
Abstract
Innate and adaptive immunity affect the pathogenesis of Parkinson's disease (PD). In particular, activation of microglia influences degeneration of dopaminergic neurons. Cell-to-cell interactions and immune regulation critical for neuronal homeostasis also influence immune responses. The links between T cell immunity and nigrostriatal degeneration are supported by laboratory, animal model, and human pathologic investigations. Immune-associated biomarkers in spinal fluids and brain tissue of patients with idiopathic or familial forms of PD provide means to improve diagnosis and therapeutic monitoring. Relationships between oxidative stress, inflammation, and immune-mediated cell death pathways are examined in this review as they are linked to PD pathogenesis. Harnessing the immune system by drugs or by vaccination remain promising future therapeutic options. Antioxid. Redox Signal. 11, 2151–2166.
Introduction
Inflammation is a self-defensive reaction against pathogenic stimuli or injury, that is, under normal conditions, reparative to the host process. A well-controlled immune response to infection, environmental toxins, or injury is helpful as it protects the host by clearance of debris or pathogens, and promotes healing. However, when chronically sustained and dysregulated, inflammation can lead to significant tissue and cellular damage (116). Such events are regulated, in large measure, through the innate immune system comprising cellular elements that include mononuclear phagocytes (MP) [monocytes, microglia, macrophages and dendritic cells]; natural killer (NK) cells; and neutrophils; as well as regulatory humoral elements such as complement, cytokines, and a host of other secretory factors that control host surveillance and homeostasis in a nonspecific manner (97). Apropos the innate immune system, microglia are the resident phagocytic cells of the central nervous system (CNS), constituting 20% of the total glial population, and as such represent the first line of immune defense in the brain. Microglia are distributed throughout the brain and are highly mobile, capable of clearing damaged neurons, plaques, and infectious agents. When microbial pathogens cross the blood-brain barrier (BBB), microglia react to destroy the infectious agents before inflicting host tissue damage. As antibodies are normally too large to cross the BBB, microglia serve to recognize foreign factors, phagocytocize and destroy them through phagolysosomal fusion mechanisms, as well as acting as immune effectors and antigen presenting cells. Although necessary for foreign cell surveillance, the microglia, once activated, may be directly involved in neurotoxicity that is linked to uncontrolled inflammatory responses. In an inflammatory state many aspects of the “immune privileged” state of the brain break down and disease can quickly ensue. Indeed, an activated microglial response is strongly associated with dopaminergic cell loss in PD and neuronal dysfunction in degenerative diseases of the CNS (27, 66, 93, 132, 154, 162).
On the other hand, the adaptive immune system is highly specialized; is comprised of cells with specific immunologic effector, regulatory, and memory capabilities (T lymphocytes and B lymphocytes) that specifically eliminate or prevent pathogenic insults; but is activated by the “nonspecific” innate immune system. The CNS has traditionally been considered “immune privileged” and protected through the BBB, which prevents toxins and infections from reaching the CNS. We now know that both the innate and cell-mediated immune processes are highly active in PD (11, 107).
Innate Immunity
Overview
Innate immunity consists of the immune mechanisms that are encoded in the germ line possessed at birth, and work in a “nonspecific” manner, for immediate defense, against microbial infection. These mechanisms include removal of foreign substances by phagocytes, recruitment of additional immune cells to the site of infection through cytokine and chemokine production, activation of the complement cascade, and processing and presentation of antigens for activation of the adaptive immune response. The innate immune system functions through the nonspecific, generic recognition of common cell signaling pathways shared through a host of endogenous and exogenous threats called pathogen-associated molecular patterns (PAMPs). These are recognized by toll-like receptors (TLRs), which are expressed by microglia, astrocytes, oligodendrocytes, and neurons (16, 17, 36, 51, 85, 107). Engagement of TLRs contributes to neuroinflammation by activating signaling cascades that result in pro-inflammatory cytokine and chemokine production, as well as affecting BBB permeability.
Microglia
Microglia actively monitor the CNS environment by continual movement of their fine processes in the healthy brain (106), key in immune surveillance functions. In the brain cortex, microglial processes and protrusions directly contact astrocytes, neuronal cells bodies, and blood vessels, suggesting close communication (106) that allows microglia to react to brain insults quickly. With a multitude of TLRs, cytokine and chemokine receptors, and ion channels, microglia are sensitive to changes in their extracellular environment from a wide variety of stimuli that range from pathogens to aggregated proteins to alterations in ion homeostasis. Detection of disturbances within the neuron microenvironment induces microglia to become activated and affect a graded response (84). Upon activation, microglia proliferate, undergo morphogenesis, and increase cell volume with extension of their processes. These morphological changes are in stark contrast to resting microglia, which have a small cell body and ramified processes (162).
Among the many innate immune functions, activated microglia are well adapted for the induction of inflammation, cytokine-mediated and antibody-dependent cell cytotoxicity (ADCC), and regulation of T cell responses through antigen presentation (3). In the alerted state, microglia demonstrate increased IgG reactivity, and upregulation of complement receptors and cell adhesion molecules such as lymphocyte function associated antigen (LFA)-1, intercellular adhesion molecule (ICAM)-1 cluster of differentiation (CD)54, vascular cell adhesion molecule (VCAM)-1 (CD106), and CD1 (84). Indeed, increased expression of ICAM-1 and its counter receptor LFA-1 were identified and revealed aggregates of reactive microglia along with infiltrating LFA-1+ leukocytes in postmortem PD brains, as well as monkeys that received MPTP-injections indicative of a sustained inflammatory process in PD (98).
