
Abstract
The NADPH oxidase (Nox) enzyme family generates reactive oxygen species (ROS) that contribute to cell signaling, innate immune responses, proliferation, and transcription. The signaling mechanisms that regulate this important enzyme family are only beginning to be understood. Evidence is emerging which suggests that phosphorylation of Nox and/or their regulatory components may be important means of modulating their activity. We describe here the evidence for Nox regulation through the action of kinases, and speculate on how such regulatory mechanisms might contribute to the development of pathological disease states. Antioxid. Redox Signal. 11, 2429–2441.
Introduction to NADPH Oxidases
Discovered only within the past 9 years, the Nox family members are now recognized as producers of low levels of reactive oxygen species (ROS) that play critical roles in maintaining normal physiologic processes and in the etiology of multiple diseases. A general effect of Nox-generated oxidants on intracellular signaling pathways is likely, as a number of important intracellular signaling proteins (e.g., tyrosine phosphatases and transcription factors) are redox sensitive, and their inhibition/activation in response to changes in oxidation state have the potential to affect cellular function broadly. ROS generated via Nox also contribute to a growing number of diseases, including atherosclerosis, hypertension, arthritis, Alzheimer disease and other neurologic disorders, stroke, respiratory syndromes, cancer, and inflammation (68, 80, 85). At this point, very little is known about how the activities of the Nox proteins are regulated under normal, much less pathologic, conditions.
Increasing evidence suggests that phosphorylation of various Nox proteins or their regulatory cofactors or both may play important roles in regulating the activity of these enzymes. This may be of particular significance for the Nox family members (i.e., Nox4, Nox5, Duox) whose activity is not regulated by Rac GTPases, as other modes of acute regulation must be postulated. At this early stage, however, no information exists, to our knowledge, of direct phosphoregulation of Nox3, Nox4, or Duox. *Note added in proof: A recent publication describes the phosphorylation and regulation of Duox1 by protein kinase A-mediated phosphorylation of Ser955, while Duox2 is phosphorylated by protein kinase C (90a). This review focuses on emerging evidence for regulation of the other nonphagocyte Nox enzymes by kinase-mediated phosphorylation. We do not, in general, include in this category the upstream kinase signaling pathways leading to downstream Nox activation, but rather focus on the phosphorylation events directly modulating Nox components or activities or both.
The best-understood example of Nox regulation remains the well-studied Nox2 phagocyte enzyme. Although it is clear that regulation of the other Nox family members differs in many ways from that of Nox2, Nox2 forms a basis for understanding Nox regulation overall, and we therefore begin by summarizing Nox2 regulation here. The regulation of the phagocyte Nox2 enzyme by phosphorylation has been extensively reviewed (24, 34, 50, 51, 88), and the interested reader is referred to these reviews.
Nox2: A Paradigm for Understanding Nox Function and Regulation
An important component of the ability of leukocytes to kill microorganisms lies in their capacity to generate reactive oxygen species via a membrane-associated NADPH oxidase (9, 104). This multicomponent enzyme uses electrons derived from intracellular NADPH to generate superoxide anion, which subsequently dismutates to H2O2 and other ROS that are used for host defense against bacterial and fungal pathogens. Genetic defects in the protein components of the NADPH oxidase result in chronic granulomatous disease (CGD), a group of inherited disorders in which innate immunity is impaired (22, 53). However, the inappropriate or abnormal action of the NADPH oxidase has been implicated in the pathogenesis of inflammatory tissue injury and a number of additional disease states, highlighting the importance of tight regulation of ROS formation (20). Leukocyte-derived ROS have been reported to damage bodily tissues, perpetuating inflammatory responses and resulting in the tissue damage observed in myocardial infarction, rheumatoid arthritis, and various cardiovascular, neurologic, and inflammatory disorders.
