airways. Within minutes of Ag-induced asthmatic response, NO3 − increases markedly in all asthmatics, while NO2 − or SNO do not change, and •NO tends to decrease. Decreasing •NO and increasing NO3 − suggests that •NO may be reacting with O2 •− to yield ONOO−, which subsequently decays to NO3 − or leads to nitrotyrosine formation (92). NO3 − may also be formed as a product of peroxidase generated RNS (29, 92). In the late asthmatic response, nitration of thiols may occur by ONOOCO2 − mediated thiol oxidation and nitration, or by free radical events such as formation of thiyl radicals (Fig. 7). Despite notable changes in asthmatic airways, healthy control individuals have no changes in levels of •NO or NO reaction products, even after challenge with aerosolized allergen.
The content of nitrotyrosine in airway proteins recovered from patients with severe asthma are an order of magnitude higher than those in healthy controls (217). It has been postulated that increased levels of HOBr production may result in increased peroxynitrite formation by interaction of HOBr with •NO, which favors nitration. Levels of nitrotyrosine have been found to be elevated in exhaled breath of asthmatics, and immunoreactivity to nitrotyrosine has also been shown to present in airway epithelial cells of asthmatics. Furthermore, increased nitration is found during an asthma exacerbation (92, 217, 347) and S-nitrosothiols concentrations are elevated in exhaled breath condensate in patients with asthma (69). Persistently increased ROS and NO in asthma leads to RNS formation, and subsequent oxidation and nitration of proteins, which may cause alterations in protein function that are biologically relevant to airway injury/inflammation. The measurement of nitration of tyrosine residues, which form from a reaction product of superoxide and NO, provides a stable and quantitative marker of tissue oxidative stress.
On the other hand, NO synthesis can also decrease airway resistance, an effect mediated in part by formation of the endogenous •NO oxidation product and bronchodilator, S-nitrosoglutathione (GSNO) (113, 115). GSNOR, glutathione-dependent formaldehyde dehydrogenase (FALDH; EC 1.2.1.1) is a ubiquitous enzyme known as a class III alcohol dehydrogenase. FALDH catalyzes the NAD+-dependent formation of S-formylglutathione from S-hydroxymethylglutathione, which forms spontaneously by condensation between formaldehyde and glutathione. Recently, it has been demonstrated that FALDH is very active in reduction of GSNO, which leads to generation of NO (48, 85). Unfortunately, airway activity of GSNO reductase (GSNOR) is increased in asthma (114). In fact, GSNOR substrate, GSNO, is undetectable in the human airway during asthmatic respiratory failure (114, 273). GSNOR-deficient mice, which cannot break down GSNO, are completely protected from methacholine hyper-reactivity following allergen sensitization and challenge (273).
GSNO inhalation increases exhaled NO in humans in part because GSNOR reduces GSNO to hydroxylamine which is converted to NO by catalase (105). Thus, increased airway GSNOR activity can lead to increased exhaled NO and methacholine hyper-responsiveness.
E. Redox imbalance in asthma
1. Oxidative stress
Homeostasis of cellular functions during oxidative stress depends on the rapid induction of protective antioxidant enzymes (240). For example, detectable concentrations of 8-isoprostane in EBC in healthy subjects are reported and suggest “physiological” levels of oxidative processes (240). Naturally occurring antioxidants exist to protect cells and tissue against the continuous production of ROS/RNS during normal metabolism (139). However, high levels of reactive species may overwhelm the antioxidant defenses, resulting in oxidant-mediated injury or cell death (15, 42). The terms “oxidant stress” or “oxidative stress” are often used to refer to this effect (132). Studies suggest that oxidant stress plays a crucial role in the initiation and progression of asthma.
2. Antioxidant deficiency in asthma
Both enzymatic and nonezymatic antioxidants are employed with the lung. The lung epithelial surface lining fluid contains several nonenzymatic antioxidants, such as glutathione, ascorbic acid, albumin, and alpha-tocopherol. Enzymatic antioxidants defenses are present in the epithelial lining fluid as well as in plasma and epithelial cells. Asthma is characterized by loss of antioxidant activities.