Activated microglia are known to produce a variety of toxic substances that, in addition to killing infectious agents, can accelerate neuronal injury and death. These toxic substances include reactive oxygen species (ROS), reactive nitrogen species (RNS), proinflammatory cytokines, and prostaglandins. Several molecules upregulate microglial secretory responses including lipopolysaccharide (LPS), interferon gamma (IFN-γ), amyloid beta (β-amyloid), CD40 ligand (CD40L), chemokines, neurotransmitters, gangliosides, and proteases such as thrombin, tissue plasminogen activator (tPA), and matrix metalloproteinase-3 (MMP-3) (1, 4, 79, 102, 135, 139, 149). Large numbers of human leukocyte antigen (HLA)-DR-positive reactive microglia are found in the substantia nigra (SN) of patients with PD and parkinsonism with dementia. Additionally, activated microglia are present in the SN and/or striatum of animals used in models of PD such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-, 6-hydroxydopamine (6-OHDA)-, and medial forebrain bundle axotomy-induced parkinsonism (15, 117, 131, 132). Uncontrolled microglial activation is toxic to neurons, due in part to release of pro-inflammatory factors including, but not limited to interleukin one beta (IL-1β), tumor necrosis factor alpha (TNF-α), IL-6, nitric oxide (NO), prostaglandin E2 (PGE2), and superoxide radical. The SN contains the highest concentration of microglia in the brain, especially in the ventral tier of the pars compacta (78, 88), making this region especially susceptible to altered microglial activation responses. Abnormally high levels of IL-1β and TNF-α found in the plasma and cerebral spinal fluid in patients with PD supports this contention (13, 56). Reactive microglia are highly localized, found close to cell bodies of dead or injured dopaminergic neurons in the SN and not to their degenerating termini (99), suggesting a retrograde mechanism for neuronal death. Cell death can also result from loss of trophic support stemming from microglia-induced neuritic beading or synaptic stripping along dendrites (125, 138). These findings suggest a direct link between dopaminergic neuronal death in PD and microglial activation. Indeed, postmortem examinations demonstrate that neuronal degeneration in PD is associated with a substantive gliosis linked to activated microglia, and has also been shown in MPTP-induced parkinsonism in primates, rodents, and humans (27, 86, 94, 96, 144). Moreover, activated microglia have been shown phagocytosing dying dopaminergic cells correlated with α-synuclein (α-syn) deposition in neuronal inclusions (49). Importantly, in vivo and in vitro studies demonstrated that microglia become activated in response to overexpression of α–syn or nitrated and aggregated forms of α-syn, a major component of Lewy bodies found in the brains of PD patients (119, 130).
Complement
Complement activation, a biochemical cascade that serves to facilitate pathogen clearance can also affect neural function. Although typically initiated through antibody deposition (classical pathway), the complement system is also considered an arm of the innate immune response. Increased mRNA levels of complement components are found in PD-affected brain regions (95) and complement components, including all constituents of the membrane attack complex (MAC) (164, 165), have been found intracellularly on Lewy bodies and on oligodendroglia in the SN in sporadic PD. IgG present in PD patients and recombinant human C5a synergistically induced dopaminergic neurodegeneration in rat mesencephalic neuron-glia cultures, while either alone was minimally toxic. IgG from unaffected individuals did not affect dopaminergic neurotoxicity (160). Such toxicity is mediated by microglia (160). Activated complement cascades promote inflammation and opsonization, facilitating phagocytosis. Complement may also be lytic to cells by inserting itself into viable cell membranes causing cytosolic leakage resulting in cell death. Cell membrane leakage could possibly increase the release of aggregated α-syn and other inflammatory mediators into the extracellular milieu to engage adjacent glial cells and propagate the inflammatory response.
Oxidative Stress, Inflammation, and Nigrostriatal Degeneration
Although the etiological event for idiopathic PD remains enigmatic, positive risk factors suggest the involvement of a multitude of factors including genetics, environmental exposure to toxins, and aging. Collectively, these influence the tempo and progression of disease (105). The single greatest risk factor currently identified is age, which suggests cumulative CNS damage for disease pathogenesis. However, considerable variation among degenerative lesions in the SN of PD patients compared to aged non-PD patients, suggests that aging and the disease processes underlying PD may occur independently (141). A significant body of evidence implicates ROS and RNS as possible initiating factors. The brain tissue itself is especially sensitive to oxidative damage, as this tissue accounts for 20% of the total oxygen demand of the body and is rich in peroxidizable fatty acids (20:4 and 22:6), while within the SN and basal ganglia, antioxidant defenses (i.e., catalase, superoxide dismutase, glutathione, and glutathione peroxidase) are the most sparse (43); and the microglia, a primary source for reactive oxygen species which contribute to neuronal degeneration, are the greatest in number. Figure 1 illustrates the consequences of microglial activation and oxidative stress on neuronal physiology in PD. In part due to dopamine metabolism by endogenous enzymes such as monoamine oxidases (MAO) or by autooxidation that can yield H2O2 and dopamine-quinones, the neurons in the SN are especially vulnerable to oxidative stress (57, 58, 129, 133). Furthermore, the presence of transition metals, such as iron, has been shown to accelerate the auto-oxidation of dopamine (133). Thus, the breakdown products of dopamine exacerbate inflammation and tissue damage by feeding H2O2 into the ROS cycle and/or by dopamine-quinone modification of protein sulfhydryl groups via nucleophilic additions. Subsequently, overproduction of free radicals such as superoxide and peroxynitrite feed into the ROS cycle to create an imbalance in the oxidation/reduction capacity of cells. Increased free radicals, without adequate antioxidant buffers in the SN, then react with proteins and nucleic acids to alter their functions, induce lipid peroxidation, or inhibit enzymes of the electron transport chain, eventually contributing to neuronal injury and death.