Components and regulation of the phagocyte NADPH oxidase
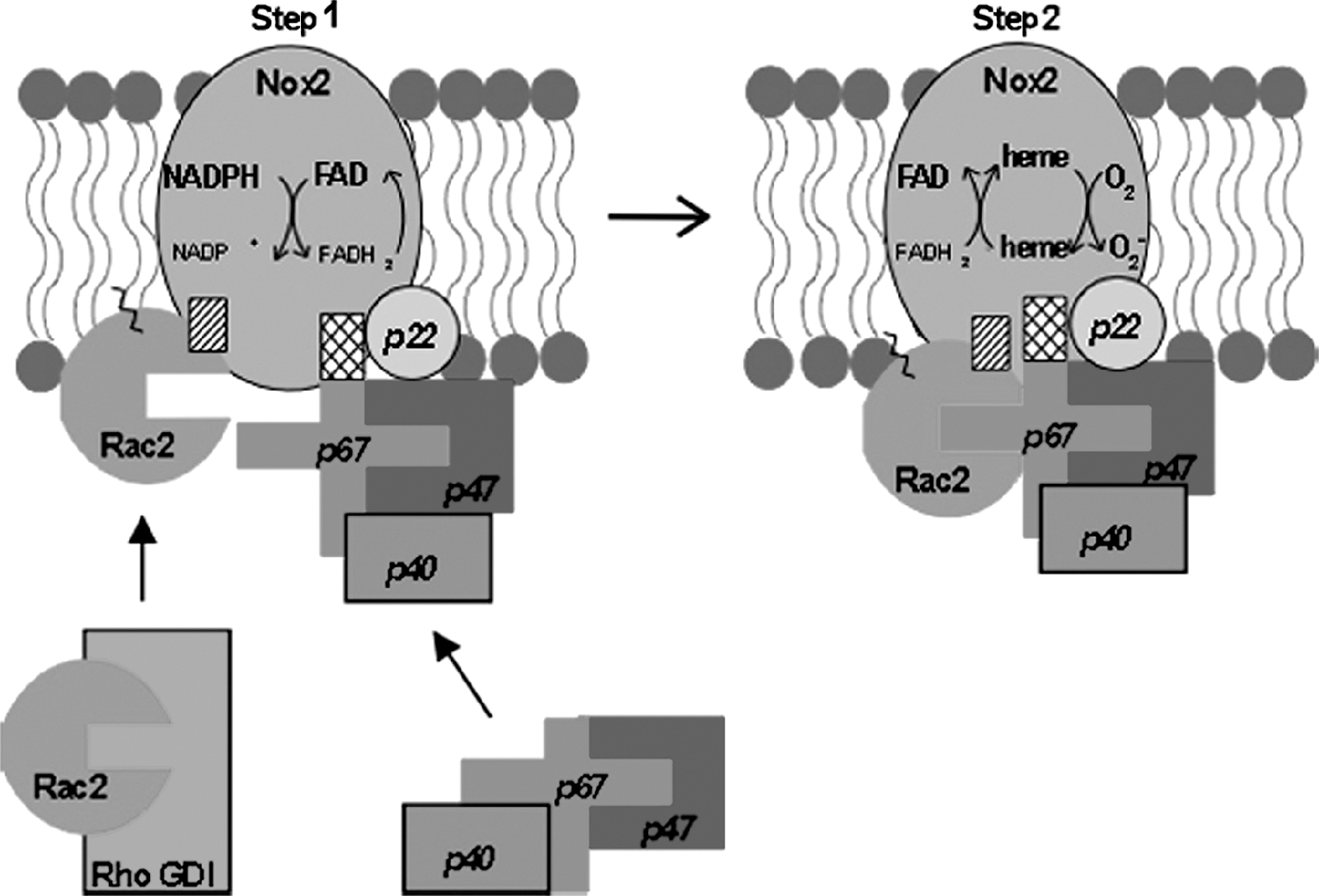
The NADPH oxidase of stimulated phagocytic leukocytes catalyzes the one-electron reduction of oxygen to produce superoxide anion by using NADPH as substrate (Fig. 1). When the phagocyte is activated through the action of soluble chemoattractants and chemokines, or phagocytic particles, cytosolic regulatory components (Rac2, p67
phox
, and p47
phox
or p40
phox
) of the
The phosphorylation of the p22 phox subunit of cyt b was observed in stimulated neutrophils long ago (46). Phosphorylation of p22 phox correlated with NADPH oxidase activity, and both phospholipase D-dependent and -independent phosphorylation pathways have been reported (90). The significance of p22 phox phosphorylation in neutrophils for Nox2 regulation remains unknown. However, it is interesting to speculate whether the phosphorylation of this common Nox membrane co-subunit may have regulatory implications for other Nox family enzymes as well.
A recent publication has described the protein kinase C-mediated phosphorylation of Nox2 itself (89). Although the sites of phosphorylation were not determined, they were present in the carboxy-terminal fragments aa. 321–405 and 466–570. PKC-mediated phosphorylation of Nox2 increased the intrinsic diaphorase (INT-reductase) activity of the recombinant Nox2 cytosolic domain, as well as its binding to the regulators Rac2, p47 phox , and p67 phox . Thus, phosphorylation of Nox2 may be an important mechanism for modulating NADPH oxidase assembly or activity or both. Whether a similar mechanism might regulate nonphagocyte Nox family members will be interesting to investigate.
p67 phox is an absolutely required component of the active Nox2 enzyme. A putative “activation domain” has been identified in p67 phox (aa. 199‱210) by deletional and mutational analysis that is required for stimulation of oxidant formation by Nox2 (52, 81). This domain interacts directly with Nox2 to regulate the transfer of electrons from NADPH to Nox2-bound flavin (Step 1) through as-yet-undefined means. A number of reports exist that p67 phox can be phosphorylated both in vivo and in vitro (2, 23, 33, 40, 41, 95); however, the physiologic significance of p67 phox phosphorylation has not been demonstrated.