a. SOD deficiency
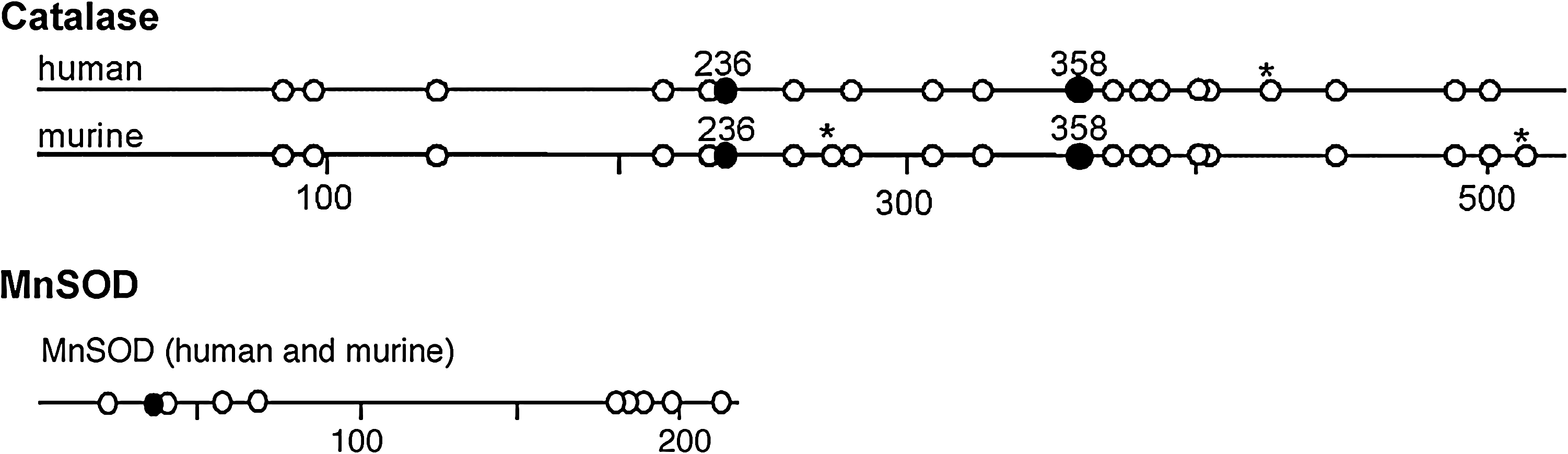
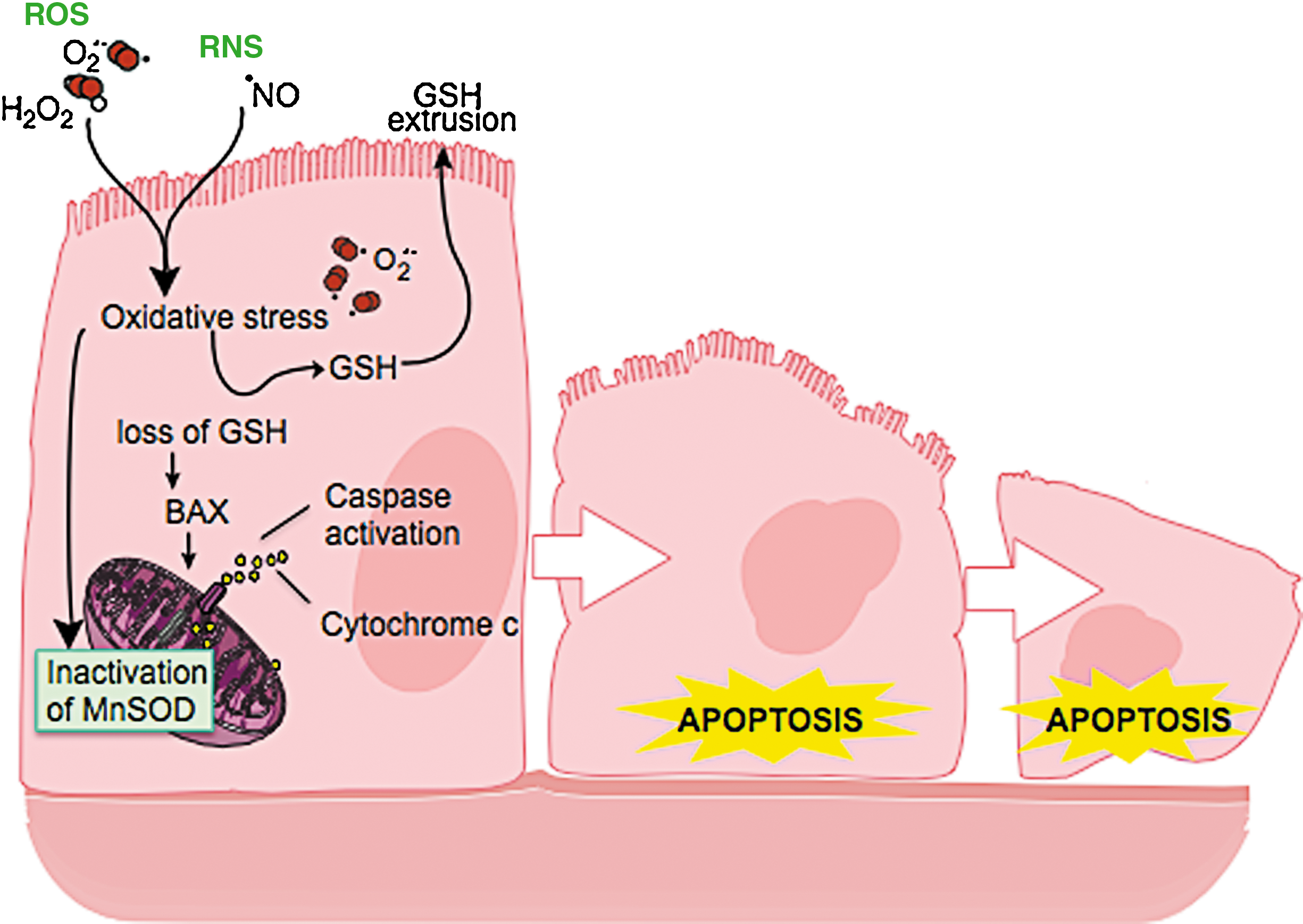
In asthma, SOD activity is significantly lower in epithelial lining fluid and airway epithelial cells as compared to healthy controls. Loss of SOD activity occurs within minutes of an acute asthmatic response to segmental antigen instillation into the lung of individuals with atopic asthma. This rapid decrease in SOD activity occurs in relation to a twofold increase in O2 •− generation after antigen instillation into airways of atopic individuals (37). DeRaeve et al. and Smith et al. initially showed a correlation between the degree of airway reactivity and SOD activity levels (82, 313). Later studies in large populations confirmed that airway reactivity is inversely related to SOD activity (58, 63, 65). Together, these findings support a link between SOD activity and physiologic parameters of asthma severity. Murine models of asthma also provide evidence of a link between antioxidants and airway hyper-responsiveness. For example, transgenic mice that overexpress SOD have decreased allergen-induced physiologic changes in the airway in comparison to controls (197). Studies indicate that the lower SOD activity in asthma is a consequence of the increased oxidative and nitrative stress in the asthmatic airway, and thus serves as a sensitive marker of airway redox and asthma severity. Reduction in SOD activity can also contribute to oxidative stress and perhaps asthma severity. Oxidatively modified and nitrated MnSOD is present in epithelial cells recovered during bronchoscopy from asthmatic airways (65, 119). Stable isotope dilution tandem mass spectrometry of MnSOD isolated from human asthmatic airways reveals the presence of oxidation of phenylalanine and tyrosine residues. Dominant modifications include nitration of tyrosine, nonphysiologic tyrosine isomers [m-Tyr (meta-tyrosine) and o-Tyr (ortho-tyrosine)] that typically occur with exposure to hydroxyl radical-like oxidants, chlorination of tyrosine (a specific molecular marker for myeloperoxidase-catalyzed halogenation), and oxidative cross-linking of tyrosine as monitored by dityrosine (a product of tyrosyl radical) (63, 119, 129, 217). This pattern of oxidative modification is consistent with MnSOD exposure to Fenton/Haber–Weiss reaction mechanisms in asthmatic airways. The presence of a diverse array of distinct oxidative modifications indicates functional impairment of activity due to oxidative processes. Generation of reactive oxygen and nitrogen species is greatly increased during acute asthma attacks (37, 217, 347). Thus, loss of SOD contributes to oxidative stress during acute asthma exacerbations (37, 58, 217, 347). Other reports have shown that MnSOD is a target for tyrosine nitration and oxidation (213, 215), which leads to loss of enzyme function, and tissue injury (214, 215). Based on the reported quantitative data on MnSOD oxidation and nitration in human asthmatic lungs, up to 10% of MnSOD recovered from asthmatic airway epithelial cells possess at least 1 oxidative modification (65) (Fig. 13) (Table 3). Although it is unclear whether this average amount of modification of MnSOD can affect redox and cell functions in vivo, oxidative modification/inhibition of MnSOD triggers apoptosis in airway epithelial cells in vitro. Cleavage fragments of caspase-9 (35 kDa) and PARP (85 kDa) are present in asthmatic epithelial cells and are correlated with airflow in asthma. Apoptosis and shedding of epithelial cells are also observed in asthmatic patients (31, 89, 253, 331, 332) (Fig. 14). Thus, the redox modifications of SOD may contribute to a major component of asthmatic airway remodeling, airway epithelial apoptosis, which leads to denudation of the airway surface and predisposes to greater airway hyperreactivity. Recent studies also report a loss of circulating SOD activity in asthmatics. However, the isoform of SOD responsible for the loss is not known. The intracellular enzymes CuZnSOD and MnSOD are released to the circulation during normal turnover of cells and account for serum SOD activity. Although EC-SOD is found in extracellular matrix space, it is bound to heparan sulfate proteoglycans of endothelial cell surfaces and <1% of EC SOD is found in the serum (171, 301). Thus, EC-SOD contributes very little to serum SOD.
Ranges in values of oxidative modifications observed in MnSOD and catalase from epithelial cell brushings from mild asthmatic subjects. The numbers are normalized to the content of the precursor amino acid (mmol oxidation product/mol precursor tyrosine or phenylalanine), which is monitored within the same injection. All data are representative of 4 asthmatic individuals. BrY, bromotyrosine; ClY, chlorotyrosine; DiY, dityrosine; mY, m-tyrosine; NO2Y, nitrotyrosine; oY, o-tyrosine; Phe, phenylalanine; Y, tyrosine. From Ghosh et al. (102) and Comhair et al. (56).
Recent studies also report a loss of circulating SOD activity in asthmatics. Similar to the correlation of airway SOD to lung function, serum SOD activity is related to asthma lung function, and this relationship appears to be unique to asthma since serum antioxidant capacity in chronic obstructive pulmonary diseases is unrelated to airflow limitation (62, 63, 263, 282). In vitro studies have shown that reactive oxygen and nitrogen species lead to oxidative and nitrative modification of tyrosine and inactivation of MnSOD and ECSOD, while Cu,ZnSOD can be inactivated by ROS and RNS through targeting of critical histidine residues and formation of histidinyl radicals (7, 213, 216).