Activation of mitochondrial-dependent programmed cell death pathways are found in postmortem PD brains and in rodent models of PD (113, 114, 156). The mitochondrial apoptotic pathway involves mitochondrial outer membrane permeabilization leading to the release of cytochrome c, apoptosis-induced factor (AIF), endonuclease G, second mitochondria-derived activator of caspases (Smac), and high temperature requirement protein A2 (HTRA2)(151). Inhibition of complex I of the electron transport chain (ETC), for instance with MPTP, results in a time-dependent and region specific increase in the soluble pool of cytochrome c in the mitochondrial intermembrane space that can be released into the cytosol by programmed cell death agonists such as Bax (113). This occurs together with the release of caspase-9 and -3. Bax regulates SN pars compacta (SNpc) dopaminergic cell death associated with these caspases since their release coincides with Bax upregulation and translocation to the mitochondria, and caspase activation is prevented by genetic ablation of Bax (114, 155). Blocking caspase-9 or Apaf-1 also provides some degree of dopaminergic neuroprotection (101, 157). The importance of these events have been confirmed in PD as activation of Bax, caspase-9, and caspase-3 are detected in SNpc dopaminergic neurons of postmortem PD brains (64, 142, 157). Additionally, reduction of complex I activity by 30% was described in idiopathic PD patients (110, 124). Complex I inhibition results in depletion of ATP and the inevitable impairment of all ATP-dependent cellular processes, as well as blocking the flow of electrons along the ETC which increases generation of free radicals that increase oxidative stress. Specifically, complex I inhibition causes oxidative damage by peroxidation of the inner mitochondrial lipid cardiolopin which affects the binding of cytochrome c to the inner mitochondrial membrane, leading to increases in the soluble cytochrome c pool of the mitochondrial intermembrane space. These factors leading to complex I inhibition most probably sensitize neurons to cell death agonists such as Bax.
Oxidative stress also damages mitochondria and proteosomes, while functional loss of the proteosome can also contribute to increased oxidative stress and induce neural apoptosis. Both oxidative stress and proteosome inhibition act in concert to promote protein fibril formation and accumulation of protein aggregates. Indeed, misfolded proteins are found in inclusion bodies associated with several neurodegenerative diseases (40, 63, 74, 121). In PD, the normally soluble, unfolded protein α-syn is found in intraneuronal cytoplasmic inclusions or “Lewy bodies” in aggregated form along with ubiquitin, and lipids. Alpha-synuclein structure contains a central hydrophobic region that contributes to its propensity to aggregate, but oxidative stress induced nitration also contributes to α-syn aggregation and protofibril formation. Dopamine stabilizes α-syn protofibrils by forming a dopamine-α-syn adduct (24). Normally, misfolded proteins are ubiquitinated and degraded by the proteosome, but inhibition of this mechanism by oxidative stress allows greater accumulation of aggregated proteins. Furthermore, oxidative modification of α-syn can lead to self-aggregation and aggregation of other proteins, as well as damage to the ubiquitin–proteosome system. Importantly, aggregated α-syn has been shown to activate microglia leading to enhanced secretion of ROS (118, 145, 167). Activation of microglia by nitrated α-syn (N-α-syn) may also diminish protective mechanisms against oxidative stress by lowering the cellular glutathione buffering capacity as demonstrated by diminished GSH levels, GSH/GSSG ratios, and total glutathione levels from microglia stimulated with N-α-syn (119). The role of proteolytic stress in PD pathology is further supported by the observation that excess levels of parkin substrate proteins are found in a nonubiquitinated state in PD patients with mutations in the parkin gene (encodes an ubiquitin E3 ligase), and these mutations are associated with autosomal recessive juvenile parkinsonism (104). Overexpression of α-syn in models and duplication or triplication in the wild-type gene in PD patients are associated with neurodegeneration and microglial activation, possibly because of the inability of the proteosome to handle the increased number of misfolded proteins. Taken together, these findings illustrate the close relationship between oxidative and proteolytic stress, microglial activation, and inflammation that may contribute to neuronal injury and cell death in PD.
Adaptive Immunity
The adaptive immune system provides the ability to recognize specific pathogens and mount stronger responses with each encounter due to immunological memory. An adaptive immune response ensues when the innate immune system encounters a pathogen, recognized by PAMPs, and links the adaptive immune response through antigen presentation of the foreign protein. Antigen presenting cells (APC) phagocytose or endocytose foreign pathogens, then process and present foreign antigen complexed with surface major histocompatibility complex (MHC) molecules that are recognized by CD4+ (helper) or CD8+ (cytotoxic) T cells. T cells become fully activated when the APC provides appropriate co-stimulatory molecules. Activated CD4+ T cells can in turn recruit other T cells and B cells to sites of inflammation propagating the immune response.
Naïve T cells and B cells are normally precluded from entry into the CNS; however, in a neuroinflammatory state, activated glial cells secrete factors which disrupt the BBB and allow entry of adaptive immune components (11, 42, 69). Increased expression of cellular adhesion molecules, and the induction of chemokine gradients by glia, direct leukocytes to sites of inflammation (7). Indeed, T cell infiltration has been found in CNS tissues of PD patients (98), and adoptively transferred immune splenocytes into MPTP-treated mice results in significant infiltration into the brain and localization within the inflamed SN (11).
