
Abstract
Oxidative stress has been implicated in the pathogenesis of neurologic and psychiatric diseases. The brain is particularly vulnerable to oxidative damage due to high oxygen consumption, low antioxidant defense, and an abundance of oxidation-sensitive lipids. Production of reactive oxygen species (ROS) by mitochondria is generally thought to be the main cause of oxidative stress. However, a role for ROS-generating NADPH oxidase NOX enzymes has recently emerged. Activation of the phagocyte NADPH oxidase NOX2 has been studied mainly in microglia, where it plays a role in inflammation, but may also contribute to neuronal death in pathologic conditions. However, NOX-dependent ROS production can be due to the expression of other NOX isoforms, which are detected not only in microglia, but also in astrocytes and neurons. The physiologic and pathophysiologic roles of such NOX enzymes are only partially understood. In this review, we summarize the present knowledge about NOX enzymes in the central nervous system and their involvement in neurologic and psychiatric diseases. Antioxid. Redox Signal. 11, 2481–2504.
Introduction
The ROS-generating NOX enzymes are electron transporters. The electron from cytoplasmic NADPH is transferred across biologic membranes through NOX enzymes and bound to oxygen in the extracellular space or in the lumen of intracellular organelles, via intermediate flavin adenine dinucleotide (FAD) and heme prosthetic groups. Superoxide (O2 −) is generally thought to be the primary product of the electron transfer, but other downstream ROS, in particular, hydrogen peroxide (H2O2), also are generated (16).
Seven NOX genes have been identified: NOX1 to 5 and DUOX1 and 2. Because little is known about the role of NOX5 and DUOX1 and 2 in the CNS, in this review, we focus exclusively on NOX1, NOX2, NOX3, and NOX4. The first NOX enzyme to be discovered was NOX2 in phagocytotic cells, and, for this reason, it is also known as gp91
phox
(phox:
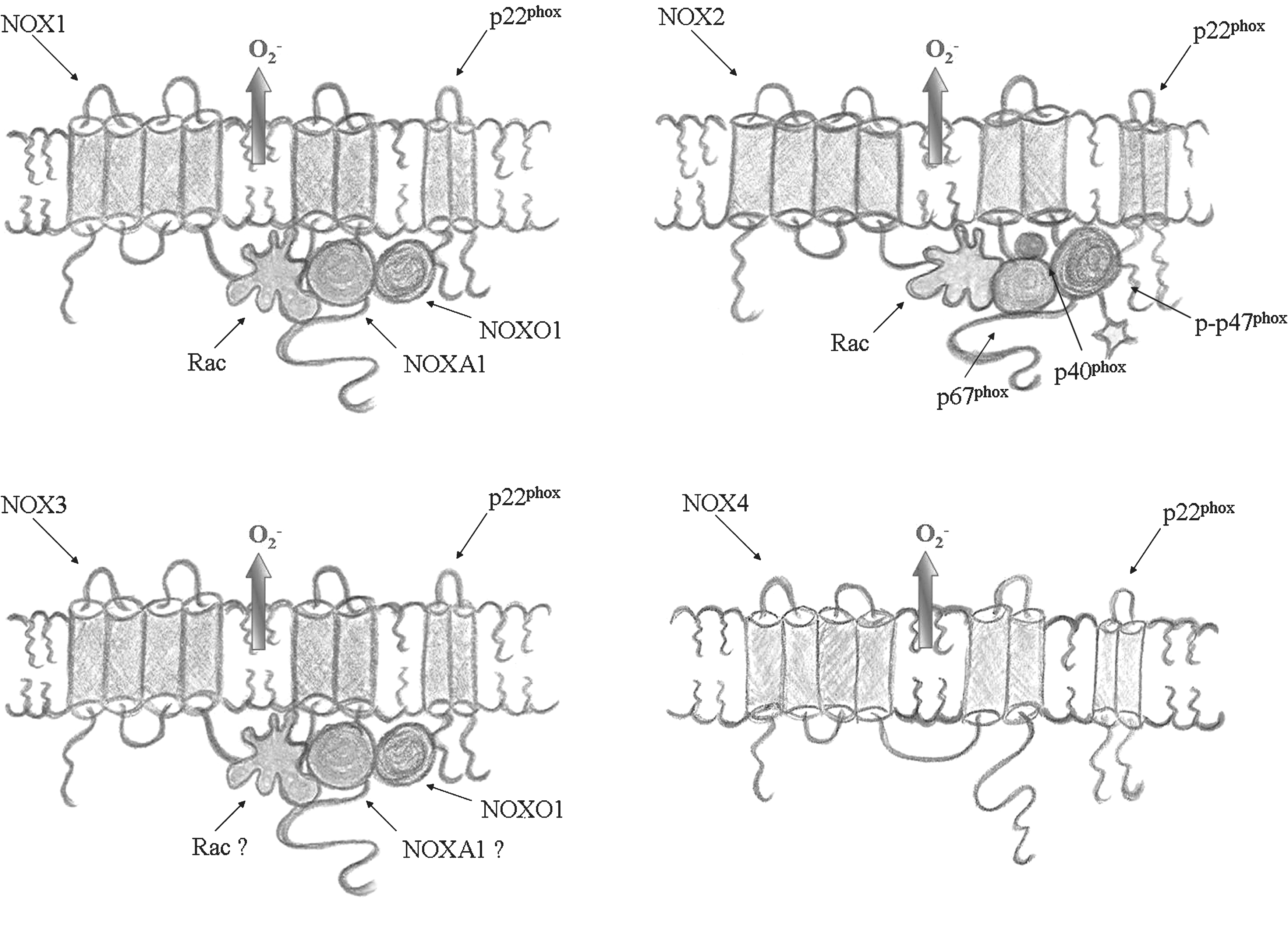
The conditions in which NOX is regulated/activated in the CNS are not elucidated. Indeed, the specificity of NOX regulation in the CNS could come from putative NOX-interacting proteins, which have a preferential expression in the CNS, such as NOXO1γ and Rac3.
The subcellular localization of NOX enzymes can also influence the ROS-mediated signaling and cellular functions. However, in most cases, the question of the subcellular localization of NOX isoforms is still a matter of debate. This uncertainty is, at least in part, due to the lack of high-quality antibodies. Most of the data concern NOX2 in granulocytes, where the enzyme localizes predominantly to intracellular granules in quiescent cells, but is translocated to the plasma membrane or the phagosome or both in response to cell activation (47). As seen for phagocytes, it appears that in neurons, a membrane translocation of cytosolic NOX2 subunits exists with cell activation (76, 235). In addition, it has been observed that in hippocampal neurons, NOX2 enzyme is present in synaptic sites (235). To our knowledge, no studies have been published about the localization of other NOX isoforms in CNS cells. However, based on observations in vitro on other cell types, it has been suggested that NOX4 might localize to the endoplasmic reticulum (37, 95, 210) or vinculin to focal adhesions (100), whereas NOX1 might preferentially localize to the plasma membrane (95, 100), possibly to caveolae (100). Very little is known about NOX3 localization.
In this review, we summarize the current knowledge in the field of NOX enzyme in the CNS. In the first part, we describe the expression and functions of NOX enzymes in the CNS under physiologic conditions. In the second part, we discuss the possible implications of NOX enzymes in several CNS pathologies.
Expression of NOX Enzymes in the CNS and in Its Cells
The family of NOX enzymes is widely distributed in a variety of tissues, but very high expression levels can be found in specific organs or cell types (e.g., NOX1 in the colon, NOX2 in phagocytes, NOX3 in the inner ear, and NOX4 in the kidney). It also appears that many cells can express several NOX enzymes with nonredundant functions. This nonredundancy of NOX function is most likely explained by different subcellular localization and activation mechanisms of the different NOX isoforms (16, 134, 175).
