
Abstract
The superoxide-generating NADPH oxidase NOX1 is thought to be involved in signaling by the angiotensin II–receptor AT1R. However, underlying signaling steps are poorly understood. In this study, we investigated the effect of AngII on aortic smooth muscle from wild-type and NOX1-deficient mice. NOX1-deficient cells showed decreased basal ROS generation and did not produce ROS in response to AngII. Unexpectedly, AngII-dependent Ca2+ signaling was markedly decreased in NOX1-deficient cells. Immunostaining demonstrated that AT1R was localized on the plasma membrane in wild-type, but intracellularly in NOX1-deficient cells. Immunohistochemistry and immunoblotting showed a decreased expression of AT1R in the aorta of NOX1-deficient mice. To investigate the basis of the abnormal AT1R targeting, we studied caveolin expression and phosphorylation. The amounts of total caveolin and of caveolae were not different in NOX1-deficient mice, but a marked decrease occurred in the phosphorylated form of caveolin. Exogenous H2O2 or transfection of a NOX1 plasmid restored AngII responses in NOX1-deficient cells. Based on these findings, we propose that NOX1-derived reactive oxygen species regulate cell-surface expression of AT1R through mechanisms including caveolin phosphorylation. The lack cell-surface AT1R expression in smooth muscle could be involved in the decreased blood pressure in NOX1-deficient mice. Antioxid. Redox Signal. 11, 2371–2384.
Introduction
A major source of ROS is the NOX family of superoxide-generating NADPH oxidases (3). To date, seven members are known in mammals. At least two NOX enzymes, NOX1 and NOX4, are found in vascular smooth muscle (18). Recently, we developed NOX1-deficient mice and showed that in response to AngII, NOX1-deficient mice have an almost complete loss of the sustained blood-pressure response, whereas the initial increase was conserved. In addition, NOX1-deficient mice have a marked reduction in aortic media hypertrophy (16). Activation of the AT1R through AngII is intimately linked with ROS generation (15, 20). ROS trigger downstream signaling of AT1R through various mediators, such as inhibition of tyrosine kinases (29, 45), MAP kinases (39, 40), Ca2+ fluxes (12, 33), or nitric oxide inactivation (35).
Recent evidence points to a role of caveolae in AT1R processing and signaling. Caveolae are specialized regions of the plasma membrane, composed mainly of caveolins (31). They have been implicated in endocytosis, transcytosis, Ca2+ signalling, and numerous other signal-transduction events. Caveolae are very abundant in smooth muscle cells (32). AT1R depend on caveolae for efficient surface localization (19, 22, 47) and present a caveolin-binding–like domain in their cytoplasmic tail (28). NOX1 colocalizes with caveolin1 (21). Phosphorylation of caveolin has been shown to be related to budding and fusion of caveolae and thus could be responsible for the transport of proteins (1, 31).
In this work, we used aortic primary smooth muscle cells derived from wild-type and NOX1-deficient mice to study the role of NOX1-derived ROS on AngII signaling and on the AT1R expression and localization. AngII-induced ROS formation and Ca2+ release is blunted in NOX1-deficient mice. Immunostaining showed that AT1R is not properly targeted to the plasma membrane in NOX1-deficient cells. Our data suggest that decreased phosphorylation of caveolin contributes to the lack of plasma-membrane targeting of the receptor and the subsequent decrease in the Ca2+ release in response to AngII. Thus, NOX1-derived ROS play an important role in AT1R sorting and hence represent a novel cellular mechanism to explain how oxidative stress sensitizes the vascular system to the effects of AngII.
Materials and Methods
NOX1-deficient mice
NOX1-deficient mice were generated as described previously (16).
Tissue preparation
Two-month-old C57BL/6J wild-type mice and C57BL/6J NOX1-deficient mice were used. Thoracic aortas were removed, placed in Hank's Balanced Saline Solution (HBSS; Gibco, CA), and connective and fat tissues were removed. For ex vivo studies, harvested aortas were directly stimulated for 1 h with H2O2 diluted in HBSS.
Cell culture
Primary smooth muscle cells were isolated with enzymatic dissociation. In brief, cleaned aortas were transferred in HBSS containing trypsin (Sigma, Buchs, Switzerland; 1.25 mg/ml), elastase (Sigma; 0.5 mg/ml), and collagenase type 2 (Sigma; 1 mg/ml) to remove the adventitia. Then tissues were incubated in HBSS containing papaïne (Sigma; 0.3 mg/ml) and dithioerythritol (Sigma; 1 mg/ml) and then transferred to HBSS containing collagenase. Isolated primary smooth muscle cells were plated and cultured in DMEM containing 10% fetal calf serum and used directly for each experiment after 7–10 days.
For H2O2 treatment, cells were incubated overnight at 37°C with the indicated concentrations.