T cells and neuropathobiology
Several studies support a role for an adaptive immune response in the etiology of PD. Recent evidence from our laboratory suggests that nitrated α-syn activate peripheral leukocytes in draining lymphoid tissue (11). The aforementioned study demonstrated in the MPTP model the necessity for T cells in dopaminergic neurodegeneration and that dopaminergic neuronal loss was exacerbated by T cells for induced adaptive immune responses towards nitrated but not native α-syn, thus suggesting a causal link between T cell infiltration and sustained microglial activation associated with PD. Figure 2 illustrates a possible mechanism for N-α-syn-mediated adaptive immune responses in potentiating microglial activation and exacerbating neuronal death. Increased numbers of CD8+ T cells are found in close proximity to activated microglia and degenerating neurons within the SN in PD patients (94). The presence of T cell subsets at levels exceeding those typically found in the CNS and in lower ratios found in the periphery suggests a role in PD more profound than that associated with surveillance. In addition, aberrations in peripheral lymphocyte subsets are detectable in PD patients. Total numbers of lymphocytes in PD cohorts have been shown to be diminished by 17%, while CD19+ B cells were diminished by 35% and CD3+ T cells were diminished by 22% (10). Among CD3+ T cells, numbers of CD4+ T cells were diminished by 31% whereas numbers of CD8+ T cells were not significantly changed. A greater loss of naïve helper CD4+ T cells (CD45RA+) and either unchanged or increased effector/memory helper T cell subset (CD29+ or CD45RO+) was observed (10). A selective loss of CD4+CD45RA+ cells was also observed in diseases such as multiple sclerosis and Down's syndrome, suggesting a common immunological abnormality in neurological disorders (26, 41). Increased mutual co-expression of CD4 and CD8 by CD45RO+ T cells, as well as upregulation of CD25 (α-chain of the IL-2 receptor), TNF-α receptors, and significant downregulation of IFN-γ receptors suggested that these T cell subsets from PD patients are activated. In addition to CD4+ and CD8+ T cells that express αβ T cell receptors, another T cell subset exists with pathological relevance to PD. Elevated T cell populations expressing γδ T cell receptors have been described in the CSF of patients with PD (42) and are thought to play a regulatory role in CNS inflammation (115). Moreover, a greater proportion of the γδ T cells were CD25+ in the CSF suggesting a preferential activation of this T cell subset within the CSF compartment in PD patients (42). Increased proportions of memory and activated T cell subsets observed in PD patients relative to controls suggest a causal role in PD etiology.
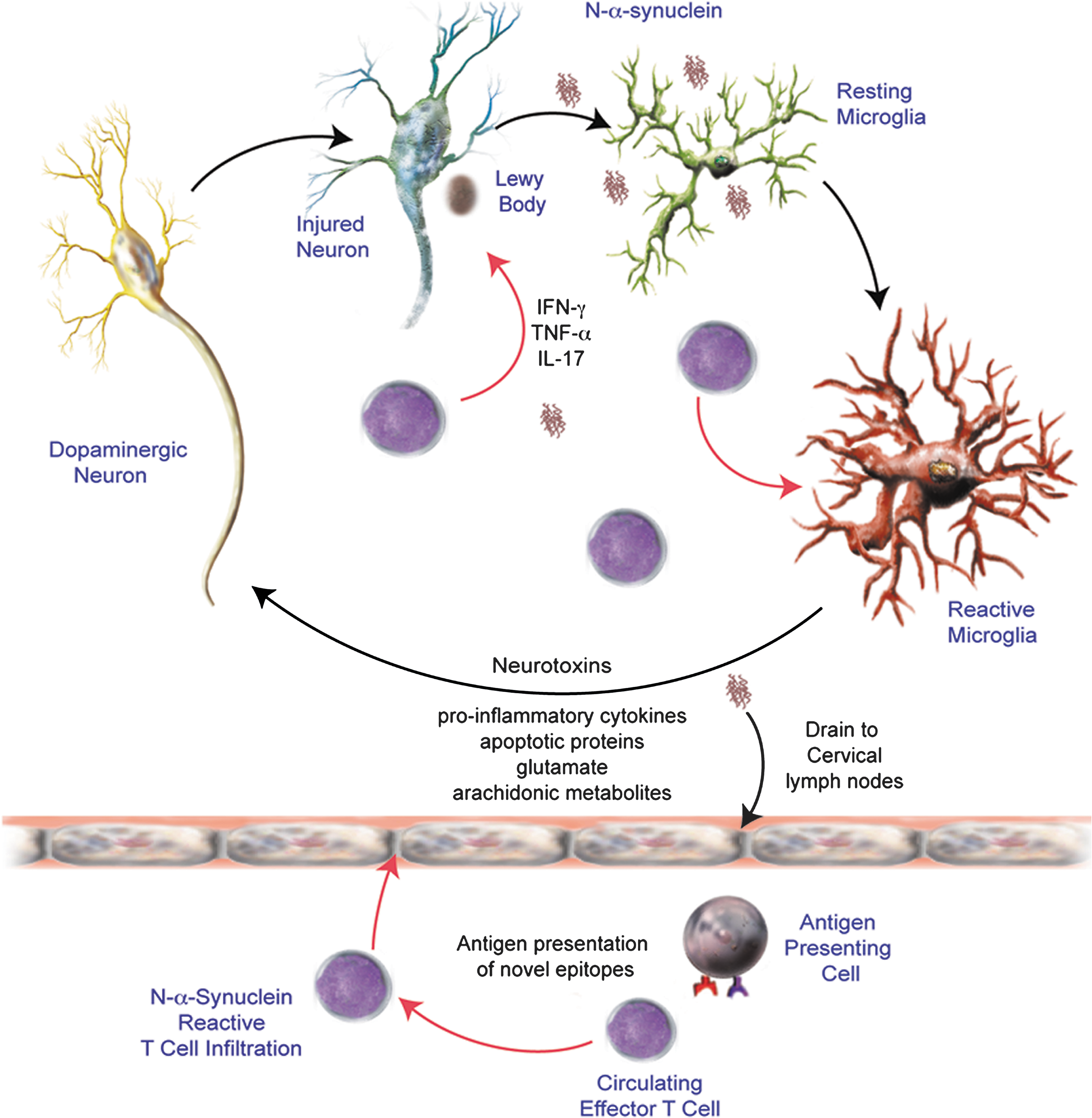
Humoral immunity
Humoral immunity may play a role in the initiation or regulation of inflammation in PD. Opsonization of a cell or damaged neuron can more easily target it for phagocytosis and degradation by phagocytic macrophages and can activate the complement system, a major mediator of immune/inflammatory reactions. In both idiopathic and genetic cases of PD, IgG immunolabeled dopaminergic neurons were found associated with an increased number of activated microglia expressing the high affinity IgG receptor, FcγRI and were strongly associated with a more progressive state of neurodegeneration (108). IgG is closely associated with Lewy bodies. Microglia in the SN expressing the FcγRI receptor phagocytose IgG-immunopositive neurons (108). In addition, deletion of the FcγR by genetic ablation protects mice from microglial activation and dopaminergic cell death (67).