Expression of NOX enzymes in the brain and in specific CNS areas
The presence of NOX1, NOX2, NOX3, and NOX4 transcripts has been identified in total brain samples (11, 108, 242). In addition, several studies have investigated the expression of NOX isoforms in specific CNS regions. Most data concern NOX2; however, evidence exists on the CNS localization of NOX1, NOX4, and possibly also NOX3 (Table 1). Although available studies do not provide a comprehensive description of the CNS distribution of NOX enzymes, it appears that (a) several NOX isoforms are coexpressed in various CNS regions; and (b) at least under some circumstances, NOX expression in certain CNS regions might be inducible, rather than constitutive.
H, human; M, mouse; R, rat; Rab, rabbit.
Expression of NOX enzymes in specific cells of the CNS
Evidence of the expression of NOX isoforms in specific cell types of the CNS comes mostly from in vitro studies on derived primary cultures. It appears that NOX1, NOX2, and NOX4 are present in neurons, astrocytes, and microglia, whereas the localization of NOX3 is not known (Table 2). Unfortunately, the relative amount of different NOX enzymes and their functional activity in different brain cells has not been comparatively studied.
H, human; M, mouse; R, rat; Chimp, chimpanzee.
Physiologic Functions of NOX Enzymes in the CNS and in Its Cells
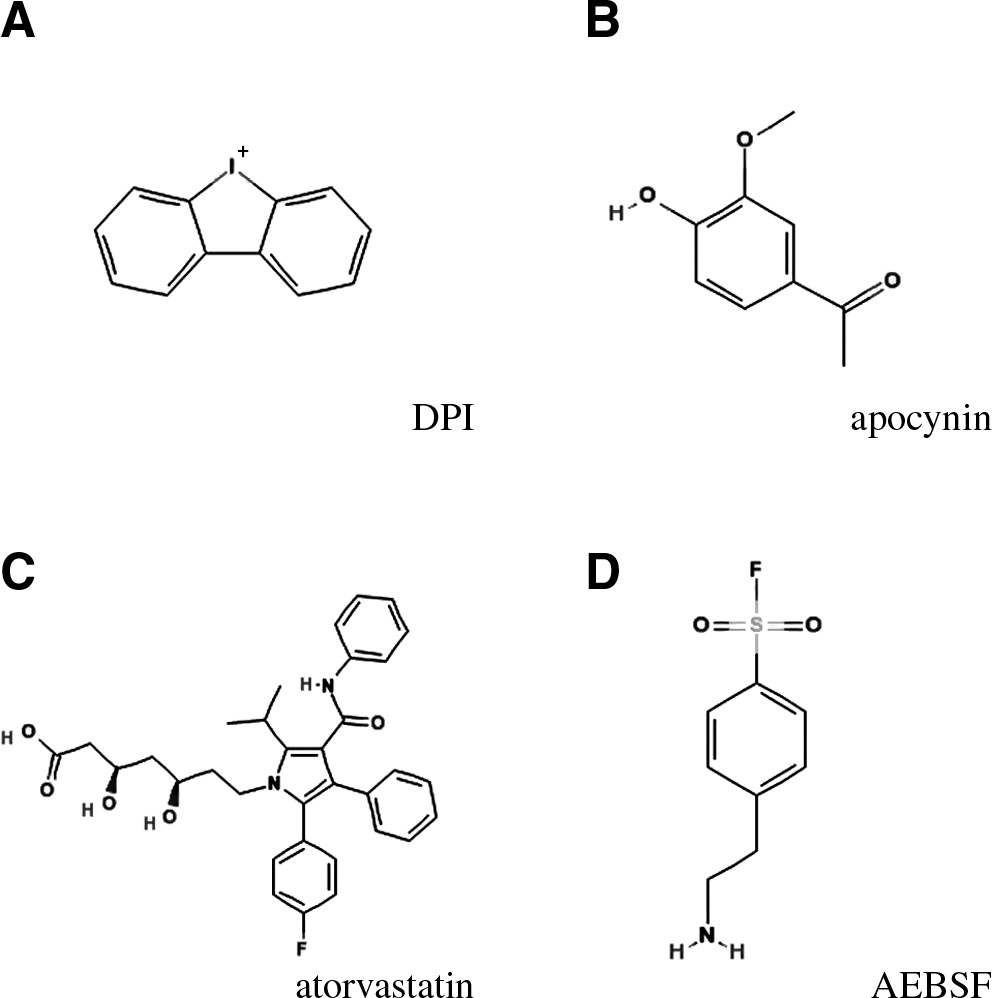
Physiologic concentrations of ROS are necessary for the proper function of several biologic processes. Activation of NOX enzymes is required in different processes, such as host defense, biosynthesis, or cellular signaling cascades (16). Genetic and pharmacologic studies have allowed the identification of mechanisms regulated by NOX-dependent ROS production. Genetic deletions or mutations in NOX enzymes occur naturally in humans or can be generated in animals and have helped us to understand some of the roles of certain isoforms. Most of the chemical inhibitors used currently in pharmacologic studies targeting NOX activity are not NOX specific [for a review, see (135) and Jaquet et al., in this issue]. Some of the compounds that have been used in various studies, described in this review, are as follows (Fig. 2):
Diphenyleneiodonium (DPI) is a potent blocker of electron transporters. In addition to inhibiting NOX enzymes, DPI inhibits xanthine oxidase, nitric oxide synthetase, and mitochondrial electron transport.
Apocynin is an antioxidant, which under certain circumstances can be metabolized into a NOX inhibitor (135). In most in vitro studies with CNS cells, high concentrations of apocynin (>100 μM) have been used and have displayed antioxidant effects. However, in animal models, apocynin appears to be efficacious even at relatively low concentrations [i.e., 2.5–5 mg/kg; (18, 234, 258)], which argues against a ROS scavenger effect and in favor of an inhibitor effect.
Atorvastatin, in addition to its effect on cholesterol synthesis, inhibits isoprenylation of Rho GTPases and, through this mechanism, decreases the activation of Rac-dependent NOX enzymes.
AEBSF is a serine protease inhibitor, which also inhibits NOX enzymes, albeit with a relatively low potency. It is not clear whether AEBSF acts directly or indirectly on NOX enzymes.
gp91ds-tat is an inhibitory peptide based on an analogue sequence within gp91
phox
/NOX2. gp91ds-tat appears efficacious in several animal models; however, its specificity has not been rigorously proven.
Neurologic functions of NOX enzymes in the CNS
Most of the information on the physiologic functions of NOX enzymes in the CNS has been obtained by using NOX-deficient mice and can be summarized as follows: NOX1: In mice, genetic deletion of NOX1 leads to decreased blood pressure and protects against aortic dissection in response to angiotensin II (73, 74, 163). With respect to neurologic functions, it has been reported that NOX1 deficiency does not alter locomotor activity or movement coordination. Nevertheless, NOX1 enhances the sensitivity to inflammatory pain. Thus, NOX1 deficiency decreases thermal and mechanical hyperalgesia during inflammation but does not affect nociceptive responses to heat and mechanical stimuli (107). It is not clear whether these effects on pain are due to NOX1 in the central or the peripheral nervous system. NOX2: The best-documented human pathology linked to a loss of function of an NOX enzyme is chronic granulomatous disease (CGD). It is an immunodeficiency caused by mutations in one of the genes of the NOX2 enzyme complex, such as NOX2/gp91
phox
, p47
phox
, p67
phox
, p22
phox
(221). Because mice that are deficient for the NOX2 enzyme complex display pathologic features similar to those of CGD patients, they have been widely used as experimental models (174, 208). Data about the possible role of NOX2 in CNS functions arise from studies on CGD patients and CGD mice. Among CGD patients, an elevated prevalence rate of cognitive deficits is found (183). It has been argued that the chronic disease in CGD patients (in particular, recurrent infections) is not the cause of the cognitive deficits (183). This is also in line with the observation that NOX2- and p47
phox
-deficient mice, which under SPF conditions generally do not display signs of the disease, show impaired memory (125). Thus, it is likely that NOX2 plays a role during CNS development or in CNS function or both. Supporting this hypothesis, it is known that ROS are important signaling molecules involved in mechanisms underlying synaptic plasticity and memory formation (99). NOX3: Mutation of NOX3 in mice causes vestibular defects due to altered otoconia formation in the inner ear (182). A similar effect also has been observed in NOXO1- (128) and p22
phox
-mutated mice (174). To our knowledge, analyses of neurologic function have not been performed in these mice. NOX4: No information has been reported on the effects of genetic alteration of NOX4 in humans or animals.