Cell transfection
Isolated NOX1-deficient primary smooth muscle cells were cultured for 7 days and then transfected by using JetPEI (Polyplus Transfection, Polyplus, Illkirch, France) with a pCDNA3.1 plasmid–expressing mouse NOX1 (mNOX1) (2) by following the manufacturer's protocol. Transfected cells were selected with 0.5 mg/ml G418 and were used 3 days after transfection.
SDS-PAGE and immunoblotting
Cleaned aortas (see tissue preparation) were mechanically dissociated by using Tissue Lyser (frequency, 30 Hz; Qiagen, CA). Once dissociated, the resulting mix was further digested on ice in lysis buffer containing 1% Triton X-100 in 50 mM NaCl, 10 mM MgCl2, 1 mM EGTA, 50 mM Tris-HCl, pH 7.4, supplemented with a protease inhibitor cocktail (Complete mini; Boehringer-Mannheim, Mannheim, Germany) for 30 min and sonicated. Protein concentration was determined with protein assay (BioRad, CA). Proteins were separated on a 12% SDS-PAGE gel. Immunoblotting was conducted according to standard techniques and probed with rabbit polyclonal AT1R (Santa Cruz Biotechnology, Heidelberg, Germany; sc-1173), total caveolin, and phospho (Y-14)-caveolin antibodies (BD Transduction Laboratories, Allschwill, Switzerland). Antibody binding was visualized with horseradish peroxidase (HRP)-labeled antibodies and chemiluminescence reagents (ECL reagent; Amersham Biosciences, PA). α-Tubulin (Santa Cruz Biotechnology) was used as a standard.
Cytosolic Ca2+ measurement
The cytosolic Ca2+ concentration was monitored with the fluorescent Ca2+ indicator Fura-2 (Invitrogen) as previously described (14). In brief, after 7–10 days in culture, cells were incubated for 1 h in HBSS containing 2 μM Fura-2 AM (Invitrogen) and 0.1% pluronic acid (Sigma). To minimize compartmentalization of the dye, loading and experimentation were performed at room temperature. Fluorescence was processed by calculating 340:380-nm fluorescence ratios by using the Metafluor system (MDS Analytical Technologies, CA).
RT-PCR
RNA was extracted from purified primary smooth muscle cells with Qiagen RNA Micro kit (Qiagen). After standard reverse transcriptase reactions, specific smooth muscle markers were examined with PCR. Primers are described in Table 1.
Quantitative real-time PCR
RT-PCR was conducted as described (17). AT1R primers were as follows, forward: 5′-CCA TTG TCC ACC CGA TGA AG-3′ and reverse: 5′-TGC AGG TGA CTT TGG CCA C-3′. Two constitutively expressed reference genes, EEF1A1 (eukaryotic elongation factor 1A1) and TBP (Tata box‱binding protein) were selected for normalization of candidate gene-expression levels.
ROS production
We used DHE (Invitrogen), a widely used probe, to detect ROS generation (48). After 7–10 days in culture, primary smooth muscle cells were loaded in HBSS containing 5 μM DHE for 30 min at 37°C. Nuclear ethidium fluorescent signal (excitation, 480 nm; emission, 590 nm) was detected and analyzed with Metafluor.
Immunostaining
For histology, aortas were used in a free-floating tissue configuration. In brief, cleaned aortas were opened and fixed with 4% paraformaldehyde for 2 h. AT1R was stained with a rabbit polyclonal anti-AT1R diluted in saline solution containing 0.3% Triton X-100. An HRP-coupled antibody (Cappel, OH) was used for the revelation. Specific staining was revealed by addition of 3,3′-diaminobenzidine (DAB, Dako, Glostrup, Denmark) to produce a brown coloration, and nuclei were counterstained with cresyl violet. Primary smooth muscle cells were fixed with 2% paraformaldehyde and permeabilized with 0.25% Triton. Antibody raised against AT1R was added and revealed by Alexa Fluor 488-conjugated antibody (Invitrogen). For calreticulin (primary antibody gift of Prof. M. Michalak, University of Alberta, Calgary, Alberta, Canada), revelation was conducted with Alexa Fluor 555-conjugated antibody (Invitrogen). Specific AT1R-blocking peptide (Santa Cruz Biotechnology; sc-1173P) was used for each technique to assess the quality and reliability of the signal obtained with the AT1R antibody. The concentration of the peptide was 5 times the antibody concentration, and they were incubated for 2 h at room temperature for absorption. Confocal imaging was performed on a LSM 510 Meta confocal scanner mounted on an Axio Imager Z1 microscope (Carl Zeiss, Feldbach, Switzerland).
Electron microscopy
Thoracic aortas from 2-month-old wild-type and NOX1-deficient mice were fixed for 2 h in 3% glutaraldehyde (Sigma). The fixed tissues were embedded in epoxy resin and processed for electron microscopy, as described previously (13).
Statistical analysis
Data analysis was performed by using the software GraphPad Prism 4 (GraphPad Software, San Diego, CA). Results are expressed as mean ± SEM. The p values >0.05 were considered not significant; p values ≤ 0.05 were considered significant.