Oxidative modification/inactivation of MnSOD is present in asthmatic airway epithelial cells (65). Altogether, the global loss of SOD activity reflects the increased oxidative and nitrative stress in asthmatic patients. This suggests that SOD may serve as a surrogate marker of oxidant stress and asthma severity (63, 65, 313).
Recent studies suggest that angiogenesis occurs in asthma (205, 295) and indicate a relation between the numbers of blood vessels in the bronchial wall and the severity of asthma (12, 260, 295, 338). VEGF-transgenic mice have an asthma-like phenotype with Th2-type inflammation, parenchymal and vascular remodeling, edema, mucus metaplasia, myocyte hyperplasia, and airway hyper-responsiveness (199). Reactive oxygen species such as superoxide and hydrogen peroxide enhance VEGF expression (192), while exogenous SOD prevents VEGF expression (192). These data suggest that the increased vascularization found in asthma may be due to the involvement of oxidative stress, perhaps via effects on hypoxia inducible factors.
b. Catalase inactivation
Red blood cells of asthmatic children were shown to have lower catalase activity than healthy children >15 years ago (251). Recently, catalase activity was found to be 50% lower in bronchoalveolar lavage of asthmatic lungs, as compared to healthy controls (120). The reduced catalase activity is not due to lower protein levels. Rather, catalase isolated from asthmatic airway epithelial cells has increased protein oxidation markers, including nitrotyrosine and chlorination and oxidation of sulfhydryls, linking oxidative modification to the reduced activity in vivo. Tyrosine oxidant modifications of catalase occur in asthma: chlorination of tyrosine by peroxidase-catalyzed halogenation, and oxidative cross-linking of tyrosine as monitored by dityrosine, a product of tyrosyl radical (120). The most extensive modification found in asthmatic lungs is tyrosine chlorination, which is 20-fold more extensive than tyrosine nitration (120). Unlike MnSOD, oxidation of phenylalanine to the nonphysiologic tyrosine isomers, m-Tyr and o-Tyr, is rare, indicating that exposure to hydroxyl radical-like oxidants through Fenton/Haber–Weiss reaction mechanisms is not prevalent in the oxidation of catalase.
Interestingly, catalase contains a recently identified putative chlorination site (KXHY) at Tyrosine 236, which may influence the susceptibility of the enzyme to peroxidase activity (26). On the other hand, tyrosine modification itself is not likely the complete cause of the loss of catalase activity. Other oxidative modifications, specifically oxidation of the cysteine 377 to cysteic acid, contribute to activity loss of the enzyme (120). Nevertheless, altogether the studies provide strong evidence that loss of antioxidant activities occur by multiple different oxidant mechanisms in asthmatic airways, which may be related to the enzyme structure and function and/or intracellular localization in different compartments of the cell.
c. Glutathione systems in asthma
In contrast to SODs and catalase, extracelluluar GPx (eGPx) is present at higher than normal levels in lungs of individuals with asthma (59, 60, 62, 64). The increase is due to induction of eGPx mRNA and protein expression by bronchial epithelial cells in response to increased intracellular or extracellular reactive oxygen species (59, 64). Not all oxidative stress will lead to increase of eGPx, for example, exposure to ozone decreases levels of eGPx protein and activity, whereas no change is detected with exposure to NO2 (14). It has been known for some time that alterations of GSH and GSSG levels and the ratio of GSSG/GSH are present in asthmatic airways (58, 59, 82, 312). Levels of glutathione in exhaled breath of children with asthma during acute asthma exacerbation are lower than control subjects (68), and the glutathione levels in exhaled breath of subjects with asthma increase after oral steroid treatment compared with pretreatment levels (68). Asthma and asthma exacerbations lead to rapid changes in intracellular as well as extracellular GSH and GSSG. Rapid changes in redox potential occur immediately after antigen challenge in epithelial lining fluid of asthmatics (58). Minutes after challenge, GSH levels drop and GSSG increases in the lung epithelial lining fluid, which verifies loss of reducing potential in asthmatic airways (58). GSH depletion in vivo and/or in vitro leads to inhibition of Th1-associated cytokine production and/or favors Th2-associated response (265). Thus, GSH facilitates a Th2 phenotype, and reduction in GSH levels supports the maintenance of Th2 response in asthma (265). Shifts of intracellular/extracellular pools of glutathione alter intracellular redox balance; efflux of GSH reproducibly activates BAX and cytochrome c release in cell lines (Hela cells and U937, monocyte cell line) and is one established mechanism for induction of apoptosis (117, 118, 167) (Fig. 14). Hence, alterations in the GSSG/GSH ratio and intracellular/extracellular distribution likely also contribute to the airway epithelial cell apoptosis in asthma.