The cytosolic p47 phox and p40 phox regulatory components exist both in free forms and in complexes with p67 phox in resting neutrophils (69). p40 phox and p47 phox serve in the intact cell to (a) provide regulatory response elements for NADPH oxidase assembly induced by extracellular activators; (b) act as “organizers” to facilitate membrane compartment-localized binding of p67 phox with Nox2 (42, 67); and (c) promote full Nox2 activity (30, 31). The function of the p47 phox organizer is critical for normal stimulated NADPH oxidase activation in intact leukocytes, as evidenced by the marked impairment in ROS formation in CGD patients lacking p47 phox (22). Similarly, p40 phox deficiency in a mouse gene knockout model elicits a dramatic defect in stimulus-induced oxidase activity, albeit accompanied by decreases in p67 phox expression (36). However, neither p40 phox nor p47 phox is absolutely required for NADPH oxidase activity under cell-free assay conditions (42, 67).
The phosphorylation of p40 phox during neutrophil activation has been reported in several studies (14, 44, 96). Phosphorylation takes place on Thr154 and Ser315, and most likely is mediated by a PKC-family enzyme (14). The significance of p40 phox phosphorylation is unknown, although it has been reported to promote negative regulation of NADPH oxidase function by p40 phox (71).
Regulation of Nox2 by phosphorylation of p47phox
During neutrophil activation, the phosphorylation of p47 phox is critical to NADPH oxidase priming and activation. p47 phox is phosphorylated on at least eight individual serine residues through the action of multiple kinases, including several PKC family members, ERK1/2, p38 MAPK, Pak1, and Akt (34, 38). The stepwise phosphorylation of p47 phox induces an initial translocation of the p47 phox /p67 phox complex to the plasma membrane, where it “primes” the oxidase for activation (34). Subsequent complete p47 phox phosphorylation, including serines 303 + 304, 359 + 370, and 379, enables the interaction via multiple binding sites with both subunits of the integral cyt b membrane protein to form an active enzyme complex (24, 51). The Src-mediated phosphorylation of p47 phox on tyrosine residue(s) has been reported both in vitro and in hyperoxia-stimulated human pulmonary artery endothelial cells (19). Inhibition of Src prevented the hyperoxia-induced assembly of p47 phox with membrane-associated cyt b.
Available biochemical and genetic evidence indicates that phosphorylation of p47 phox induces conformational changes that disrupt autoinhibitory intramolecular interactions between internal SH3 (Src homology 3) domains, the PX domain (phox homology domain), and a C-terminal polyproline domain. This exposes the SH3 domains in p47 phox , which are now capable of binding to proline-rich regions of other NADPH oxidase components (50, 51). This is supported by the findings of the x-ray structure of both autoinhibited and active p47 phox by Rittinger and associates (50, 51). They demonstrated that the tandem SH3 domains of p47 phox form a “super” SH3 domain in which the ligand-binding surface is formed by a combination of the two individual SH3 ligand-binding domains. The flexible linkage of the two SH3 domains ensures a high local domain concentration that drives an otherwise low-affinity interaction, whereas a conserved “GWW” motif in each SH3 domain contributes unique interdomain interactions. The super-SH3 domain can then bind to the nonconventional C-terminal polyproline SH3-binding motif. Additional residues outside or at the border of the ligand-binding surface, including Ser303, Ser304, and Ser328, contribute substantially to the autoinhibited conformation. Substitution of these serine residues with glutamate residues (to mimic phosphorylation) induces a cumulative disruption of the autoinhibited conformation. The ability of multiple serine phosphorylations within p47 phox to destabilize and relieve autoinhibition by this unique super-SH3 domain provides an important control mechanism to ensure that inappropriate activation of NADPH oxidase activity does not occur.
Rac GTPase regulation of Nox2
More than 18 years ago, Rac2 was identified as a required regulatory component for the human NADPH oxidase of phagocytic leukocytes (64, 65). As a member of the Rho GTPase family (including Rho, Rac, and Cdc42), Rac2 functions as a molecular switch that is regulated by the binding of guanine nucleotides. When bound to GTP, Rac2 is active, whereas conversion of GTP to GDP by hydrolysis results in inactivation. Therefore, Rac2 must be in a GTP-bound state to stimulate NADPH oxidase activity both in vivo and in cell-free systems. The activities of the Rac GTPases (Rac1, 2, 3) are regulated by exogenous proteins that modulate their GTP/GDP state (10).