Results
Given the strong evidence for a role of NOX1 in vascular AngII responses (7, 16, 17, 20, 30), we decided to investigate cultured aortic smooth muscle cells. Purification of smooth muscle cells from aortae of wild-type and NOX1-deficient mice revealed no apparent differences in their number or in their morphologic appearance. These cells were tested with PCR for specific smooth muscle markers, smooth muscle actin, and the calponin-related protein SM22 (10), and with AT1R and GAPDH as a control. Corresponding mRNA were found in both cell preparations (Fig. 1A). Thus, we concluded that we had obtained high-quality aortic smooth muscle cells from both wild-type and NOX1-deficient mice.

(
Unfortunately in our hands, none of the commerically available NOX1 antibodies gave satisfying results in Western blots or in immunostaining (data not shown). We therefore analyzed our samples with RT-PCR. In agreement with previous observations (18), we found abundant NOX4 and NOX1 mRNA in wild-type cells, whereas only NOX4 mRNA was detected in NOX1-deficient cells (Fig. 1A).
AngII-induced ROS production is abolished in NOX1-deficient cells
We proceeded to measure ROS generation in the cultured cells. Basal ROS accumulation was ∼30% lower in NOX1-deficient, as compared with wild-type mice (p = 0.0092) (Fig. 1B and C, left panel). We then investigated stimulated ROS generation. Adenosine triphosphate (ATP) did not stimulate ROS production in wild-type aortic smooth muscle cells (data not shown), whereas AngII clearly did. This ROS production was inhibited by the flavoprotein inhibitor diphenylene iodonium (DPI). Importantly, AngII-dependent ROS generation was almost completely absent in NOX1-deficient cells (Fig. 1B and C). Allopurinol (inhibitor of xanthine oxidase) or carbonyl cyanide 3-chlorophenylhydrazone (CCCP, inhibitor of mitochondrial respiratory chain) did not have any effect on the AngII-induced ROS generation in wild-type cells (Fig. 1C, right panel). Thus, the AngII-induced ROS formation is mediated predominantly by NOX1. These data demonstrate that NOX1 deficiency decreases basal ROS generation and almost completely abolishes AngII-stimulated ROS generation in aortic smooth muscle cells.
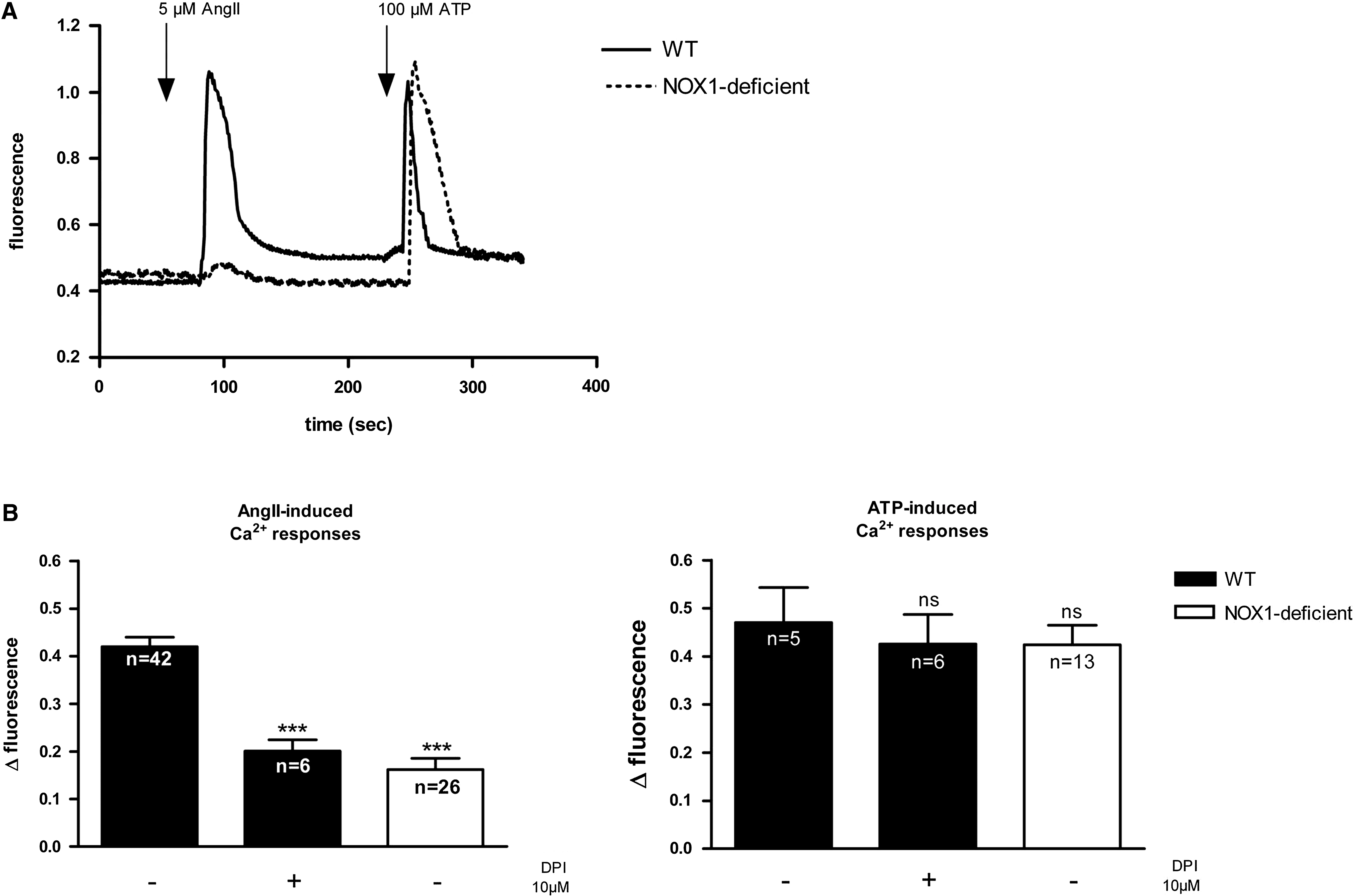