Cellular functions of NOX enzymes in the CNS
It has been reported that NOX enzymes have physiologic and pathologic effects on many cell types within the CNS. However, the precise mechanisms of action are not yet fully understood. ROS produced by NOX enzymes can directly influence cellular functions, by inducing the oxidation of proteins and subsequently their structural and functional changes (e.g., hydrogen peroxide‱dependent cysteine modifications in tyrosine phosphatases, transcription factors, or ion channels). However, other processes may also occur, including superoxide interaction with nitric oxide (i.e., nitric oxide depletion, peroxynitrite production), electrogenic effects of NOX activity (i.e., plasma membrane depolarization), or impact on pH homeostasis (16).
Neurons: differentiation, signaling, and death
Neuronal differentiation
Reactive oxygen species might be involved in neuronal differentiation during development (238, 239). In this context, NOX enzymes have been implicated in nerve growth factor (NGF)-induced neuronal differentiation of PC12 cells (106, 191, 228) and mesenchymal stem cells (255). During neuronal growth, production of H2O2 by NOX enzymes seems to influence NGF signaling through regulation of tyrosine phosphorylation and activation of the transcription factor AP-1 (228). Consequently, ROS-mediated protein activity and gene expression modulate the development of neuronal cells. In particular, it has been reported that ROS production by NOX enzymes can be involved in different aspects of neurite outgrowth (106, 172). Thus, NOX enzymes can participate in neuronal maturation and differentiation during brain development.
Neuronal signaling
Regulation of membrane potential and H+ fluxes
Production of ROS by NOX enzymes involves electron transport across biologic membranes and, hence, causes cellular depolarization. For each electron transported across the membrane, one H+ ion is left in the cytoplasm. Thus, to avoid H+ accumulation and cytosolic acidification, H+ extrusion occurs throughout proton channels. These mechanisms have been studied on NOX2 in phagocytes (12, 173, 209); however, they might be also important in neurons, because neuronal activity is dependent on the plasma membrane potential. Expression of voltage-gated proton channels was first detected in snail neurons but also is present in rat hippocampal neurons, where it contributes to regulation of pH homeostasis and action potentials (50). Interestingly, it has been observed that expression of proton channels and NOX2 often occur in parallel (50). However, further analysis must be done to understand better the involvement of NOX enzymes in the regulation of membrane potential and H+ fluxes in neurons.
Angiotensin II receptor
Angiotensin II is an oligopeptide involved in cardiovascular homeostasis and regulation of blood pressure, mainly through the interaction with the angiotensin II type 1 receptor (AT1R) (82). Release of ROS mediates angiotensin II signaling in the vasculature (83), and NOX enzymes have been implicated in such a mechanism (72). In particular, the effect of angiotensin II on NOX enzymes in cerebral vasculature has been investigated (167). It appears that angiotensin II signaling is dependent on NOX activation also in neurons. It has been demonstrated that NOX2-derived ROS are responsible for the entrance of Ca2+ currents in angiotensin II–stimulated neurons (253, 280). As a consequence, NOX2-derived ROS influence AT1R signaling cascades (34, 35, 146, 252), neuronal activity (226), and CNS-regulated cardiovascular responses (69, 70, 279). In addition, it has been observed that NOX enzymes are expressed by neurons of cerebral areas implicated in blood-pressure control [i.e., neurons of tractus solitarius (253), lamina terminalis (279), or hypothalamus and brainstem areas (226)], where they can influence angiotensin II signaling via ROS production.
If, in physiologic conditions, NOX activity serves as regulator of angiotensin II effects, in pathologic conditions, such as an excess-salt diet (118, 265) or exposure to hypertensive agents (76), an altered NOX activity and excessive ROS production induced by angiotensin II can contribute to neurogenic hypertension and related cerebrovascular diseases. Interestingly, angiotensin II–induced NOX activation can enhance neuronal death triggered by 6-hydroxidopamine (203, 204).
NMDA receptor
The ionotropic NMDA receptor (NMDA-R) is activated mainly by the neurotransmitter glutamate (52). Whereas in physiologic conditions, NMDA-receptor activity is involved in mechanisms such as neuronal growth and synaptic plasticity, its excessive excitation in several CNS diseases may cause neuronal death.
Two putative connections exist between NOX enzymes and the NMDA receptor: NOX enzymes may regulate the NMDA receptor A tight regulation of NMDA-receptor function is required for cell maintenance and survival. In addition to its ligand-dependent activation, the NMDA receptor appears to be modulated by the redox potential: the reduction or oxidation of cysteine residues on its subunits modifies channel conformation and promotes Ca2+ current entrance or blockade, respectively (145). It has also been suggested that the precise effect of ROS on NMDA-receptor activity is dose dependent (126). Relatively little is known about the role of NOX enzymes as being the source of the redox regulation of the NMDA receptor. In one study, the nonspecific NOX inhibitor DPI prevented the loss of NMDA receptor–dependent long-term potential induced by β-amyloid, thereby suggesting that NOX-inhibition enhances NMDA-receptor function (256). The NMDA receptor may regulate NOX enzymes Activation of the NMDA receptor may lead to NOX2-dependent ROS production in neurons, and thereby activates redox-sensitive signaling cascades including ERK1/2 phosphorylation (127, 212). In apparent contradiction, evidence also suggests that the NMDA-receptor antagonist ketamine increases NOX2 activity (18, 19); however, this is most likely an indirect effect mediated via neuronal production of interleukin-6 (18, 19).
Neuronal death
Most studies on the role of NOX enzymes in neurons have focused on the induction of cell death caused by excessive ROS production. As shown in Table 3, in many situations, NOX-dependent neuronal death occurs. In the majority of the cases, the role of NOX enzymes has been assessed by using nonspecific NOX inhibitors, and NOX2 has been indicated as the main source of ROS-dependent cell death. However, compounds used in these studies are not specific inhibitors of NOX activity, and they do not target NOX2 selectively. Given that neurons also express other NOX isoforms, their role in neuronal death cannot be excluded.
Glia: activation and neuroinflammation
Microglia
Microglia are resident macrophages of the CNS involved in host defense. Usually present in a ramified resting form, they become amoeboid and active in response to several insults (e.g., damaged neurons, pathogens, altered protein accumulation), and release cytotoxic and inflammatory mediators, such as ROS, nitric oxide, and cytokines (246). The production of ROS in activated microglia after many different types of stimuli was associated with NOX2 expression, but recent reports suggest that NOX1 and NOX4 can also play a role (Table 4). Several physiologic processes that are involved in the activation of microglia are regulated by NOX-dependent ROS production, including inflammatory responses (171, 188), cell proliferation (111, 156), induction of neuronal apoptosis during development (161), and release of neurotransmitters (13, 91). Thus, NOX function in microglia is important for health and normal physiology of the CNS; however, when excessively activated, it may also contribute to disease progression. A variety of CNS pathologies characterized by neuronal death seem to have a crucial participation of microglia-derived ROS, in particular, when activation of NOX enzymes is combined with generation of nitric oxide, resulting in extensive neuronal death via peroxynitrite production (29, 56). Excessive ROS generation by microglia contributes to the aggravation of neuronal damage after a stroke or in neurodegenerative diseases (24, 29, 56) and plays a possible role in the development of psychiatric disorders (14, 222, 243). In addition, evidence exists for a role of microglial ROS generation in neurotoxicity associated with infections [e.g., human immunodeficiency virus (241), Mycobacterium tuberculosis (266), and LPS (lipopolysaccharide) (39, 64, 65, 68, 138, 141, 178, 193, 194, 227, 271)] or with chemicals such as paraquat (27, 169, 189), lindane, and dieldrin (157, 158), diesel-exhaust particles (23), or Zn2+ (116).