Natural regulatory T cells
Following an immune response, once the foreign pathogen/antigen is removed, suppression of the active effector immune response is necessary so that the cytotoxic affects of inflammation do not have a profound effect on self-tissues. Several mechanisms exist by which the immune response can be regulated. Among the various cell types involved, there exists a naturally occurring T cell subtype that functions to prevent immune responses towards self-peptides: the regulatory T cell (Treg). The naturally occurring Tregs are generated in the thymus and constitutively express CD4 and CD25 in addition to the transcription factor FOXP3 (forkhead box p3) that is required for their development, maintenance, and function (44, 72). CD4+CD25+ Tregs play an important role in preventing autoimmune diseases such as type I diabetes, and limiting chronic inflammatory diseases such as asthma and inflammatory bowel disease (25, 123, 128, 163). Tregs are widely recognized to be capable of controlling innate immune reactivity and suppressing both CD4+ and CD8+ effector T cell responses as well as B cell responses to both self and foreign antigens (122). Thus, Tregs have a critical role in immune homeostasis. Tregs exhibit regulatory activity by suppression of immune responses by secretion of anti-inflammatory cytokines such as IL-10 and TGF-β (65, 76), by cytolysis (61), by metabolic disruption (109), and by modulation of dendritic cell maturation or function through CTLA4 ligation (136, 140) (Fig. 3). Effector cells such as B cells or T cells, myeloid and APCs, including microglia, are inactivated or neutralized through the actions of Tregs (20, 134, 146).

CTLA4 is a critical negative regulator of T cell responses and is instrumental in maintaining immunological tolerance. This co-receptor has been shown to inhibit T cell activation, IL-2 production, and cell cycle progression (147). One mechanism by which CTLA4 can mediate T cell responses is through the competition with CD28 for CD80/86 binding which leads to decreased activation. Binding of CD28 by CD80/86 in the presence of TCR engagement stimulates proliferation of T cells and production of IL-2 (91). However, in the absence of CD28 co-stimulation, activated T cells become anergic to antigens or become apoptotic (50, 166), but when CTLA4 binds these same ligands, T cell activation is restricted (112, 158). In addition, signaling through the cytoplasmic tail of CTLA4 and modulation of TCR signaling leads to cell cycle disruption and suppression of IL-2 production. Furthermore, dendritic cell function can be conditioned to express indoleamine 2,3-dioxygenase (IDO) through interactions between CTLA4 and CD80 and CD86, resulting in suppression of effector T cells (147, 158). As one might expect, CTLA4 and Foxp3 expression are strongly correlated, yet Foxp3 alone is not sufficient for Treg activity. Treatment of human CD25− T cells with IL-2 in the absence of CTLA4 upregulated Foxp3 expression in cells without suppressive capacity; however CTLA4 transfection into CD25−FoxP3− T cells produced suppressive T cells without Foxp3 (168). These data suggest that CTLA4 is required for suppressive function, but not Foxp3 even though they are both expressed after T cell activation (168). CTLA4 may also be involved in suppression through disruption of CD28 signaling at the immunological synapse and by interfering with lipid raft formation (28, 111). Furthermore, CTLA4 can reduce the contact period between T cells and APCs and lead to decreased proinflammatory cytokine production and T cell proliferation (127).
The mechanism by which Tregs suppress metabolic function in effector cells includes the induction of apoptosis by competition for and deprivation of IL-2, transfer of cAMP into effector T cells through membrane gap junctions, or by activation of the adenosine receptor 2A (A2AR), binding which has also been shown to enhance Treg generation via promotion of TGF-β expression and inhibition of IL-6 expression (82). However, much work is still needed to understand the mechanism(s) responsible for Treg induced suppression of Teffs, as it is possible that there are multiple mechanisms that are context dependent or differentially functional for each Treg subset. In addition, Tregs have also been shown to promote neurotrophic support by inducing astrocytes to increase expression of BDNF and GDNF (12, 118) and may promote glutamate clearance (48). Understanding the Treg mechanism of action is of great importance considering their capacity to control immunity and inflammation, events strongly associated with the pathogenesis of PD.