An additional level of control of the Nox2 activation process is accomplished through the regulated release of Rac GTPase from cytosolic complexes with GDI (see Fig. 1) and its subsequent translocation to and activation at the membrane. The release of Rac from RhoGDI appears to be directly regulated by the phosphorylation of RhoGDI at Ser101 and Ser174 (27). A number of other kinases (e.g., c-Src) modulate Rac activity through the phosphoregulation of GTPase-GDI cycling (25, 26, 31).
The translocation of Rac2 to the plasma membrane NADPH oxidase in human neutrophils occurs through an independent mechanism from, but in a coordinated manner with, translocation of the p47 phox /p67 phox complex (11, 30, 87). Rac2 activation can occur through the action of membrane-localized GEFs, including Vav1 (61) and P-Rex1, a leukocyte-enriched PIP3- and G protein βγ subunit-regulated Rac GEF (107, 112). Rac binds directly to p67 phox (but not p47 phox ) via the GTPase switch I domain (29). It was shown that a tetratricopeptide repeat (TPR) in the N terminus of p67 phox is the site of Rac binding (66), and this was confirmed on determination of the crystal structure of the Rac-p67 complex (70). Subsequent data showed that Rac also interacts directly with Nox2, at least partially through the Rac insert domain (28). More recently, a direct Rac interaction site on Nox2 that is also conserved on Nox1 and Nox3 has been identified (59).
Regulation of the Nonphagocyte Nox Enzymes by Phosphorylation
In general, the Nox family proteins and their respective regulatory proteins all contain sites for potential regulatory phosphorylation(s) by various kinases, as predicted by a number of sequence-analysis algorithms. Moreover, many of the Nox family proteins have been shown to be stimulated by the addition of phorbol esters such as phorbol myristate acetate to cells. As phorbol esters are known to stimulate the activity of various protein kinase C enzymes, it is likely that Nox activation is therefore due to phosphorylation of either the Nox enzymes themselves or their regulators or both by PKCs. As yet, no direct demonstration has been made that this is the mechanism of Nox activation by phorbol esters. However, increasing evidence indicates that a variety of kinases and phosphorylation-initiated mechanisms do regulate the activity of various Nox enzymes. These mechanisms include (a) direct phosphorylation of Nox or Nox regulatory components; (b) the phosphorylation-induced binding of regulatory 14-3-3 proteins; and (c) stimulation of Rac GTPase activation.
Nox1: Regulation via cAMP-dependent protein kinase (PKA) and Src tyrosine kinase
Nox1 is structurally most closely related to Nox2 (60, 98), and their regulation is also similar (Fig. 2). First, p22 phox , an essential component of the Nox2 heterodimer, is also required for function of Nox1, Nox3, and Nox4 (8). Second, homologues of the p47 phox and p67 phox cytosolic regulatory proteins were identified in colon epithelial cells and were shown to be essential for Nox1 activity (5, 47, 101). NOXO1 is homologous to p47 phox , whereas NOXA1 is homologous to p67 phox (see later). Their function as required components for Nox1 activity has been demonstrated in heterologous coexpression experiments, and subsequently verified by siRNA-mediated knockdown of the endogenous proteins. Finally, the small GTPase Rac1 is also required for full Nox1 activity: Both Nox1 (59) and NOXA1 (101) bind Rac1-GTP, and Nox1-mediated ROS formation is reduced by dominant inhibitory Rac1 and by Rac1-specific siRNA (17, 75, 76).

NOXO1 organizer component is ∼25% identical at the protein level to p47 phox and shares a number of functional domains. Each protein has a phox (PX) domain that aids in their interaction with distinct membrane phosphoinositides: PI(3,4)P2 in the case of p47 phox and PI(4)P, PI(5)P, and PI(3,5)P2 in the case of NOXO1 (18). Both also have two SH3 domains and a C-terminal proline-rich region that mediate interactions between p22 phox and NOXA1/p67 phox , respectively. A major difference lies in the absence in NOXO1 of the internal autoinhibitory domain present in p47 phox , which acts to suppress interactions with Nox2/p22 phox . The binding of this autoinhibitory region in p47 phox is normally relieved by a series of phosphorylations of the serine-rich region adjacent to this domain, as described earlier. This difference between the NOXO1 organizer and the p47 phox organizer has important regulatory consequences for Nox1 function. Unlike p47 phox , NOXO1 does not require phosphorylation-induced conformational changes for activation and is constitutively membrane localized (18). Because both p47 phox and NOXO1 serve as organizers to promote assembly of cytosolic regulatory components with the membrane-localized Nox catalytic subunits, it would appear that assembly of the functional Nox complex may be constitutive in Nox1 as opposed to Nox2. It has been shown that NoxA1 is associated with the membrane under basal conditions because of its recruitment by NoxO1 (76). Membrane binding of NoxA1 may also be influenced by Rac1 under certain circumstances (76). NoxA1 interacts through its C-terminal SH3 domain with proline-rich repeat sequences in NoxO1, as well as with Rac1 GTPase through its N-terminal TPR repeats (76, 101). These interactions are critical for Nox1 activation and are important for the recruitment of the NoxA1 activation domain to the membrane-localized Nox1.