Changes in the cellular redox status lead to formation of mixed disulfides between protein sulfhydrl groups and glutathione (S-glutathionylation) on multiple proteins. Glutathionylation of proteins is reversible, as those proteins can be reduced by glutaredoxins and thioredoxin (78, 79). Glutaredoxins are expressed in human alveolar macrophages and lung homogenates and to a lesser extent in bronchial epithelial cells (262, 279). A recent report by Reynaert et al. demonstrates that glutaredoxin 1 is upregulated in a mouse model of asthma (287). During asthma exacerbation in humans, the levels of serum TRX1 increase and are inversely correlated with airflow (350). This suggests that TRX may have a protective effect in asthma. In vitro studies have shown that exogenous TRX1 can prevent Th2 development by upregulating the expression of Th1-like cytokines, leading to a decrease in airway reactivity and airway inflammation (145). Because TRX1 reduces oxidization of proteins or the levels of hydrogen peroxide together with peroxiredoxin (247), the protective effects of TRX1 in asthma are thought to be partly dependent on its antioxidant effect (145). Through antioxidant effects, TRX1 regulates redox-sensitive signaling pathways (247), and may further affect pro-inflammatory pathways. One other target of TRX is the family of Prxs, which are reduced by TRX. Interestingly, a recent report by Avila et al, shows that Prx5 is increased in sputum of asthmatic and during viral-induced inflammation. Lehtonen et al. demonstrated that Prx1, 5 and 6 are upregulated in bronchial epithelial cells and alveolar macrophages of COPD (201).
3. Redox-dependent transcriptional regulation
Redox reactions have attracted attention as important chemical processes that regulate signal transduction. The response of a cell to a reactive oxygen- and nitrogen-rich environment often involves the activation of numerous intracellular signaling pathways, which can cause transcriptional changes and allow the cells to respond appropriately to the perceived oxidative stress. For example, at least two well-defined transcription factors, NF-κB and activation protein-1 (AP1), are regulated and influenced by the redox status and are implicated in the transcriptional regulation of a wide range of genes involved in oxidant stress and cellular response mechanisms (88, 221, 306). In addition to the activation of transcription factors, evidence suggest signaling pathways such as the family of mitogen-activated protein kinases (MAPKs) are directly or indirectly altered by redox changes (56, 209). In the nucleus, redox affects histone acetylation and deacetylation status, which at least partly regulates inflammatory gene expression by activation of the redox sensitive transcription factors (209).
a. Transcription factors NF-κB and AP1
Redox-sensitive molecular targets, such as transcription factors, usually contain highly conserved cysteine residues, and oxidation, nitrosylation, or the formation of disulfide links are crucial events in oxidant-redox signal.
Nuclear factor-κB (NF-κB) is activated in epithelial cells and inflammatory cells during oxidative stress, leading to the upregulation of a number of pro-inflammatory genes. NF-κB is a protein heterodimer made up of p65 and p50 subunits. There is evidence of activation of NF-κB in biopsies and sputum inflammatory cells such as macrophages and neutrophils of asthmatics (136). Many of the inflammatory genes responsible for the pathogenesis of asthma are regulated by NF-κB. Nitrosation of NF-κB subunits is an important mechanism for the redox sensing of NF-κB. In an elegant series of experiments, Reynaert et al. demonstrated that S-glutathionylation regulates activation of the NF-κB pathway (286, 287). Glutaredoxin-dependent reversal of S-glutathionylation of the inhibitory kappa-B kinase (IKKβ) modulates the activation of NF-κB in response to redox changes by protecting IKKβ from irreversible inactivation (286).
Activator protein-1 (AP-1) is a protein dimer, composed of a heterodimer of Fos and Jun proteins. The oxidant-sensitive cysteine in the DNA-binding site of c-Jun undergoes reversible S-glutathiolation during oxidative stress in the presence of physiologic levels of GSH (182). AP1 regulates many of the inflammatory and immune genes in oxidant-mediated diseases. Gene expression of γ-GCS, the rate-limiting enzyme for the GSH synthesis, is induced by the activation of AP1 (281). Asthmatic epithelial cells have increased expression of c-Fos. Cigarette smoke increases AP-1 DNA binding in human epithelial cells in vivo (281). High levels of NO and hydrogen peroxide cause increases in c-fos and c-jun mRNA of epithelial cells (281). The binding sites of the redox-regulated transcription factors NF-κB and AP-1 are located in the promoter regions of many antioxidant genes, such as NOSII and GPx, which are directly involved in lung diseases such as asthma (11, 221, 304, 348).
Recent evidence indicates that chromatin remodeling plays a critical role together with NF-κB and AP-1 in the activation of inflammatory genes such as NOS2, IL-8, and TNFα. Oxidative stress and other stimuli, such as cytokines, activate various signal transduction pathways leading to activation of transcription factors, such as NF-κB, and AP-1. Binding of transcription factors to DNA elements leads to recruitment of CREB-binding protein (CBP) and/or other co-activators to the transcriptional initiation complex on the promoter region of various genes. Activation of CBP leads to acetylation (Ac) of specific core histone lysine residues by an intrinsic histone acetyltransferase (HAT) activity (276). This results in the acetylation of core histones, opening up the chromatin structure to allow binding of RNA polymerase II, which initiates gene transcription. The process of acetylation and deacetylation of histone is also influenced by redox changes (274, 275). In biopsies and peripheral blood mononuclear cells from asthmatics, there is an increase in acetylation and a reduction in deacetylation activity, which upregulates some inflammatory gene expression and downregulates others (70). Redox changes also can activate members of the mitogen-activated protein kinase signaling (MAPK), such as extracellular signal-regulated kinase (ERK), c-jun N-terminal kinase (JNK), p38 kinase, and phosphoinositol-3 kinase, all of which may ultimately promote inflammation (47, 334).