Astrocytes
Astrocytes are star-shaped glial cells that provide nutrients for neurons and regulate their activity. However, in pathologic conditions, astrocytes can also contribute to inflammatory processes and neuronal cell death. Several NOX isoforms are expressed in astrocytes (Table 2). Production of ROS by NOX enzymes plays a role in astrocyte signaling (5), survival (147), and production of proinflammatory mediators (188). Nonetheless, after toxic stimuli, activation of NOX enzymes in astrocytes can also induce damage of astrocytes (36, 40, 79, 86, 121, 202) or neurons (3, 4).
Oligodendrocytes
Oligodendrocytes are cells responsible for the production of myelin around neuron axons in the CNS. Although little is known about NOX expression in oligodendrocytes, they can, in certain circumstances, be very sensitive to damage by NOX-derived ROS (139, 140).
Role of NOX Enzymes in CNS Diseases
Several studies have analyzed the involvement of NOX enzymes in ROS overproduction underlying CNS diseases. In this chapter, after a brief introduction about each specific pathology, we first discuss the evidence for an involvement of NOX enzymes derived from patient studies, then data from animal models, and finally mechanistic and molecular insights from analyses of in vitro systems.
Neurodegenerative Diseases
Alzheimer's disease
Alzheimer's disease (AD) induces cognitive dysfunctions and represents the most common single cause of dementia worldwide. The underlying mechanisms leading to the development of the disease are only partially known. Even for the small percentage of cases in which genetic factors have been identified [mutations of the β-amyloid precursor protein (APP), presenilin 1 and 2], the sequence of pathologic events is not fully understood (259). The typical microscopic hallmark of AD is the presence of intraneuronal neurofibrillary tangles and extracellular senile plaques. The neurofibrillary tangles are a compact filamentous network formed by paired helical filaments composed of abnormal phosphorylated tau protein. The senile plaques are generated by a deposition of fibrils of the β-amyloid peptide (Aβ) (154). These abnormal protein deposits are associated with an accumulation of activated microglia and astrocytes, and profound synaptic and neuronal loss. Neuronal oxidative stress represents another commonly observed feature of AD (58), due in particular to glia activation (59).
Patient studies
Several studies indicate that NOX enzymes are involved in the pathomechanisms of AD (269). Activation of NOX2 in the brain of AD patients has been demonstrated, based on the translocation of NOX2 subunits (214). It appears that Aβ can cause NOX2-dependent ROS production, because the exposure of neutrophils and monocytes of CGD patients to Aβ-peptides did not induce H2O2 production (20). In addition, increased levels of mRNA transcripts of NOX1 and NOX3 also were described in early-stage brain tissue from AD patients (49). Thus, it appears that NOX2 is activated in AD patients, but an upregulation of other NOX enzymes might contribute to oxidative stress.
Animal models
Demonstrating the crucial role of NOX2 in AD development, NOX2 deficiency improved the outcome in a mouse model of AD. Mice that overexpress the Swedish mutation of APP (Tg2576, which leads to Aβ fragments accumulations) were crossed with NOX2-deficient mice. Absence of functional NOX2 was protective and prevented the negative effects of Aβ deposits. Neuronal oxidative stress was abrogated, and behavioral deficits improved in both young (3‱4 months old) and aged (12‱15 months old) Tg2576/NOX2-deficient mice (185, 186). Lack of NOX2 did not affect the accumulation of β-amyloid fragments, indicating that the NOX2-derived ROS are important player in pathologic alterations induced by β-amyloid. Thus, the formation of senile plaques is not NOX dependent; however, the toxicity of the plaques is markedly amplified through NOX2-dependent ROS generation. In addition, a NOX2 inhibitory peptide, gp91ds-tat, reduced both oxidative stress and AD pathology in aged mice (186). This observation suggests that NOX inhibitors could act in an advanced state of the disease and, for this reason, they are possible candidates for AD therapy (21).
In vitro models
The role of NOX enzymes in AD pathology has been studied by using in vitro cultures of microglia, astrocytes, and neurons.
Microglia
Given that (a) the presence of activated microglia around Aβ represents a major characteristic feature of neuroinflammation in AD brains (224), and (b) ROS are released from activated microglia and contribute to neurotoxicity (29), the role of NOX2 in microglia has been investigated in different AD models in vitro. Exposure to Aβ fragments stimulates the assembly of NOX2 complex on the plasma membrane of microglia and consequently the production of ROS (20), which in turn causes functional alterations (256) or death (197) of neuronal cells. Similarly, by using a co-culture system with human neuroblastoma cells overexpressing APP or mutated APP and microglia/macrophages, it was shown that neurons, releasing Aβ fragments, activate NOX2 in microglia, which produces oxidative stress and leads to neuronal death (196). Also, the protective effect of melatonin in AD models was attributed to decreased ROS production after β-amyloid exposure due to inhibition of NOX2 complex formation in microglia (276). In addition to its neurotoxic effect, NOX2-dependent ROS production seems to influence microglial proliferation, which is stimulated by Aβ through the release of proinflammatory cytokines (e.g., TNF-α, IL-1β) (111). Different pathways involving NOX2 activation in microglia have been investigated: In an autocrine manner, ATP released from Aβ-stimulated microglia induces the production of ROS by NOX2 through activation of the purinergic receptor P2X7 (123, 187); The interaction of Aβ with microglial cell-surface receptor complex (10) leads to the tyrosine phosphorylation of Vav-GEF, a guanine nucleotide exchange factor for Rac1, thereby affecting the assembly of NOX2 (260), β-Amyloid causes NOX-dependent ROS generation, and subsequent oxidation-dependent activation of the chloride channel CLIC1. This oxidation is presumably due to the formation of an intrachain disulfide bond that promotes CLIC1 dimerization and activation (170). Sustained NOX activity itself in this model, however, also appears to depend on activity of the chloride channel, possibly providing a mechanisms for excessive ROS generation.
Astrocytes
An accumulation of astrocytes has been observed near amyloid plaques, and several references indicate that Aβ-induced NOX2 activation in astrocytes contributes to neurodegeneration (2 –4). In addition, Aβ-peptides, through NOX2-dependent ROS generation, in astrocytes alter their membrane structure (98) and lead to depolarization of astrocyte mitochondria (4, 277). Both these factors might therefore contribute to AD pathology.
Neurons
Contributing to neurodegeneration, Aβ-peptides induce NOX2 activation not only in glia cells, but also directly in neurons (212, 240). In addition, the expression of particular mutated form of presenilin 1 and 2 (presenilin 2 N141L mutated or presenilin 1 with mutations in the C-terminal fragment) associated with familial forms of AD, directly caused neuronal death, which was abolished by apocynin (92, 93). Thus, NOX2 might also be directly expressed in neurons of AD patients and, thereby, contribute to oxidative stress and cell death.
In summary, strong evidence indicates that oxidative stress in AD involves ROS generation by NOX enzymes, in particular NOX2. The precise mechanisms of action of NOX enzymes in AD, however, need further evaluation, and it is likely that, in addition to the well-established NOX2/microglia axis, other sites of NOX2 action exist.
Parkinson's disease
Parkinson's disease (PD) is a progressive neurodegenerative disorder, which primarily leads to motor dysfunctions due to loss of dopaminergic neurons in the substantia nigra and the consequent dopamine deficiency. With the exception of rare familial cases, the etiology of PD is still unclear, and both genetic and environmental factors have been implicated (207). Oxidative stress is thought to be an important pathogenetic factor for the degeneration of dopaminergic neurons in PD. Mitochondrial dysfunction has been proposed to be a major source of oxidative stress in PD (96); however, increasing evidence has been found for a role of NOX enzymes.
Patient studies
Several studies have shown the presence of oxidative-stress markers in samples from PD patients (274). In particular, increase in lipid peroxidation (112) and reduction of glutathione levels (217) were found to occur in substantia nigra of PD patients. However, to our knowledge, the role of NOX enzymes in patients with PD has not been investigated.