Nox1 regulation by the PKA-mediated phosphorylation of NoxA1
A mechanism for inhibition of Nox1 activity in colon epithelial cells by the cAMP-dependent protein kinase (or PKA)-mediated phosphorylation of the NoxA1 regulatory protein has been identified (62, 63). The elevation of cellular cAMP (cyclic adenosine monophosphate) by agents such as forskolin or by bacterial toxins or by both, such as cholera toxin, activates PKA (72, 105). PKA was shown to phosphorylate NoxA1 at two distinct sites, Ser172 and Ser461 (Fig. 3). The Ser172 phosphorylation site lies within a consensus 14-3-3 protein-binding motif (RRGS172LP vs. RXXpSXP) (82); see later section on 14-3-3 binding). Of note, this same site is not present in the highly homologous p67 phox Nox2 regulatory protein.
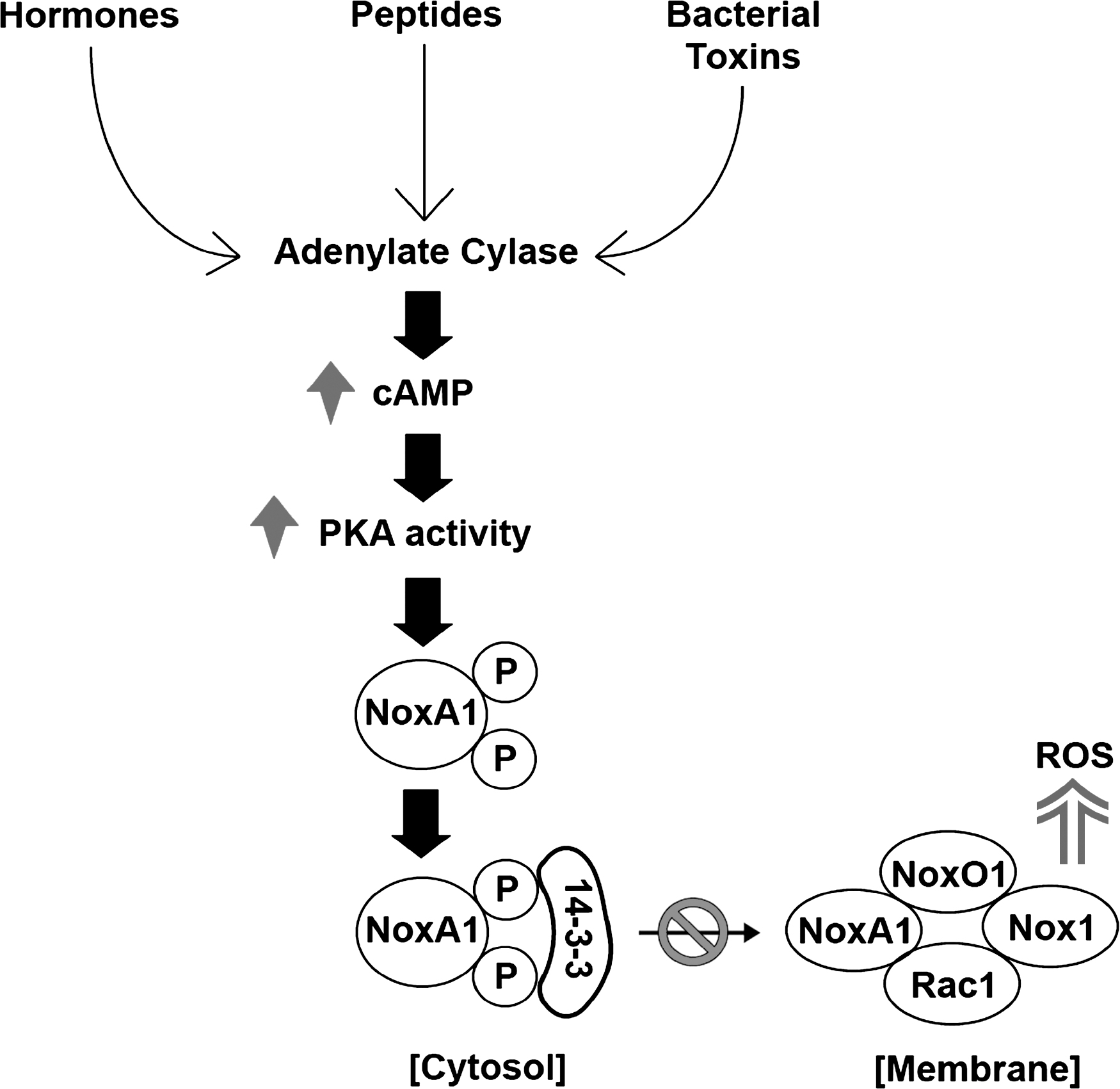
Consistently, 14-3-3ζ was observed to bind directly to phosphorylated NoxA1 in a manner that was dependent on Ser172 phosphorylation, both in vitro and in cells (63). Maximal 14-3-3 binding required both phosphorylation of Ser172 and Ser461 and was reduced by mutation of a site (K49E) in 14-3-3ζ that is required to recognize substrate-phosphorylation motifs (111). The requirement for both phosphorylation sites on NoxA1 may reflect the known propensity for 14-3-3 dimers to interact with more than one phosphorylation site within a substrate to achieve stable binding (15, 108). It is not clear whether the interaction of NoxA1 is specific for 14-3-3ζ versus other 14-3-3 proteins. In experiments with expressed 14-3-3γ protein, we did not observe significant phosphorylation-dependent binding to NoxA1, suggesting that not all 14-3-3 proteins are able to mediate this regulatory interaction (J.-S. Kim, B. Diebold, G.M. Bokoch, unpublished observations).
The interaction of NoxA1 with 14-3-3ζ has functional consequences for Nox1-dependent ROS formation. In the presence of PKA and 14-3-3ζ protein, a substantial inhibition of Nox1-mediated ROS formation occurred in both a transfected 293 cell model system and with endogenous components in HT-29 colon cells. These inhibitory effects required the presence of phosphorylatable Ser172 and Ser461 residues in NoxA1. 