b. Redox-dependent activation of JAK/STAT pathway
Binding of cytokines, including interleukin-4 and interferons, to their specific receptors leads to transphosphorylation of tyrosine residues on Janus kinases (JAK), which then recruit and phosphorylate the signal transducers and activators of transcription (STAT) family of transcription factors on tyrosine residues and result in gene expression of pro-inflammatory genes such as NOS2. Many of the inflammatory and immunomodulatory cytokines important in asthma pathogenesis signal through this canonical pathway. Multiple studies now support that STAT1 and STAT3 activation is redox regulated. Although STAT3 has not been evaluated in asthma, STAT1 is activated at high levels in asthmatic airway epithelium but not in healthy controls (129). Among the first reports, Simon et al. showed that members of the STAT family of transcription factors, including STAT1 and STAT3, are activated in response to H2O2 or GSH-depletion, and inhibited by antioxidants. STAT activation is H2O2 specific, not occurring in response to superoxide or NO (309), and effects are mediated via JAK2 and tyrosine kinase 2 (TYK2). Vanadium compounds in the particulate matter of air pollution, which are released from the industrial burning of fuel oil, activate signal transduction via the generation of H2O2; Wang et al. showed that vanadium leads to STAT-1 activation (339). Recently, the detailed redox mechanisms that regulate STAT activation by IL-4 have been identified (307). Upon ligand-receptor binding, ROS are rapidly generated by phosphatidylinositol 3-kinase-dependent activation of the NAD(P)H oxidase (NOX)1 and NOX5L. Consequently, cysteine oxidation by ROS inactivates protein tyrosine phosphatase 1B, which is then unable to inactivate/dephosphorylate the receptor. This amplifies the cytokine signal transduction and results in greater JAK/STAT activation and enhances the inflammatory response to cytokines (309). Thus, homeostatic control of cytokine-receptor activation and signal transduction occurs through ROS generation via activation of NOX enzymes; the ROS subsequently promote the receptor's own activation and the activation of other cytokine receptors in the cell, enabling cytokine signaling crosstalk through redox events.
F. Genetics of redox in asthma
Since SOD is decreased in asthma, and its activity is strongly related to asthma pathophysiology, it has been hypothesized that genetic variability of SODs may play a role in the development of asthma. Single nucleotide polymorphism Ala16Val of MnSOD is common and may change the secondary structure and mitochondrial targeting of the protein (180). A polymorphism (R213G) of EC-SOD causes more than ninefold higher levels of EC-SOD in plasma due loss of anchoring to heparin in the interstitium (301). The EC-SOD R213G polymorphism is associated with reduced exacerbations in COPD and lower rates of hospitalization (168). However, the functionally important genetic variants Ala16Val (MnSOD) and R213G (ECSOD) are not associated with genetic susceptibility to develop asthma (180). Genetic variation in the untranslated regions of transcripts, as well as intronic polymorphisms, can be functionally important, both of which have been described in the SOD gene loci. For example, two novel polymorphisms occur in the noncoding 5' untranslated region (Exon 1) and first intron (Intron 1) of the SOD3 gene (77). Both polymorphisms are situated in a conserved mammalian interspersed repetitive element, and may be functionally relevant to lung disease. Although not evaluated in asthma, a recent report by Dahl et al. found that EC-SOD homozygous for the Exon1/Intron1 polymorphism associates with reduced lung functions in individuals with chronic obstructive lung disease (77). This supports a role for EC-SOD in oxidant-mediated events influencing airway diseases and lung function (77).
The glutathione S-transferase superfamily includes a number of subclasses including glutathione S-transferase P1 and glutathione S-transferase M1 which are expressed in the lungs and have been implicated in asthma pathogenesis (317). The deletion allele of GSTM 1 (null-genotype) has been associated with increased risk of asthma and lower lung function (121, 325). A functional sequence variant in GSTP1 at codon 105 (Ile105Val- rs1695) (148, 323) has also been associated with asthma in some studies (249, 325). This variant has been reported to be both protective (249) or a risk factor (200, 325) for asthma. There are reports indicating a gene-pollution interact with asthma pathogenesis (210). A recent study by Islam et al. (155), reports that children with a Val105 mutation in GSTP1 variant allele may have a lower risk of asthma associated with exercise, especially with high ozone levels.
V. Clinical Implications
Inhaled glucocorticosteroids are the mainstay of therapies for most adult asthmatics (154). Other drugs such as cromolyn sodium, nedocromil, and leukotriene modifiers can also be used. Oral corticosteroids are used when inhaled therapies are inadequate. However, subsets of patients with severe asthma are refractory to treatment or develop significant side effects to the medications (154). Antioxidant therapy may be a safe and effective alternative. However, there is no evidence to support a benefit of antioxidants in severe asthma.