AngII-induced Ca2+ responses are blunted in NOX1-deficient cells
The absence of AngII-induced ROS generation in NOX1-deficient cells might indicate that NOX1 is the only superoxide-generating system activated by the AngII receptor. It might, however, also be due to a lack of signaling through the AngII receptor in NOX1-deficient smooth muscle cells. We therefore measured the cytosolic free Ca2+ concentration, which is an important second messenger for AngII responses. Changes in intracellular Ca2+ were reported by the fluorescence ratio (F340/F380) of the Ca2+ indicator, Fura-2.
ATP induced comparable Ca2+ elevations in both types of cells (Fig. 2). This suggests that the Ca2+ signaling and Ca2+ stores are intact in NOX1-deficient cells. However, on stimulation with AngII, a marked increase in cytosolic Ca2+ was observed only in wild-type, but not in NOX1-deficient cells (Fig. 2). AngII-induced Ca2+ responses in wild-type cells were inhibited by DPI, whereas the ATP-induced Ca2+ responses were not (Fig. 2B). These data demonstrate that in NOX1-deficient cells, not only ROS generation, but also AngII-induced Ca2+ signaling were greatly diminished. Thus, a global AT1R signaling defect, rather than a selective lack of AngII-induced ROS generation was observed.
Expression and localization of the type 1 angiotensin II receptor
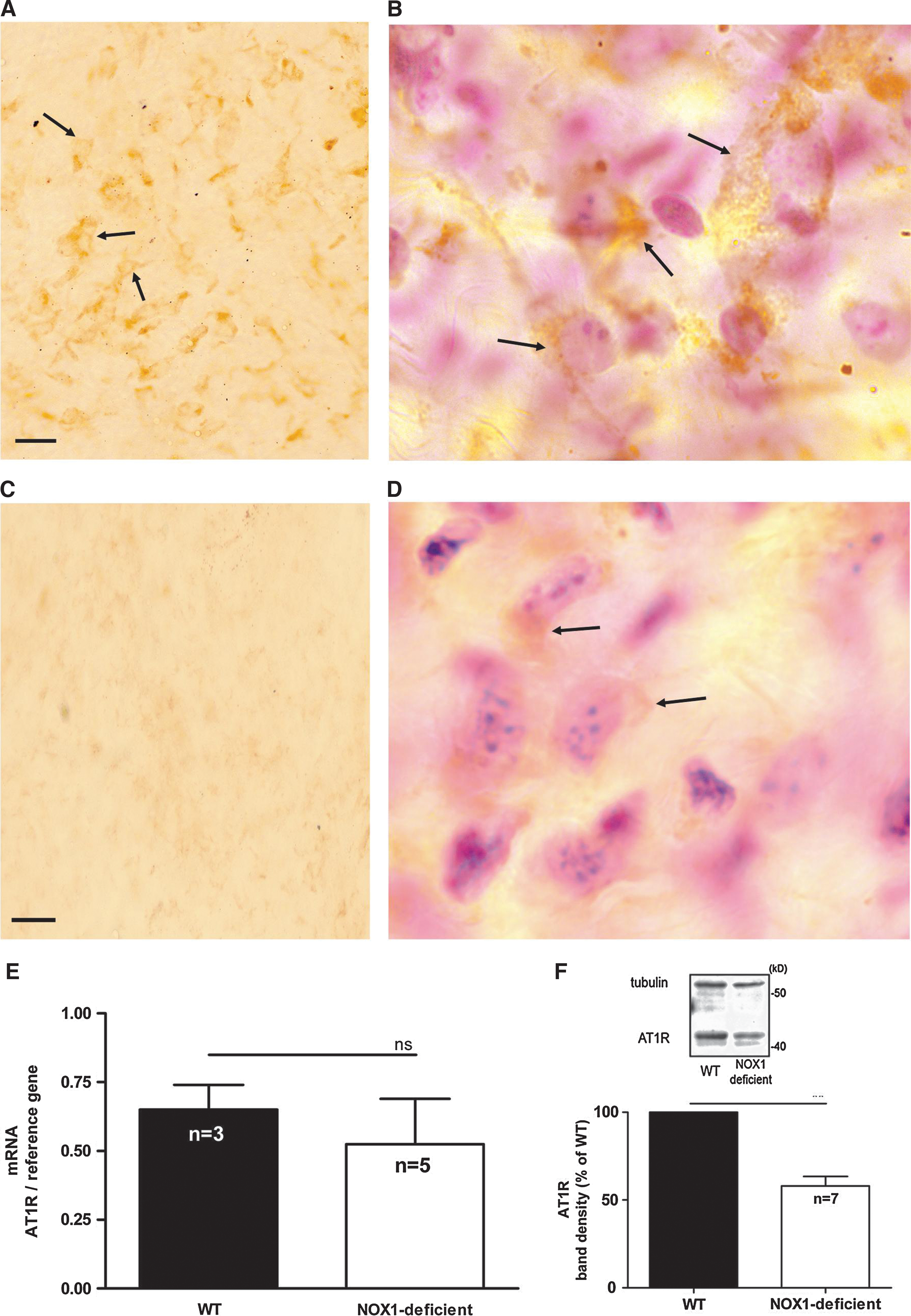
AT1R is the major AngII receptor in vascular smooth muscle cells (8). We therefore investigated the localization of the AT1R in wild-type and NOX1-deficient cells. AT1R antibody specificity was tested by using a specific blocking peptide. The addition of blocking peptide suppressed the signal in both immunohistochemistry and Western blot, demonstrating the specificity of the staining (supplementary Fig. 1). In wild-type cells, AT1R was almost exclusively observed in regions corresponding to the plasma membrane (Fig. 3A, green). In contrast, in NOX1-deficient cells (Fig. 3B), AT1R did not localize to the cell-surface region, but was diffusely found in the cell. AT1R only partially colocalized with the endoplasmic reticulum marker calreticulin (red fluorescence, Fig. 3) and with the Golgi marker GM130 (data not shown). The observed pattern demonstrates an increased intracellular localization of AT1R in NOX1-deficient smooth muscle cells. This altered localization might be, in part, due to retention in the endoplasmic reticulum and the Golgi. Interestingly, nuclear staining also was observed in NOX1-deficient cells, but not in wild-type cells. We have not further investigated this point, but nuclear translocation of AT1R has been described previously (5, 11).