14-3-3ζ protein induced some inhibition when expressed in cells alone; however, this effect was probably due to the presence of some phosphorylated NoxA1 under basal conditions, as it was reduced by the addition of the PKA inhibitor H89. Inhibition by PKA and 14-3-3ζ together was evident when Nox1 was activated by either PMA, Rac1Q61L, or through the angiotensin II type I receptor, but was most effective with the receptor-mediated activation. This may be because the first two stimuli represent nonphysiologic activating conditions.
How does the phosphorylation of NoxA1 and the subsequent induction of complex formation with 14-3-3 modulate NoxA1 (and Nox1) activity? Under these conditions, a substantial dissociation of NoxA1 from the membrane fraction where it normally is found in cells. The NoxA1 interaction with membrane-assembled Nox1 is critical for Nox1 activation via the NoxA1 activation domain. Because the Ser172 and Ser461 sites are near the Rac1-binding TPR domain and NoxO1-binding SH3 domain, respectively, it is possible that the induced binding of 14-3-3 protein(s) to NoxA1 would sterically hinder these interactions. Because NoxA1 is required for Nox1 activity, an important consequence of 14-3-3 binding appears to be the loss of NoxA1 from the Nox1 membrane-associated complex, resulting in the inhibition of Nox1 activity (Fig. 3). Whether other kinases (and phosphatases) are able to modulate the activity of Nox1 or other Nox family members by regulating phosphorylation of the same or nearby sites on NoxA1 would be an interesting area for further investigation.
These findings suggest that Nox1-expressing cells would be susceptible to negative regulation of ROS production by hormonal stimuli able to stimulate cAMP formation and consequent PKA activation. Interestingly, a number of bacterial toxins that may be generated by gut-localized bacteria (e.g., Vibrio cholerae, Bordetella pertussis) are effective inducers of cAMP formation (72, 105). We established that cholera toxin effectively activates PKA to stimulate NoxA1 phosphorylation in CCD841 colon epithelial cells (63). Similarly, we also showed that the B. anthracis edema toxin, a bacterial adenylate cyclase, invokes a similar Nox1 regulatory pathway (62). The resulting inhibition of Nox1 activity may provide a mechanism for downregulating the innate immune response of the gastrointestinal epithelium, providing the means for these virulent microorganisms to establish their presence in the host.