In addition to natural CD4+CD25+ Tregs, effector T cells can be converted into inducible regulatory T cells capable of controlling peripheral T cell responses, largely based on their potential to produce regulatory cytokines following antigen priming (65). These inducible Tregs include type 1 Treg (Tr1), in which the suppressive function is cell-contact independent and involves IL-10, and Th3 cells that produce primarily TGF-β (23, 120). Induced Tregs may also work by a granzyme B and perforin-dependent manner, as evidenced by human induced Tregs expressing these cytolytic factors (61). These populations do not typically express Foxp3. Other T cell populations with immune-regulatory properties have been described and include IFN-γ producing Th1 cells and IL-4 producing Th2 cells (80). In mice, adaptive Tregs can be induced via the upregulation of Foxp3 in the presence of cytokines, especially TGF-β, however, whether this also true in humans is controversial, but recent data suggests that CTLA4 expression is required (168).
Neuroimmune Pharmacology
Several lines of evidence in both animal studies and clinical trials suggest that manipulation of various aspects of the immune response may provide substantial neuronal protection (12, 23, 32, 118, 154). The possible mechanisms include decreased microglial activation, increased neurotrophic support, inhibition of pro-inflammatory T cell responses, and enhanced clearance of aberrant proteins (Fig. 4).
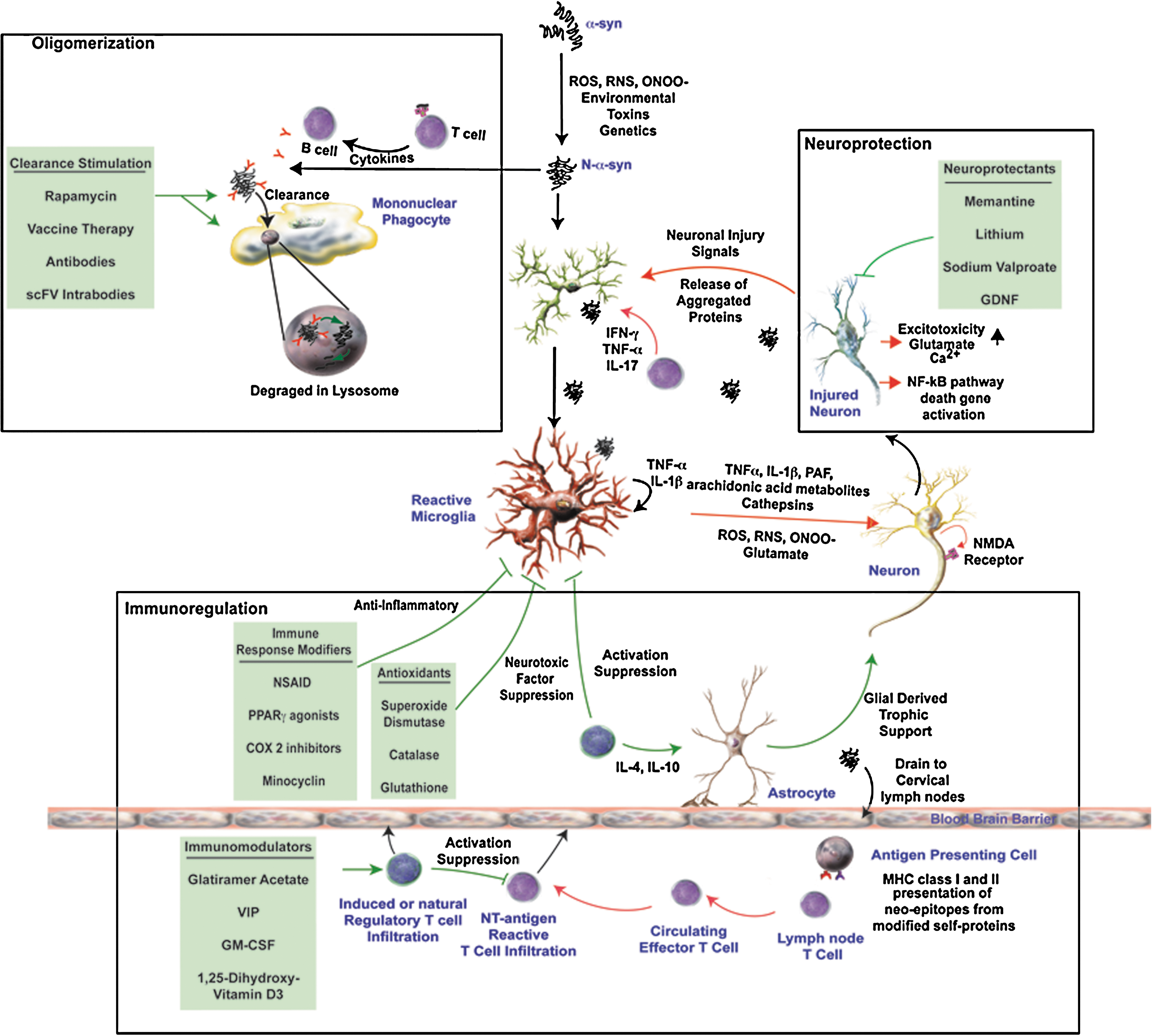
Immune modulation as therapy
One potential target in PD is cyclooxygenase type 2 (COX-2). COX-2 has been shown to be upregulated in SN pars compacta dopaminergic neurons in both PD patients and in animal models. Pre-treatment with nonsteroidal anti-inflammatory drugs (NSAIDs) is protective against MPTP and 6-OHDA-induced nigrostriatal dopaminergic degeneration in mice (66, 143). Furthermore, recent studies have shown that the risk of developing PD is reduced in humans who use daily NSAIDs, particularly ibuprofen. However, the mechanism by which this protection occurs and what specific drug and dosage regimen that is best for prevention of PD is still not completely understood. Like other tissues in the body, the COX isoforms have a heterogeneous distribution in the brain. COX-1 and COX-1b are detected in microglial cells, while COX-2 is found in neuronal and glial cells, and significant levels of COX are expressed by astrocytes (71, 81). The normally low levels of COX-2 expressed in the nigral dopaminergic neurons is upregulated in both PD patients and experimental models of PD (29, 37, 38, 144, 150, 154, 159). Furthermore, knockout mice that do not express COX-2 exhibit less neuronal death following ischemia, challenge with NMDA (73), and MPTP (37, 38, 154, 159). Moreover, COX-2 inhibition through pharmacologic and genetic manipulation has been shown to protect the entire nigrostriatal pathway (154).