Thus, isolated vascular smooth muscle cells from NOX1-deficient mice show an intracellular localization of AT1R, which is likely to explain the signaling deficit shown earlier. To investigate AT1R localization in vivo, we investigated AT1R in explanted aortas from wild-type and NOX1-deficient mice with immunohistochemistry. An intense immunoreactivity for AT1R was observed in wild-type aortas (Fig. 4A and B, arrows). In NOX1-deficient aortas, almost no signal was observed (Fig. 4C and D, arrows). We therefore applied quantitative methods to investigate AT1R expression. AT1R mRNA levels, as detected by real-time PCR, were not changed in NOX1-deficient mice (Fig. 4E). In contrast, AT1R protein levels, as detected with Western blotting, were markedly decreased in NOX1-deficient mice (Fig. 4F). These data demonstrate that, in addition to the decreased cell-surface AT1R expression, decreased protein levels are present. The latter was not due to decreased mRNA synthesis.
Low levels of exogenous hydrogen peroxide rescue AngII responses and AT1R cell-surface expression in NOX1-deficient cells
If the cell-surface localization of AT1R depends on NOX1-dependent ROS generation, it might be possible to rescue the phenotype by exogenous addition of ROS. We therefore treated NOX1-deficient vascular smooth muscle cells with increasing H2O2 concentrations (from 1 to 500 nM) and measured AngII-induced Ca2+ responses. A dose-dependent rescue of AngII-induced Ca2+ responses occurred on addition of H2O2 (Fig. 5A); the amplitude of the recovered Ca2+ responses reached roughly two thirds of the one observed in wild-type cells. To investigate whether this rescue was due to a retargeting of AT1R to the cell surface, we performed immunofluorescence experiments. As shown in Fig. 5B, a partial retargeting to the plasma membrane was observed when cells were treated with 200 nM H2O2. These experiments indicate the importance of ROS in the AT1R localization.