It is intriguing to speculate here on the possibility that Nox1 activity could be upregulated under pathologic circumstances by interference with the PKA-mediated phosphorylation of NoxA1 (Fig. 4). It was recently established that PKA itself undergoes a redox-mediated enhancement of its own activity under mild oxidizing conditions (54, 55). This appears to be the result of ROS-mediated inactivation of an inhibitory Ser/Thr phosphatase activity. However, at high levels of ROS, direct oxidation of reactive thiol groups in PKA inhibit its activity. Such biphasic regulation of PKA activity by ROS formation provides an intriguing self-limiting feedback mechanism that could determine Nox1 activity and output. One could envision a scenario in which, under normal physiologic conditions, an initial low level of Nox1 activity generates sufficient ROS to enhance PKA activity. This would feed back through NoxA1 phosphorylation to limit Nox1 activity and ROS output. However, if conditions occur that result in very high levels of oxidative stress (e.g., through inflammatory mechanisms or Nox1 upregulation or both), as reported in Crohn disease (100), then PKA would be effectively shut off. Nox1 activity (and ROS formation) would go unchecked by the phosphorylation of NoxA1. It would be of interest to determine whether such a mechanism contributes to various inflammatory diseases (e.g., of the colon or colon cancers or both), because Nox1 is abundant in the gastrointestinal system.

14-3-3 proteins and regulation of Nox by phosphorylation
Phosphorylation of Nox isoforms or their regulatory subunits suggests the possibility that these protein(s) will interact with other signaling proteins that act to modulate the function of phosphorylated proteins in a variety of biologic processes. One of the most intriguing classes of signaling molecules that engages with phosphoproteins is the 14-3-3 protein family (43). 14-3-3 proteins lack any areas of conserved structural homology to other proteins (i.e., no SH3 domains, PRR domains), yet they are able to bind to a variety of proteins and regulate diverse processes such as apoptosis, cell-cycle progression, intracellular trafficking, cytoskeletal rearrangement, transcription, and neural development. Two recent proteomics studies revealed >200 proteins as confirmed and new potential clients of 14-3-3 proteins in HEK 293 cells (58, 86).
The protein 14-3-3 was initially discovered in 1967 (3) as a 30-kDa, abundant bovine brain protein that eluted in the 14th fraction of DEAE chromatography and in the 3.3 fraction of starch gel electrophoresis (hence the name). The cloning of 14-3-3 in the 1990s led to the finding that a family of 14-3-3 proteins exist ubiquitously in all eukaryotic organisms (3). Seven isoforms are found in mammals (β, γ, ζ, ɛ, η, τ/θ, σ), two in yeast and Drosophila, and 15 in Arabidopsis plants. 14-3-3 proteins are usually cytoplasmic, but can also be detected in the plasma membrane, Golgi apparatus, and the nucleus. The crystal structures of 14-3-3 proteins have revealed that these proteins are highly helical, dimeric molecules made up of two antiparallel ∼30-kDa monomers that are composed of nine α-helices (91). In vitro and in vivo studies demonstrated that the 14-3-3 isoforms tend to differ in their selectivity and ability to heterodimerize with other isoforms (16, 103). These differential combinations, in addition to the variable C-terminal regions of the monomers, may determine the specificity of 14-3-3 interactions with their clients (16). The 14-3-3 dimer forms a binding pocket lined by several conserved basic residues, K49, R567, R127, and Y128 (numbering based on zeta isoform) that bind to specific residues in the client protein (110). In 1996, a novel phosphoserine/threonine-containing motif was identified as a predictor of putative 14-3-3 binding sites (77). This motif has two modes: R-S-X-pS/T-X-P (mode I) and R-X-(Y/F]-X-pS/T-X-P (mode II).
Whereas the number of 14-3-3/client protein interactions is quite large, the results of these interactions appear to fall within common themes among the different signaling pathways (Fig. 5; reviewed in ref. 73). One purpose of 14-3-3 proteins is to keep enzymes in specific phosphorylation-induced conformations. For example, binding of 14-3-3 to phosphorylated AANAT, an enzyme that regulates the synthesis of melatonin, restricts movement of a floppy loop and keeps the enzyme's active site in an open conformation (45).

Another function of 14-3-3 proteins is to promote or prevent translocation of phosphoproteins between cell compartments. For example, phosphorylation of cytosolic cyclin-dependent kinase inhibitor p27 (Kip) results in binding of 14-3-3, which masks the nuclear-localization signal sequence of p27 and prevents binding of importin, a protein that chaperones p27 into the nucleus (94). 14-3-3 proteins also may compete with other proteins for binding to target proteins. For example, binding of 14-3-3 to the phosphorylated, proapoptotic protein BAD prevents BAD from binding to and inactivating the antiapoptotic protein, Bcl-2/.Bcl-XL (74). Thus the BAD/14-3-3 interaction promotes cell survival. Finally, 14-3-3 proteins may also protect phosphoproteins from dephosphorylation (49, 83) or ubiquitination (78).