Animal models
Because the toxic compound MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) and its derivate MPP+ (1-methyl-4-phenylpyridinium) cause effects similar to PD symptoms, in animals they are widely used to study PD (249). Translocation of p67 phox was induced by MPTP in mouse brain and prevented by minocycline, an inhibitor of microglia activation (261). In addition, degeneration of dopaminergic neurons after administration of MPTP was attenuated by ∼20% in NOX2-deficient mice as compared with wild-type controls, suggesting the contribution of NOX2 in this process (273). In line with these findings, LPS injections in the substantia nigra caused reduced death of dopaminergic neurons in NOX2-deficient mice as compared with wild-type animals (198). Thus, NOX2 activation could play a role in microglia-mediated loss of dopaminergic neurons in PD.
In vitro models
Most of the information pertaining to the role of NOX enzymes in PD pathogenesis emanates from in vitro studies. In particular, the involvement of NOX2 in microglia-dependent dopaminergic neurotoxicity has been consistently observed. Reactive microgliosis is a hallmark of PD brains (165). Exposure to rotenone (an herbicide), LPS, MPTP/MPP+, fMLP (formyl-methionyl-leucyl-phenylalanine), 6-OHDA (6-hydroxydopamine), and angiotensin II induce selective death of tyrosine hydroxylase (TH)-positive neurons in primary mesencephalic cultures. For the following reasons, it is thought that these compounds exert their toxicity at least in part through microglia NOX2 (33, 63
–68, 71, 198, 203, 204): The toxicity toward dopaminergic neurons of these compounds was markedly enhanced in neurons/microglia mixed cultures, as compared with neuron-enriched cultures. Apocynin or NOX2 deficiency attenuated the deleterious effect of microglia on TH-positive neurons. In a similar manner, aggregated α-synuclein and substance P, two major components of protein aggregates in PD, activate NOX2 in microglia, thereby contributing to dopaminergic neurodegeneration (22, 272).
These results are corroborated by the analysis of the neuroprotective mechanisms of several compounds: dextromethorphan, sinomenine, FLZ (a squamosamide derivative), nimodipine, TGF-β1 (transforming growth factor-β1), and pituitary adenylate cyclase-activating polypeptides (PACAP38, 27, and 4 to 6). Indeed, these compounds are thought to inhibit activation of microglial NOX2 (138, 141, 194, 195, 267, 271). However, microglia are probably not the primary cause of neuronal damage in PD, but rather a positive-feedback loop that amplifies neurotoxic stimuli. For example, MPP+-stressed neurons release mediators, such as matrix metalloproteinase-3, which in turn activates microglial NOX2, causing oxidative stress and enhanced neuronal death (124). Moreover, the macrophage Ag complex-1 (MAC1), a microglial surface receptor, mediates microgliosis also by inducing the translocation of p47 phox and NOX2-dependent ROS production (104).
The role of NOX enzymes in PD might not be limited to microglial NOX2. NOX1 may also be expressed in microglia and contribute to neurotoxicity (39). A direct expression of NOX enzymes in dopaminergic neurons might play a role (7).
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a progressive degenerative disease affecting motor neurons. First described by Jean-Martin Charcot in 1874, ALS is characterized by muscle weakness and atrophy that lead to paralysis and death, usually within 5 years from disease onset. The majority of ALS cases are sporadic, but familial forms have been also identified (5‱10%). Sporadic and familial forms seem to be characterized by similar neurotoxic mechanisms and, in particular, by oxidative stress (60). However, the primary cause of this pathology is largely unknown. Point mutations in the gene coding for the copper/zinc superoxide dismutase (SOD1) are associated with ∼15–20% of familial forms of ALS (i.e., ∼1% of total cases). It appears that the induction of the disease by SOD1 mutations is not due to a loss of SOD1 function, but rather to the neurotoxicity of the mutant protein (25).
Patient studies
Oxidative stress plays a central role in the development of ALS. Oxidative stress markers are elevated not only in cortex and spinal cord of ALS patients, but also in cerebrospinal fluid, plasma, and sera (218). Expression of NOX2 at both messenger and protein levels was increased in the spinal cord of sporadic ALS patients. In particular, expression of NOX2 was specifically localized in microglial cells and led to ROS production and oxidative damage (262).
Animal models
The most widely used experimental model to study ALS is a mutant SOD1 transgenic mouse. This mouse develops motor neuron degeneration and neurologic symptoms comparable to those observed in both sporadic and familial ALS patients (87). In this animal model, microglia activation contributes highly to neurodegeneration. Indeed, restricted expression of SOD1 mutant in astrocytes (78) or in motor neurons (143, 190) does not lead to motor neuron degeneration, whereas specific reduction of mutant SOD1 in microglia slows the course of the disease and increases survival (26). Because oxidative damage in spinal cord and cerebral cortex is one of the main features of ALS progression in SOD1 mutant mice (97), the involvement of NOX enzymes has been investigated in this model.
A crucial role of NOX2 was demonstrated by two studies on SOD1-mutant mice crossed with NOX2-deficient mice. In both studies, NOX2 deficiency increased lifespan, improved symptoms, and decreased histologic severity (160, 262). However, the magnitude of the effect was different. In one study, the mean increase in lifespan was 13 days, whereas in the other, it was ∼100 days. It has been argued that differences in genetic background could account for these differences (160).
The involvement of NOX2 in the ALS mouse model was confirmed in other unrelated models: Deletion of the prostaglandin receptor EP2 in aging SOD1 mutant mice improved motor strength and survival. The effects of this EP2 deletion included a reduction of NOX2 subunits p47
phox
, p22
phox
, p67
phox
, and p40
phox
(142). In the same manner, the lack of functional T cells or CD4+ T cells in SOD1 mutant mice leads to accelerated disease progression accompanied also by elevated NOX2 expression (17).
In addition to NOX2, NOX1 might also play a role in ALS pathogenesis. NOX1 deficiency significantly increased lifespan in SOD1 mutant mice (160).
In contrast, pharmacologic studies using the antioxidant/NOX inhibitor apocynin in the mouse ALS model produced contradictory results. In one study, the administration of high doses of apocynin (30‱300 mg/kg) increased survival and slowed ALS progression in SOD1 mutated mice (90). Conversely, a more recent analysis with similar apocynin concentrations did not indicate such effect (1). To our understanding, these contradictions do not raise doubts about the participation of NOX2 in ALS pathology, but rather demonstrate technical issues with the relatively nonspecific inhibitor apocynin (see earlier).