Transfection of NOX1 rescues AngII responses and AT1R cell-surface expression in NOX1-deficient cells
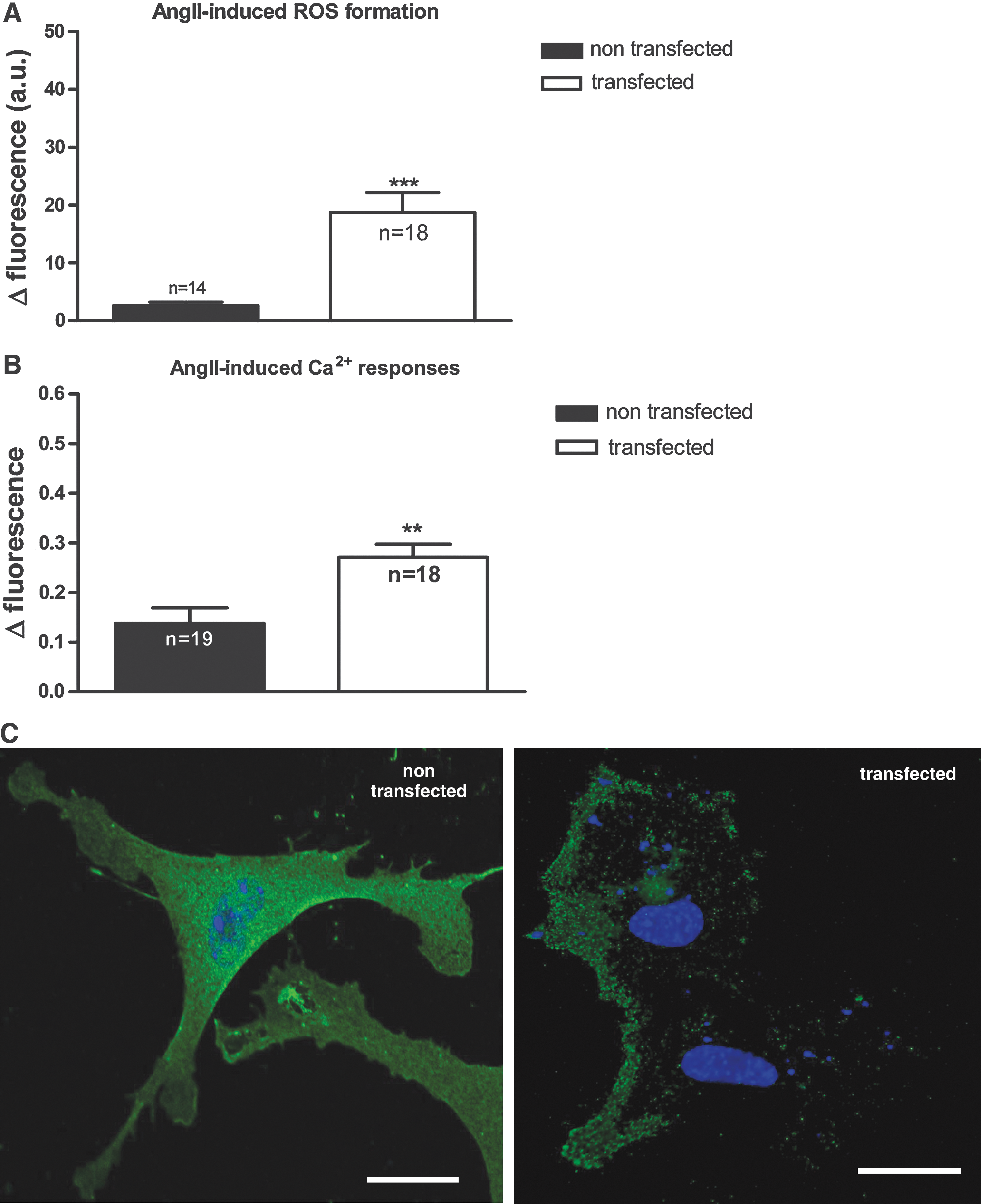
Exogenous H2O2 has been shown to rescue AngII responses and AT1R cell-surface expression in NOX1-deficient cells. This result suggests that ROS are involved in the AT1R localization and AngII signaling. To investigate this point further, we transfected NOX1-deficient primary smooth muscle cells with a mouse NOX1 plasmid and performed ROS and Ca2+ measurements. As shown in Fig. 6A, reexpression of NOX1 in NOX1-deficient cells lead to a reappearance of AngII-induced ROS formation. AngII-induced Ca2+ responses were doubled, compared with nontransfected NOX1-deficient cells (Fig. 6B). Similarly, increased targeting of AT1R to the membranes was observed in the NOX1-transfected cells (Fig. 6C). These results might indicate that NOX1 transfection allows the rescue of the phenotype of NOX1-deficient smooth muscle cells.