A. Clinical monitoring of redox in asthma
The redox abnormalities found in asthma result in distinct oxidant species. Protein bromination, lipid peroxidation, and NO production represent distinct biochemical pathways that have all been associated with the pathophysiology of asthma (217, 280, 347). Although the labile nature of oxidant species makes them difficult to quantify, the stable end-products of distinct oxidation pathways may be used as reliable indices of airway oxidative stress.
Elevated levels of 3-bromotyrosine and F2-IsoPs have been detected in both urine and exhaled breath condensates of asthmatics and are being evaluated as noninvasive biomarkers of asthma (9, 18, 95, 237, 240, 241, 259).
Numerous studies have demonstrated increased NO production in the airways of asthmatics, due at least in part to upregulation of inducible nitric oxide synthase (iNOS) in cells like bronchial epithelial cells (92, 93, 172 –174). The technology for exhaled NO measurement has developed rapidly over the last 2 decades and uniform global standardization of techniques has enabled the use in clinical practice.
Exhaled NO is widely accepted as a measure of inflammation, in particular eosinophilic inflammation. NO monitoring is not likely helpful for smoking asthmatics, since smoking reduces exhaled NO levels. NO also reflects the airway redox in asthma; as the airway environment becomes more oxidizing and acidic; greater release from storage pools of GSNO also leads to high level of exhaled NO (113, 115). Exhaled breath condensate (EBC) pH is low in acute asthma exacerbations and increases with corticosteroid therapy. EBC-based pH assays may be of value in monitoring airway redox stress in the ambulatory environment. The rapid advances in the technologic processes for measure of stable oxidant endproducts provide confidence that many more biomarkers (or panels of biomarkers) with diagnostic and prognostic utility for evaluating the presence and activity of asthma will be available in the near future.
B. Antioxidant therapeutic strategies
1. Redox-sensitive transcription factors
NF-κB and AP1, are activated and contribute to the pathogenesis of asthma. Thus, several therapeutic strategies have been used to develop small antioxidant molecule inhibitors of these redox-regulated transcription factors. One small molecule inhibitor of AP1 transcription, PNRI-299, was discovered using a template that screened for molecules that bind to redox active substances. PNRI-299 selectively inhibits AP1 transcription but not NF-κB or thioredoxin (248); its intracellular molecular target is the oxidoreductase redox effector factor-1 (REF-1). In the murine model of asthma, PNRI-299 effectively reduces airway eosinophil infiltration, mucus hypersecretion, and IL-4 levels (248). Another small antioxidant molecule inhibitor of NF-κB and AP1 transcription, MOL 294, also successfully blunts airway inflammation and hyperreactivity in a mouse model of asthma. MOL 294 inhibits both NF-κB and AP1 via inhibition of thioredoxin. Intranasal administration of MOL 294 markedly reduces airway eosinophilia and mucus hypersecretion. Although not tested in humans, these studies suggest that therapies that target redox-regulated signal transduction pathways are feasible and can reduce allergic airway inflammation and reactivity (140). A limitation of this strategy is the relative lack of selectivity of transcription factor inhibition.
2. SOD therapies
Antioxidants have been proven beneficial in control of ischemia/reperfusion injury (76), shock and lung injury related to radiation therapy and chemotherapy (99), and in chronic inflammatory disorders such rheumatoid arthritis and osteoarthritis (298). Asthma is associated with decrease in antioxidant SOD, thus strategies aimed at increasing airway SOD levels may be a rational approach to more effective therapy for asthma. Further support for SOD therapy comes from murine models of asthma, which provide a link between antioxidants and airway hyper-responsiveness. For example, transgenic mice that overexpress SOD have decreased allergen-induced physiologic changes in the airway in comparison to controls (197). In other acute and chronic inflammation models, SOD mimetics reduced PARP immunofluorescence, providing evidence of a role for SOD in inhibition of apoptosis and inflammation (296, 297). Similarly, treatment with SOD mimetics reduces the magnitude of ovalbumin-induced airway hyper-responsiveness to methacholine in murine models of asthma (52). Exogenous EC-SOD given intratracheally to mice treated with asbestos, decreases neutrophil influx and oxidative matrix degradation (186). However, manipulation of endogenous SOD for therapeutic purposes has been problematic due to its short half-life and large molecular weight. A number of SOD mimetics based around organomanganese complexes have been developed, which retain their antioxidant properties in vivo. SOD mimetics include manganese (II) penta-azamacrocyclic complex (M40401, M40403, and M40419) (17, 231, 289, 298), manganese (III) (salen) (EUK134) (22, 23, 315) complexes, and manganese metaloporphyrin class 9 AEOL-10113, AEOL-10150 of SOD. Masini et al. demonstrated that SOD mimics given before antigen challenge of sensitized guinea pigs attenuate allergen-induced asthmatic bronchospasm (223). These animal studies collectively provide proof of the concept that SOD mimetic may be effective in treating asthmatic airway inflammation. However, clinical trials on the effects of SOD mimetics in patients with asthma have not been performed and thus efficacy of SOD mimetics in treatment of asthma is yet to be tested.