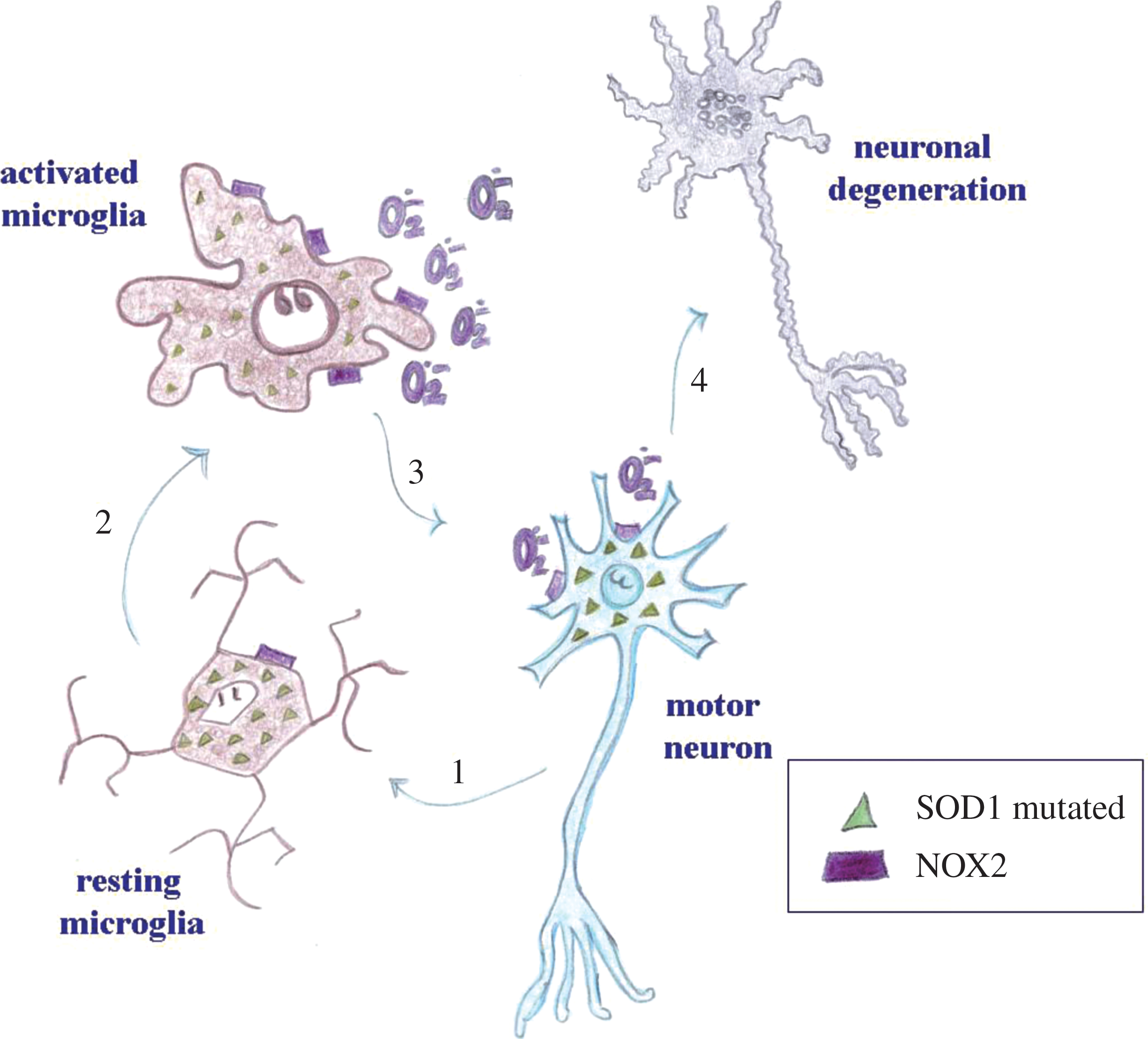
Thus, it appears that activation of NOX2, and to some extent also NOX1, is a relevant neurotoxic factor in ALS. Strong evidence suggests that microglial NOX2 is crucial; however, experimental conditions do not allow discriminating the specific cellular type involved and whether other cellular sources of NOX enzymes (e.g., neurons) might also play a role (Fig. 3).
In vitro models
A recent study suggests that SOD1, interacting with the small GTPase Rac protein, might be involved in the regulation of NOX2 activity. For this reason, SOD1 mutations, by altering the correct binding with Rac, increase NOX2 activation (90). Even if microglia activation appears to be involved highly in ALS pathogenesis, it has been observed that SOD1-mutated astrocytes can also contribute to motor neuron loss, via activation of NOX2 (159).
As mentioned earlier, SOD1 mutations account for approximately only 1% of ALS cases. It may, however, be suspected that persistent NOX2 activation is a common downstream pathway triggered by other possible mechanisms underlying ALS pathology. In particular, inflammatory processes also contribute to disease development (25). Li et al. (137), recently showed that in organotypic spinal cord slices, high doses of LPS induced NOX2-dependent death in motor neurons.
Cerebrovascular Diseases
Several NOX isoforms are highly expressed in the cerebral vasculature. For this reason, the role of NOX enzymes in cerebrovascular pathology, such as in cerebral stroke, has received wide attention. Stroke is the third leading cause of death and one of the major causes of disability worldwide. A stroke can be triggered either by the occlusion of a vessel in the brain (ischemic stroke) or by an intracranial hemorrhage (hemorrhagic stroke). Almost 85% of the strokes are ischemic, and the remainder are hemorrhagic. Symptoms may vary and depend on the brain area affected by the stroke, the extent of the insult, and the vulnerability of neurons (199). One of the molecular events that follow stroke is the generation of ROS (144). In this context, several lines of evidence indicate an involvement of NOX enzymes in the mechanisms underlying the development of brain damage.
Ischemic stroke
Ischemic stroke is caused by decreased blood supply in a defined brain region due to the occlusion of a vessel. This occlusion can be thrombotic (caused by blood clots formed locally) or embolic (caused by emboli arising from the heart or other part of the body). Atherosclerosis is one of the major risk factor for thrombotic stroke, whereas endocarditis and myocardial infarction are common causes of embolic stroke (199). After ischemia, necrosis occurs in the infarcted area, while a series of complex molecular pathways are activated in the neighboring region (the penumbra). The penumbra is functionally impaired but potentially salvageable (166). Thus, therapeutic concepts for the treatment of stroke should be designed to reduce the progression of brain damage and, hence, target the penumbra.
Patient studies
The pathogenesis of ischemic stroke in humans is thought to comprise genetic, environmental, and lifestyle factors. Genetic factors may increase the risk of developing a stroke (51) or affect the outcome in response to a given ischemic lesion (155). Among NOX subunits, p22 phox presents the highest density of single nucleotide polymorphisms (SNPs) (16). For this reason, investigations on the relation between genetic variability of NOX enzymes and cardiovascular diseases have focused on p22 phox (206). However, the results are debatable. In two cases, the T allele of the C242T polymorphism of p22 phox was significantly associated with an increased occurrence of cerebrovascular diseases (75, 109), but these data were not confirmed in other studies (133, 213). These discrepancies most likely reflect the fact that, in a strongly polymorphic gene, such as p22 phox , the effect of haplotypes (i.e., the constellation of SNPs in a given allele) rather than a single SNP should be studied.
Animal models
Permanent middle cerebral artery occlusion and transient middle cerebral artery occlusion followed by reperfusion are widely used in experimental animal models to investigate ischemic stroke. After ischemia/reperfusion in adult mice, the absence of NOX2 significantly reduced infarct size and blood–brain barrier disruption (113, 251). However, the protective effect of NOX2 deficiency was not observed in newborn pups after hypoxia/ischemia (55). Based on bone marrow–transplantation experiments, it has been suggested that, after stroke, NOX2 is a major source of ROS, not only in peripheral leukocytes (invading the CNS with the reperfusion), but also directly in CNS cells (251). The expression of NOX2 after stroke has been observed in activated microglia of the penumbra region (80). However, NOX2 in astrocytes and neurons might also play a role. In addition to NOX2 expression, increased NOX4 mRNA levels have been measured in the cortex after stroke (164), in particular in neurons and in newly formed capillaries of the periinfarct region (242).
Several pharmacologic studies demonstrated that NOX enzymes are involved in the progression of brain damage after ischemic stroke. By using the nonselective electron-transporter inhibitor DPI, superoxide production was reduced ex vivo in arteries of the penumbra area and of the contralateral part to the lesion (168). Also, the administration of the antioxidant/NOX inhibitor apocynin before ischemia exerted a protective effect (113, 233, 234, 258, 265). In particular, it has been suggested that apocynin decreases the activity of matrix metalloproteinase-9 (149), reducing the relative disruption of the blood–brain barrier and improving neurologic outcome. It has been argued that the protective effect of apocynin can be observed only within a narrow range of concentrations and that it is lost at 5 mg/kg (234). However, some studies successfully used higher concentrations (113). Whether apocynin can be effective if administered after stroke remains to be tested.
Other arguments indicating the deleterious effect of NOX-dependent ROS production in the development of postischemic brain lesions derive from study on: Atorvastatin, which decreases NOX activity through reduction of Rac isoprenylation, and reduces brain damage after stroke (101, 113). Valsartan, an inhibitor of the angiotensin AT1 receptor, which attenuates NADPH oxidase activity and superoxide production in the brain of salt-loaded stroke-prone hypertensive rats, and decreases neuronal apoptosis and inflammation (265). Normobaric hyperoxia treatment, which inhibits NOX2 expression and activity in cerebral microvessels after transient ischemia and reduces matrix metalloproteinase-9 induction, blood–brain barrier damage, and cerebral edema (149). The protective effect of postischemic treatment with adrenomedullin—a CNS peptide hormone—which has also been associated with decreased NOX activity (263, 264).