Expression of caveolin, phosphorylated caveolin, and caveolae number
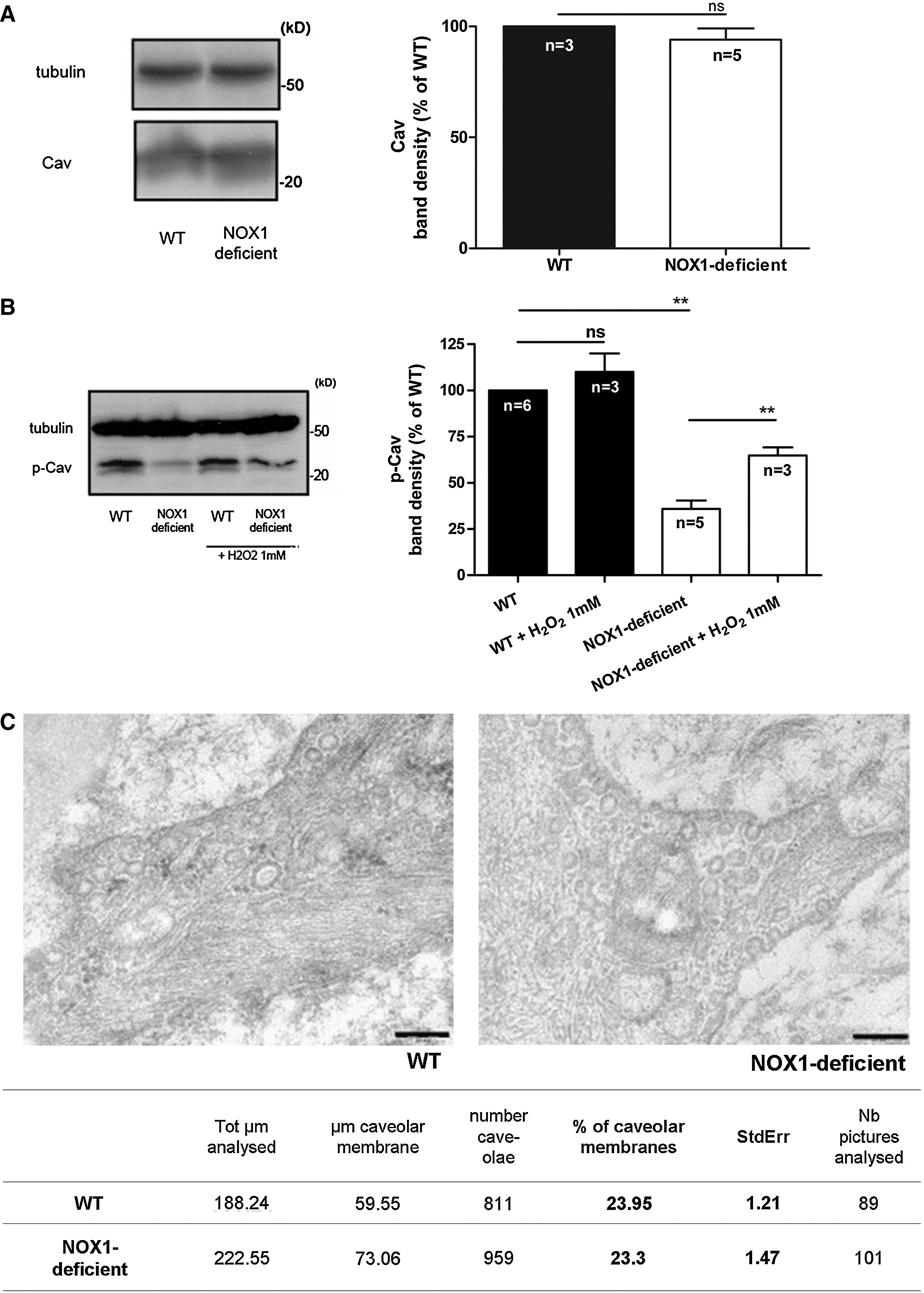
Caveolae and caveolin have been shown to be involved in AT1R plasma membrane sorting and in AT1R signaling (43). To investigate possible disorders in the anchoring of the AT1R to the plasma membrane, we studied with Western blot the expression of the major component of caveolae, caveolin, and phosphorylated caveolin. The expression of total caveolin (Cav) in whole lysate of aortae from wild-type and NOX1-deficient mice was comparable in both types of mouse (Fig. 7A). Electron microscopy suggested that the integrity and total number of caveolae were not changed in aorta from both mice (Fig. 7C). However, phosphorylated caveolin (p-Cav) expression was drastically reduced in aorta from NOX1-deficient mice, and addition of H2O2 increased the phosphorylation of caveolin in these mice, suggesting a role of NOX1-derived ROS in this reaction (Fig. 7B). Altogether, these results suggest that caveolin phosphorylation is altered in NOX1-deficient mice. This could be involved in the altered localization of the AT1R.
Discussion
Our study investigated the relation between AngII signaling and NOX1-dependent ROS generation in vascular smooth muscle. NOX1 deficiency prevented AngII-induced ROS generation and Ca2+ responses. We demonstrated that the lack of AngII responses in NOX1-deficient cells is due to a lack of plasma-membrane localization of the AngII receptor AT1R. The mechanism by which NOX1 determines plasma membrane localization of AT1R may include ROS-dependent phosphorylation of caveolin.