Prostaglandin E2 (PGE2) production and free radical generation is a prominent feature in COX-2 neurotoxicity of SN dopaminergic neurons as evidenced by their detection in experimental models and in PD postmortem tissues (33, 34, 144, 150, 154). PGE2 is thought to mediate COX-2 neurotoxicity through activation of EP1/EP3 receptors leading to disruption of Ca2+ homeostasis and excitotoxicity (19, 77). Indeed, the concentration of PGE2 has been shown to almost double following MPP+ (active metabolite of MPTP) induced COX2 upregulation, a process dependent on microglial interaction with dopaminergic neurons (144, 159). The microgliosis induced by MPP+ involves the release of inflammatory factors, including PGE2 that enhances COX-2 activity in dopaminergic neurons leading to further neurodegeneration; and increased gliosis (131, 144, 154, 159). Overall, these data suggest that COX-2 may play a significant role in microglial activation and amplification of the inflammatory response that leads to a fierce cycle of neurodegeneration.
Inhibition of prostaglandin synthesis is an action of many NSAIDs, but not all of NSAID's therapeutic affects are well understood. NSAIDS have also been shown to inactivate nuclear factor-kappa B (NF-κB) and factor activator protein-1 (AP-1) (5, 35, 60, 83, 100, 152) and may be able to scavenge ROS and RNS, thus blocking their detrimental effects (6, 60). Moreover, ibuprofen and indomethacin are activators of the peroxisome proliferator-activated receptor-γ (PPARγ), a transcription factor that antagonizes the activity of NF-κB, AP-1, signal transducer, and activator of transcription-1 (STAT-1) and nuclear factor of activated T cells (NFAT) (75, 90), and thus is associated with regulation of inflammatory cytokines (75). Furthermore, the NSAIDs acetylsalicyclic acid (ASA) and paracetamol were shown to be able to block MPP+-induced inhibition of complex I and mitochondrial ETC activity with subsequent reduction of superoxide generation (92). However, some NSAIDs (indomethacin, ibuprofen, ketoprofen, or diclofenac) may enhance MPP+ induced neurodegeneration under some conditions, possibly by inhibition of multidrug resistance proteins and blocking the efflux of MPP+ (103). The stark differences in the effects of these NSAIDs may be due to the differences in dose and duration of treatment, observational window, toxins used, or the differences in experimental conditions. Still, there is strong evidence that ASA, salicylic acid (SA), ibuprofen, and COX-2 selective inhibitors exert an overall neuroprotective effect, although the mechanism(s) through which they act remains obscure. Despite evidence of inflammation in the brains of PD patients and strong evidence in animal models, NSAIDs have not yet been formally tested in humans and the data from epidemiological studies analyzing the association between NSAID use and PD have shown conflicting results (14, 21, 22, 68, 126, 148).
Immunomodulation of adaptive immunity and therapeutic responses
While research continues into the possible use of NSAIDs for treatment of PD, much interest lies in the mechanism through which Tregs modulate inflammation and whether they can be harnessed as a cell-based treatment modality. Others and we demonstrated that in models of neurodegeneration, induction of a regulatory T cell response attenuates microglial activation and promotes neuronal survival (8, 9, 12, 18, 45, 118). Based on these data, we suggest that induction of Tregs may be used to modulate immune responses, possibly through interactions with the peripheral and CNS immune systems to provide neuroprotection. We previously demonstrated that immune cells from mice immunized with Copolymer-1 (Cop-1; Copaxone, glatiramer acetate) attenuate microglial responses and protect against MPTP induced dopaminergic neurodegeneration (12, 87). T cell depletion abrogated this protection, and later works showed that indeed CD4+ cells were responsible for the observed neuroprotection (87). Cop-1 is thought to preferentially induce Th2 and Th3 Tregs that secrete anti-inflammatory cytokines such as IL-4, IL-10, and TGF-β (2), and to promote the conversion of CD4+CD25− T cells to CD4+CD25+ Tregs through induction of Foxp3 (70).