3. Glutathione system
Other potential untested strategies include the glutathione peroxidase mimetic, Ebselen, which is a nontoxic seleno-organic drug and an effective reductant of hydroperoxides (303). Ebselen inhibits airway inflammation by reducing neutrophil recruitment and chemokine expression in various animal models of inflammation (4, 225, 279). Resveratrol (3,5,4’-trhihydroxystilbene), a phytoalexin that is found in seeds of grapes, has been reported to have antioxidant, anti-inflammatory, and anticarcinogenic properties (188). Studies have shown that resveratrol effectively inhibits oxidative damage and scavenges free radicals such as lipid hydroperoxyl, hydroxyl (· OH), and superoxide (203). Resveratrol induces GSH synthesis and attenuates oxidative stress and depletion of GSH in lung epithelial cells (188). In primary lung epithelial cells, resveratrol (10 μM) attenuates cigarette smoke-mediated GSH depletion by inducing GSH synthesis which protects the epithelial cells (164) This compound may be benefical for patients with asthma (75).
4. Dietary antioxidants
Dietary antioxidants may improve asthma control or reduce its incidence. Epidemiological studies suggest associations between low dietary antioxidant intake, reduced lung function, and increased respiratory symptoms in asthmatics (34). The most recent Third National Health and Nutrition Examination Survey (NHANES III) findings indicate an association of some antioxidants with asthma in children who have environmental tobacco smoke exposure asthma (292). A large cross-sectional study in NHANES III shows that selenium and serum vitamin C are lower in young asthmatics as compared to nondiseased controls (292). Serum vitamin C and selenium were inversely associated with asthma, and the association was most evident in those exposed to the added oxidant stress of environmental cigarette smoke (292). The provocative findings suggest that dietary supplementation with selenium in those individuals with cigarette smoke exposure should be considered (292). Likewise, studies have revealed that asthmatics have lower than normal levels of coenzyme Q (CoQ) (116). Also known as ubiquinone, CoQ participates in electron transport in cellular respiration in the mitochondria. In its reduced form, ubiquinol (QH2) also serves as an antioxidant. One study in asthma and others in cardiovascular diseases has shown that CoQ increases SOD activity and thus therapy with CoQ may benefit in asthma (25, 130, 330). In a cross-over randomized study of 41 asthmatics who were all receiving corticosteroids, supplementation with CoQ [Q-Gel® (120 mg), 32 weeks] improved asthma control and enabled reduction of corticosteroid dose (130). These studies all support the concept that antioxidant supplementation and/or reduction in oxidant production or exposures will be beneficial in the treatment of asthma (Table 4).
VI. Conclusions and Future Directions
Asthma is a chronic inflammatory airway disease, and it is clear from multiple lines of evidence that the airway inflammation is defined by alterations of the airway redox. Redox-mediated post-transcriptional modifications lead to protein structure–function changes that are present even in mild asthmatics who are well-controlled. The abnormalities in redox are magnified in the asthmatic airway in response to exacerbating factors, including microbial infection, exposure to inhaled oxidizing pollutants, or allergen triggers in atopic individuals. During leukocyte activation, such as following allergen exposure, a respiratory burst occurs, generating O2 •− and its dismutation product H2O2. Fenton/Haber–Weiss reactions affect endogenous proteins, such as MnSOD. Oxidative modifications of MnSOD amplify the oxidative milieu in the mitochondria, with potential adverse consequences on cellular respiration and cell survival. As eosinophils and/or neutrophils enter the inflamed airway, H2O2 is used in eosinophil peroxidase and/or myeloperoxidase-mediated reactions that oxidatively modify susceptible proteins in the airway environment. Among those proteins is catalase, the antioxidant enzyme that would otherwise act to control the amounts of H2O2 present in the airway. This enables more H2O2 to accumulate at the site of inflammation and further promotes peroxidase systems to produce high levels of nitrating, halogenating, and oxidizing injurious species. The greater toxic nitrogen oxides and airway acidity is accompanied by loss of beneficial nitrogen oxides, in particular nitrosothiols, which have adverse effects on smooth muscle relaxation and airway reactivity. In addition to injury of macromolecules, RNS and ROS amplify specific cytokine signal transduction by processes that include inhibition of deactivating signals. The loss of downregulatory signal transduction events further amplifies the inflammatory milieu and may contribute to Th2 lymphocyte polarization and development of the atopic environment typically seen in the asthmatic airway. Thus, alteration of redox participates in the pathophysiology of asthma. Finally, future therapy targeting redox will require the definition of the clinical pharmacology of antioxidant compounds. Furthermore, identification of noninvasive biomarkers of oxidative stress in patients with asthma will be critical for enabling assessment of treatment outcomes. Nevertheless, the cumulative data provide a compelling rationale to develop these types of therapeutic strategies for asthma that aim to correct the redox abnormalities.
Footnotes
Acknowledgments
This study was supported by Grants HL081064, HL69170, AI70649, from the National Institutes of Health.
Abbreviations Used
Reviewing Editors: Andrew Ghio, Vuokko Kinnula, Corrine Kliment, Paolo Montuschi, Sekhar Reddy, and Carl White