However, NOX2 may not only be detrimental in the context of stroke. Indeed, ethanol preconditioning, which induces a mild oxidative stress through activation of NOX2, prevents excessive brain damage in experimental stroke (257).
In vitro models
Insights into the mechanisms underlying cellular death after stroke come from in vitro studies using neuronal cultures exposed to oxygen/glucose deprivation and reoxygenation. It appears that NOX2 in neurons can contribute to ROS accumulation and neuronal death during the reoxygenation phase, whereas after anoxia, the first source of ROS are mitochondria and xanthine oxidase (6).
Hemorrhagic stroke
Hemorrhagic strokes are often due to intracerebral or subarachnoid hemorrhage. Hypertension, bleeding disorders, or amyloid angiopathy can cause intracerebral hemorrhage, whereas trauma or rupture of an aneurysm may lead to subarachnoid hemorrhage (199). Generally, the mortality rate is higher after hemorrhagic stroke than after cerebral ischemia. In particular, the most severe and frequent complication of aneurysmal subarachnoid hemorrhage is cerebral vasospasm, due to altered vasoconstrictor and vasodilator mechanisms in cerebral vessels. These vasospasms lead to a high mortality and morbidity (61).
Patient studies
Signs of reduced antioxidant defense and of oxidative damage have been observed in cerebrospinal fluid, brain, and serum of patients after subarachnoid and intracerebral hemorrhage (9, 54, 62, 117). In particular, it seems that oxidative stress could be responsible for development of vasospasms (129, 192). So far, patient-oriented research on the role of NOX enzymes in ROS production after hemorrhagic stroke has not been performed. The only available study reports no association between p22 phox polymorphisms and frequency of aneurysms (131).
Animal models
Studies on the role of NOX enzymes in intracerebral and subarachnoid hemorrhage by using pharmacologic inhibition or genetic deletion have produced conflicting results.
Rat model of subarachnoid hemorrhage
Published data have consistently shown a role of NOX enzymes in this model. Increased expression levels of NOX2, as well as decreased oxidative stress, vasospasms, and neurologic deficits after treatment with the inhibitors, DPI or apocynin, were reported (119, 215, 275). It has also been argued that the diminished neuronal injury after treatment of subarachnoid hemorrhage with hyperbaric oxygen was due to reduced NOX2 expression (180, 181). However, because inhibitors are nonspecific and no NOX2-deficient animals were tested, the evidence should, at this point, be considered circumstantial.
Mouse model of subarachnoid hemorrhage
NOX2-deficient mice after subarachnoid hemorrhage failed to show a reduced mortality rate, brain-water content, or intensity of oxidative stress (148). Inhibitors have not been tested in this model, nor have mice deficient in other NOX enzymes.
Rat model of intracerebral hemorrhage
Treatment with apocynin did not improve the outcome of rats after intracerebral hemorrhage. Hemorrhage volume, neurologic score, and brain edema were not reduced in rats injected with different doses (3‱30 mg/kg) of apocynin (236).
Mouse model of intracerebral hemorrhage
After collagenase-induced intracerebral hemorrhage, NOX2-deficient mice displayed diminished bleeding, brain injury, neurologic deficits, and mortality as compared with wild-type mice (232). An increased activity of NOX enzymes was also found in hypertensive mice with spontaneous intracerebral hemorrhage (250).
The question whether NOX enzymes are involved in the pathogenesis of hemorrhagic stroke remains open. In the context of an intracerebral hemorrhage, the protection of NOX2-deficient mice is of interest, and the absence of an effect of apocynin is only of relative weight. In the case of subarachnoid hemorrhage, the data are largely based on inhibitors and contradicted by the data from the NOX2-deficient mouse.
Demyelinating Diseases
Multiple sclerosis
Multiple sclerosis (MS) is a demyelinating neurodegenerative disease that leads to severe chronic disability and gradually to death. It is a neurologic disorder with an autoimmune component. It involves, however, other factors, including altered inflammatory response, neuronal loss, and axonal injury processes (94). Because environment and genetics strongly influence disease outcome, disease progression may vary greatly among patients (179). Several general lines of evolution of the disease have been described. It could be characterized by acute attacks (relapses) followed by remission (relapsing‱remitting multiple sclerosis) or by a progressive neurologic decline (115). Unfortunately, no curative treatment exists, but partially effective therapies are currently available, which are generally based on antiinflammatory treatment and immune modulation or suppression. The observations on the role of NOX enzymes in MS are puzzling. NOX2 activity appears damaging in some studies, but protective in others. Although this is only partially understood, it is possible that NOX2-dependent suppression of T-cell activation at early disease stages is protective, whereas NOX2-dependent ROS release from microglia has a detrimental effect.
Patient studies
Evidence of the involvement of oxidative stress in the progression of MS has been reported in several studies on MS patients [e.g. (30, 229, 248)]. Activation with PMA (phorbol-12-myristate-13-acetate) triggered higher ROS production in monocytes from MS patients than in controls (247), which might suggest a role of NOX2. Also, a diminished NOX-dependent ROS generation was measured in monocytes from interferon-β–treated patients (151). However, as discussed earlier, some patient data suggest a protective effect of NOX2 in the context of MS. A recent genetic study raised the possibility that the individual genomic pattern of p47 phox is related to the age of disease onset in MS patients. The ratio between the two types of p47 phox pseudogenes [one containing the deletion (ΔGT) and one not (GTGT)] was analyzed. It was found that a ΔGT/GTGT ratio of 2:1 or more is associated with lower oxidative burst and earlier disease onset as compared with a ΔGT/GTGT ratio of 1:1 or less (81).
Animal models
Experimental autoimmune encephalomyelitis (EAE) is widely used in animals to model and investigate multiple sclerosis (223). The data on the effects of NOX2 activation in this model have given complex results. Depending on the size of the antigen that was injected to trigger EAE, p47 phox -deficient or p47 phox -mutant mice developed either a less severe or a more severe disease (105, 245). In rats with a low ROS-generating variant of p47 phox , experimental autoimmune encephalitis was aggravated (15).
In vitro models
Several observations indicate that NOX enzymes could be involved in mechanisms of myelin loss and inflammation mediated by macrophages and microglia. Phagocytosis of myelin by microglia/macrophages leads to NOX2-dependent ROS production (150, 244). However, it is not clear whether this increased ROS generation is detrimental and causes death of oligodendrocytes (139, 220), or whether it is neuroprotective because of decreased production of proinflammatory mediators (150).
Psychiatric Disorders
Most research on psychiatric disorders has focused on abnormalities in different neurotransmitter systems. However, increasing evidence suggests a role for oxidative stress in the pathogenesis of mental diseases, in particular schizophrenia, bipolar disorder, anxiety, or autism. It seems that oxidative stress is a common characteristic of these pathologies (176). Thus, to what extent NOX enzymes are involved in the pathogenesis of mental diseases is becoming increasingly relevant. In particular, recent studies suggested a possible role for NOX enzymes in the pathophysiology of schizophrenia and anxiety.
Schizophrenia and drug-induced psychosis
Approximately 1% of the world's population is affected by schizophrenia (28). It usually appears in the late second and third decades of life and manifests with altered mental functions (hallucinations and disturbance in cognitive functions, attention, and memory) and abnormal behavior (loss of motivation or excessive emotionality). Both genetic and environmental factors have been strongly correlated with the development of schizophrenia. Affected individuals have a lifetime of disability, and ∼10% will eventually commit suicide (136). Presently, therapeutic options for schizophrenia are limited and, in general, consist of drugs that act mainly on symptoms. Moreover, they have significant adverse effects and must be taken always.
Patient studies
Evidence of the involvement of oxidative stress in the mechanisms of schizophrenia was first proposed in 1954, and subsequent studies confirmed the contribution of ROS in mechanisms underlying this mental disease. Specifically, analyses on schizophrenic patients have revealed decreased antioxidant defense, such as glutathione (53), and signs of oxidative membrane lipid damage (237). Microglia activation has been detected in brain regions of schizophrenic patients (14, 222, 243), raising the possibility that microglial NOX enzymes might play a role. In addition, an increased superoxide production in neutrophils from schizophrenic patients was observed as compared with controls (219).