Furthermore, adoptive transfer of CD3-activated CD4+CD25+ Tregs to MPTP-intoxicated mice provided >90% protection of the nigrostriatal system (118). This protective response was cell number dependent, and paralleled the modulation of microglial responses and upregulation of glial cell-derived neurotrophic factor (GDNF) and brain-derived neurotrophic factor (BDNF) and TGF-β, as well as downregulation of proinflammatory cytokines such as TNF-α. The neuroprotective effect by Tregs was shown in laboratory tests to function through modulation of microglial oxidative stress and cytotoxicity and was also operational following removal of Tregs from culture prior to stimulation, suggesting the afferent modulation of the microglial phenotype is plausible (118). Treg infiltration into the midbrain was implicated with the observation of enhanced expression of anti-inflammatory cytokines IL-10 and TGF-β that paralleled increased Foxp3 expression within the SN. Preliminary studies using single photon emission computerized tomography (SPECT) confirmed infiltration of Tregs into the SN following MPTP intoxication (unpublished observation). Adoptive transfer of activated CD4+ T cells induced neuroprotective activities in a mouse model of amyotrophic lateral sclerosis (ALS) (9).
Methods of generating Tregs for therapeutic modalities are of great interest for use in autoimmune diseases, for induction of transplantation tolerance, or in diseases that have an inflammatory component such as PD, ALS, Huntington's disease (HD), and HIV-associated dementia (HAD). In addition to Cop-1 which has already been proven to be effective in multiple sclerosis, other tolerogenic molecules exist that have been demonstrated to promote production of Tregs and may be useful as a therapeutic tool to control immunity. One such molecule is 1a, 25-dihydroxyvitamin D3, shown to activate lymphocytes and induce T cell tolerance. Furthermore, co-administration of specific antigen results in antigen-specific tolerance when administered with 1α, 25-dihydroxyvitamin D3 (59). Granulocyte macrophage colony-stimulating factor (GM-CSF) also has similar effects (46, 153). Both molecules are thought to generate Tregs and stimulate the production of TGF-β and IL-10 (137). Recent data supporting the importance of CTLA4 in Treg function demonstrated that enhanced selective engagement of CTLA4 on T cells by antigen-presenting DCs resulted in the induction of antigen specific CD4+CD25+FoxP3+ and CD4+CD25−TGF-β1+ adaptive Tregs. Moreover, generation of adaptive Tregs has been demonstrated through the in vivo delivery of a CTLA4 specific ligand along with self-antigen, resulting in protection against autoimmune disease (52).
Other molecules that may be useful in Treg generation are neuropeptides. These are also produced by lymphocytes, especially Th2 cells, in response to mitogenic and inflammatory stimuli, and promotes Th2 responses in vivo (31, 53, 54, 89). They can expand Tregs ex vivo and convert CD4+CD25− effector T cells to Tr1 regulatory T cells. Indeed, neuropeptides have been utilized to generate antigen-specific Tregs for promotion of self-tolerance and suppression of autoimmune disorders in different animal models (30, 39, 55).
In our prior works, transfer of spleen cells from mice immunized with nitrated α-syn (N4YSyn) to MPTP-treated mice demonstrated an increased loss of dopaminergic cell bodies in the SNpc and nerve termini in the striata (11). In contrast, induction of a regulatory T cell response following MPTP-intoxication was shown to promote neuroprotection and was associated with reduced microglial activation either by adoptive transfer of CD3-activated Tregs (118) or induction of a regulatory T cell response by glatiramer acetate (12, 87) or by vasoactive intestinal peptide (VIP) (32) in the MPTP mouse model of PD. Neuroprotection provided by neuropeptides may be due to suppression of the adaptive immune response following MPTP intoxication. Indeed, T cells of the adaptive immune system significantly contributed to neurodegeneration in MPTP-intoxicated mice (11), whereas the severe combined immunodeficient mouse is not susceptible to MPTP-induced neuronal degeneration. The interaction of Tregs with effector T cells by direct cell contact or via soluble factors may boost attenuation of microglial function, thus providing increased neuroprotection. This protection may be due to increased regulatory cytokines or induction of new mediators and cellular functions related to suppression, but only secreted when stimulated by Teffs.
Conclusions
Evidence for an active immunological role in PD abounds. The causal role of microglia and inflammation in the loss of dopaminergic neurons was shown to be a key player in this process. Furthermore, the role of the immune system in regulation of homeostasis in the brain has come to the forefront in the quest to understand the mechanism behind the localized microglial activation and its detrimental effect on dopaminergic neurons in the SN. Oxidative stress is involved in the perpetuation of inflammation and may be a potential trigger of the initial inflammatory response that eventually leads to a vicious cycle of gliosis associated with dopaminergic cell death. There also appears to be many different sources of oxidative stress associated with PD. The effect of oxidation on α-syn has also been shown to contribute to this process through aggregation and introduction of neo-epitopes on self-proteins. Links between the innate and adaptive arms of the immune system are active in PD, and various immune processes may contribute to dopaminergic cell death and microglial activation. Moreover, infiltration of the brain by activated immune cells and the secretion of inflammatory cytokines have been shown to be associated with PD.
There are many mechanisms that may be involved in the death of dopaminergic neurons within the inflammatory environment of the parkinsonian SN in PD that appear to involve mitochondrial dysfunction, inflammation, oxidative stress, and proteasome dysfunction, any of which may be the initial trigger, but many or all may eventually become engaged at some point in the disease process. NSAIDs such as ASA, SA, ibuprofen, and COX-2 inhibitors may still hold promise for prevention of PD, although more research is warranted to demonstrate their efficacy. The use of antioxidants, neuroprotectants, therapies that improve clearance of modified and misfolded proteins, or immunomodulatory compounds may still provide possible treatment options for PD. Of considerable interest are the immunomodulating abilities of Tregs. We propose that modulation of Treg cells can be utilized as a therapeutic modality in PD and warrants further investigation. Thus, further advances in understanding the entire picture of PD immunopathology are necessary for the development of new treatments.