To our knowledge, this is the first description of ROS-dependent regulation of AT1R targeting. This mechanism is clearly demonstrated by the lack of plasma membrane localization of AT1R in NOX1-deficient cells and the rescue through incubation with exogenous H2O2 or reexpression of NOX1. However, although exogenous H2O2 is able to target AT1R to the membrane, it appears that NOX4, which is abundantly expressed in vascular smooth muscle, is not able to do so. Thus, NOX4 is not able to compensate for the absence of NOX1. This could be explained by a localized site of action of NOX enzymes. It has been suggested that NOX1 localizes to caveolae and/or lipid rafts within the plasma membrane (21, 41), whereas for NOX4, a number of distinct localizations have been suggested (4, 21, 24, 37). The proposed association of NOX1 with caveolae provides a possible explanation for how the relatively low basal ROS generation by NOX1 may be sufficient to drive the association of AT1R with the plasma membrane. A local “ROS signaling cloud” generated by basal NOX1 activity around the region of the caveolae might be sufficient to maintain the anchoring of AT1R at the plasma membrane.
The intracellular AT1R in NOX1-deficient mice did not fully colocalize with calreticulin (Fig. 3) or Golgi (data not shown). Based on these observations, we think that the loss of plasma-membrane localization in the absence of NOX1 is more likely due to altered trafficking. However, we cannot exclude a deficient maturation process, because immunoreactivity for AT1R was found in the ER. Electron microscopy showed that NOX1 deficiency does not change the number of caveolae in the cell. Thus, it appears that the decreased caveolin phosphorylation in NOX1-deficient cells does not lead to global changes in the number of caveolae. The phospho-caveolin–dependent association of AT1R with the plasma membrane does not prove a direct association with caveolae. A previous study suggested that AT1R requires receptor occupation for the association with caveolae (22, 43). Thus, it is possible that, in a first step, phospho-caveolin is important for the association of AT1R with the plasma membrane, and in a second step, AngII is necessary for its association with caveolae. Previous studies suggested that phosphorylation of caveolin is required for the formation of caveolae (1, 31). The magnitude of the reduction in caveolin phosphorylation observed in NOX1-deficient mice did not decrease the total number of caveolae; however, it was sufficient to alter the localization of AT1R. Thus, whereas our data do not exclude a role of caveolin phosphorylation in the formation of caveolae, they favor the concept of its involvement in protein trafficking (1, 23). AT1R has a caveolin-binding–like site, and mutations of this motif considerably reduce the plasma-membrane expression of the receptor and lead to its accumulation in intracellular compartments (28). Note also that caveolin1-deficient mice show a decrease in AngII-induced contractile responses in the vascular system (9), but AT1R localization in caveolin1-deficient vascular smooth muscle has so far not been studied.
Recently, Ritsick et al. (33) showed that, in Drosophila ovarian smooth muscle, NOX5 is required for agonist-induced Ca2+ release and subsequent contraction; the same group mentioned unpublished data showing that NOX1 suppression interferes with Ca2+ signaling in human smooth muscle cells (25). These data have been interpreted as a role of NOX-derived ROS in the regulation of Ca2+ fluxes.
Redox-sensitive regulation of intracellular and plasma membrane Ca2+ channels has been described (3). In our system, the lack of Ca2+ signaling is, however, due to a different mechanism: ROS indirectly influence AngII-mediated Ca2+ signaling through regulation of receptor localization.
The mechanism described (ROS-dependent regulation of plasma membrane localization of AT1R) has potential physiological implication for the AngII response and for blood-pressure regulation. Under stress conditions, one would expect an increased AngII response of the organism. In acute stress situations, this could be considered a beneficial physiologic response. In chronic stress, however, that should contribute to development of hypertension and vascular disease. Thus, the intracellular AT1R localization might contribute to the decreased blood-pressure response in NOX1-deficient mice (16, 30).
It also should be mentioned that the main function of AT1R in aortic vascular smooth muscle is probably not blood-pressure regulation. Key players in blood-pressure regulation are peripheral resistance vessels, the kidneys, and the central nervous system (6). Thus, the mechanisms described here should not be extrapolated to the overall blood-pressure response, but only to the AngII responses in the aortic vascular wall, as, for example, cell proliferation and gene expression.
Taken together, NOX1 is an important modulator of AT1R in aortic smooth muscle, and NOX1 represents an interesting drug target for the treatment of vascular diseases (25, 46).
Footnotes
Acknowledgments
We thank Dr. Michel Dubois-Dauphin for excellent technical assistance and Drs. Daniel Hössli and Pierre Cosson for helpful discussions. This work was supported by Swiss National Foundation grant 100A0-103725 awarded to K.H.K. and 310000-120280/1 to M.F.
Author Disclosure Statement
K.H.K. and V.J. own stock in the start-up company GenKyoTex, which is developing NOX inhibitors.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.