Animal models
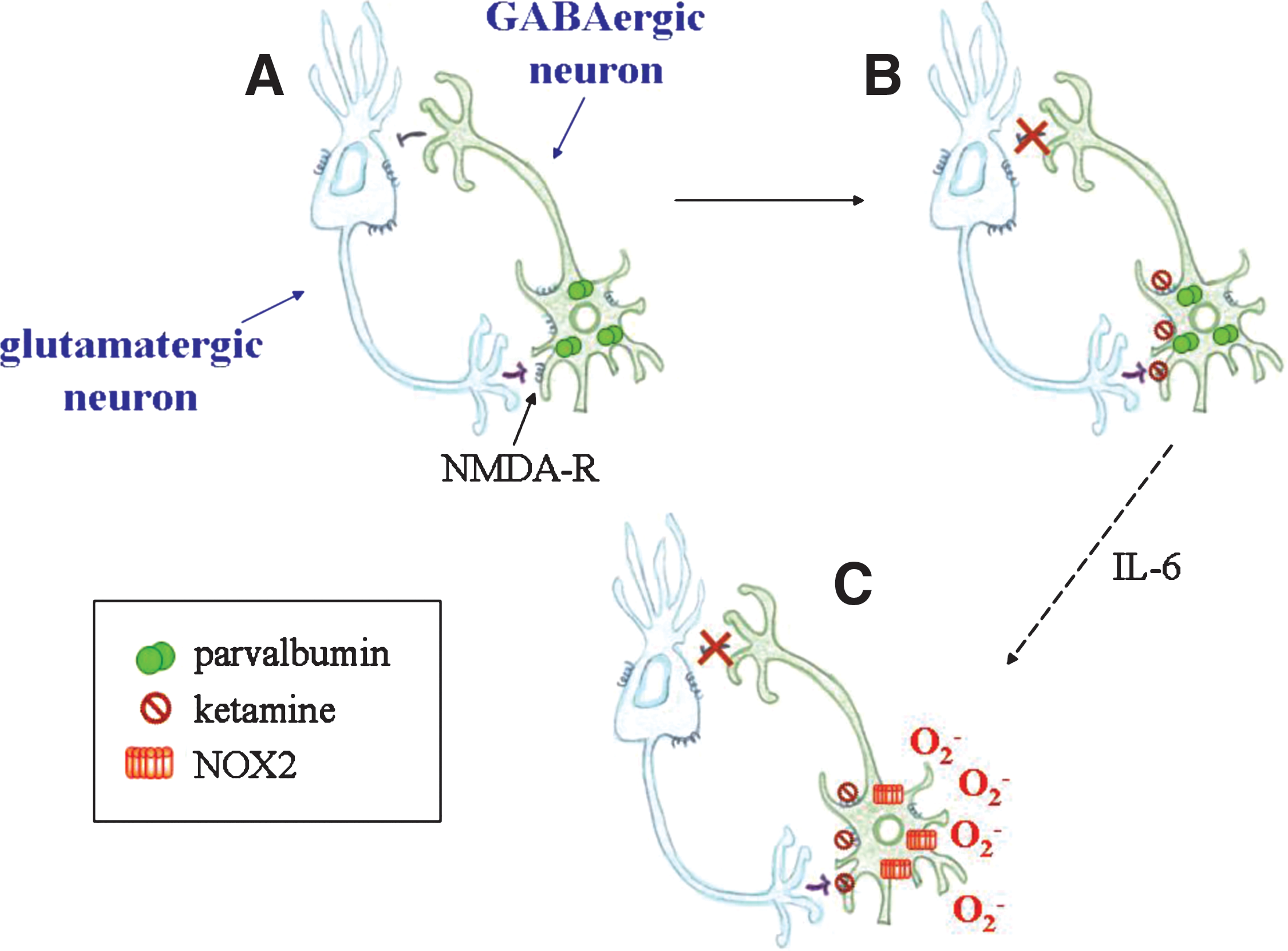
Certain drugs, in particular ketamine and phencyclidine, may cause schizophrenia-like symptoms in humans and in animals (57). Recent reports investigated the role of NOX enzymes in ketamine-induced psychosis. In this experimental system, prolonged administration of subanesthetic doses of ketamine led to oxidative stress and decreased the number of parvalbumin-expressing GABAergic interneurons, causing excessive activation of pyramidal neurons and related psychosis (18). Given that (a) ketamine augmented NOX2 expression in the mouse brain, and that (b) both apocynin pretreatment and NOX2 deficiency prevented ROS generation and the decrease of parvalbumin-expressing interneurons, NOX2 activation was indicated as a major contributor to the pathogenesis of ketamine-induced psychosis and possibly also to schizophrenia (18, 19). In this context, it was suggested that enhanced neuronal production of interleukin-6 is responsible for NOX2 upregulation or activation or both in the brain after ketamine injections (19) (Fig. 4).
In vitro models
With isolated primary neurons culture, it was observed that exposure to ketamine leads to oxidative stress, and parvalbumin-expressing neurons decrease. Because apocynin reversed ketamine effects, the role of NOX enzymes was suggested (18). As observed in vivo interleukin-6 mediated NOX2 activation also in purified primary neurons (19).
Anxiety
Anxiety is basically a physiological reaction that can affect anybody. It is characterized by behavioral and physiological alterations, such as accelerated heart rate, increased breathing, and muscle tension. However, anxiety symptoms can become pathologic when they manifest without any obvious stimuli and begin to interfere with normal life. In those cases, anxiety represents the predominant feature of a more severe mental disorder, which includes phobia, social anxiety, and posttraumatic stress or panic disorder. Genetic and environmental factors influence moderately the development of anxiety as compared with other mental diseases. Nevertheless, they can affect cerebral maturation during childhood and cause anxiety-related behavior in adults (84). Neurochemical alterations and anatomic structures related to anxiety symptoms are not yet completely clear, but increasing evidence suggests a role of oxidative damage in the pathogenesis of anxiety.
Patient studies
Some data from epidemiologic studies suggest that anxiety-related pathologies are linked to altered antioxidant defense and increased oxidative lipid damage (8, 132). However, no information on the involvement of NOX enzymes in patients with pathologic anxiety could be retrieved.
Animal and in vitro models
In mice, ROS contribute to anxiety-related behavior (103), and signs of oxidative stress are found not only in the brain (200), but also in peripheral blood cells (201) of animals with anxious behavior. One report suggests that NOX enzymes activation could induce anxiety symptoms. Oxidative stress induced with BSO (
Conclusion and Perspectives
Evidence is emerging for a role of NOX-derived ROS in neuron development and signaling, in particular through angiotensin II and NMDA receptors. ROS might also be important for astrocyte and microglia physiologic functions. However, an increasing number of studies also demonstrate the involvement of NOX enzymes in CNS pathologies. In addition to the diseases mentioned in this review, several reports suggest that NOX enzymes might also be implicated in alcohol-induced neurodegeneration (44, 45, 89), HIV-associated dementia (110, 216), neuronal loss after sleep apnea (270, 278), and neuronal death induced by high blood glucose levels after hypoglycemic coma (225).
Why are we interested in understanding the role of NOX enzymes in the CNS? The most immediate and directly relevant interest is to understand the implication of redox biology and pathobiology in brain function and disease. However, in the long run, two additional aspects might become important. (1) Genetic variations in NOX enzymes might account for sensitivity or resistance to human diseases. Thus, such variants might become important for the interpretation of clinical studies, but possibly also be a tool for the prediction of individual disease risk. (2) NOX enzymes might become drug targets in the CNS. So far, antioxidant drugs have been proposed to reduce oxidative stress for treatment of neurodegenerative diseases, such as AD and PD. Not unexpectedly, such a nonspecific scavenging approach did not yield promising results (114). Conversely, inhibition of specific ROS-generation enzymes (i.e., NOX isoforms) holds higher promise.
Footnotes
Acknowledgments
We thank Dr. Vincent Jaquet, Dr. Stefania Schiavone, and Dr. Charles E. Jefford for comments and editing.