As described earlier, the Nox1-activating protein, NOXA1, contains two PKA phosphorylation sites, one at Ser172 and another at Ser461 (63). Both of these phosphoserines participated in binding 14-3-3, even though only Ser172 was located within a canonic 14-3-3 binding motif. This exemplifies the proposed “gatekeeper” model of 14-3-3 binding in which a second phosphoserine/threonine secures the interaction between 14-3-3 and the serine within the binding motif (109).
Phosphorylated NOXA1 WT, but not the double Ser->A mutant, co-immunoprecipitated with ectopic, as well as with endogenous, 14-3-3ζ. The binding pocket mutant, 14-3-3ζ K49E, did not interact with NOXA1 when used in place of wild-type 14-3-3ζ. The functional consequence of the interaction of 14-3-3ζ with phosphorylated NOXA1 was a decrease in PMA-elicited ROS-generation by Nox1. This inhibitory effect was greatest when phosphorylated NoxA1 was present along with 14-3-3ζ.
In all three cell types studied, a decrease in the ratio of plasma membrane–to–cytoplasmic localization of NOXA1 was detected, suggesting that 14-3-3ζ sequesters NOXA1 and dissociates it from the membrane where it is normally localized. In HEK293 cells transfected with NOXA1, NOXO1, constitutively active Rac1, and 14-3-3ζ, a decrease was noted in the interaction between NOXA1 and constitutively active Rac1, as well as a slight decrease in the interaction with NOXO1 under those conditions in which NOXA1 was shown to be phosphorylated by PKA and to interact with 14-3-3ζ. Taken together, these results showed that 14-3-3ζ binds to NOXA1 in a PKA-dependent manner and inhibits Nox1 activity by limiting the interactions of NOXA1 with active Rac1, and with NOXO1 in the membrane.
Whereas a majority of well-established 14-3-3 ligands contain the canonic mode I or mode II binding motifs, some client proteins do not, indicating that some proteins may have evolved different ways of binding 14-3-3. For example, proteins with a novel carboxy-terminal motif pSpXTX1–2-CO2H (mode III) (i.e., SWTY) in which X is not proline are also able to bind 14-3-3 (21). In addition, R18, a 20-a.a. peptide with high affinity (nM) for the binding pocket of 14-3-3 was identified by phage display library (106). The sequence of this peptide is unique from modes I, II, and III. This raises the possibility that noncanonic, phosphorylated, or nonphosphorylated 14-3-3 binding motifs exist, possibly among Nox isoforms or subunits that are yet undiscovered. Currently, very little information is available about the role(s) of 14-3-3 proteins in the regulation of ROS production. In addition to Kim et al. (63), a report exists on 14-3-3 and ROS production in tobacco (37). The protein NtrbohD has been identified as the plasma membrane oxidase responsible for ROS production in elicited tobacco cells. Its C-terminus was used as a bait in a two-hybrid screen to identify putative regulators of this system, leading to the isolation of a cDNA coding for a member of the plant 14-3-3 protein family. The corresponding transcript was induced after infiltration of tobacco leaves with the fungal elicitor cryptogein. Tobacco cells transformed with an antisense construct of this 14-3-3 no longer accumulated ROS on infection with cryptogein, suggesting that 14-3-3 regulates ROS production in these cells. We speculate that these two examples are indicative of more-widespread roles for 14-3-3 proteins in the regulation of nonphagocytic and phagocytic Nox family enzymes.
Nox1 regulation via c-Src-mediated Rac1 activation
Nox1 is most abundant in the colon epithelium (8). It is interesting to note that the cytoplasmic tyrosine kinase c-Src is also highly expressed and active in human colon epithelial tumors (13, 56). Indeed, c-Src has been suggested to play key roles in the development and progression of colon cancers, including acquisition of the metastatic phenotype (99).
Nox1 and the formation of ROS have also been linked to colon cancer (92, 102), suggesting potentially interesting regulatory interactions between Src signaling and Nox1 activity. A pathway linking c-Src signaling to Nox1 activation through the tyrosine phosphorylation-mediated activation of the Rac1 exchange factor Vav2 has been described (48) (Fig. 6). ROS formation by HT29 colon carcinoma cells is Nox1 dependent and was shown to be inhibited by the Src kinase inhibitor PP2 (but not the inactive analogue PP3), as well as by dominant negative Src. Similarly, expression of active c-Src increased Nox1-dependent ROS production. Some correlation was found between Nox1 activity in various colon cell lines and levels of active c-Src.
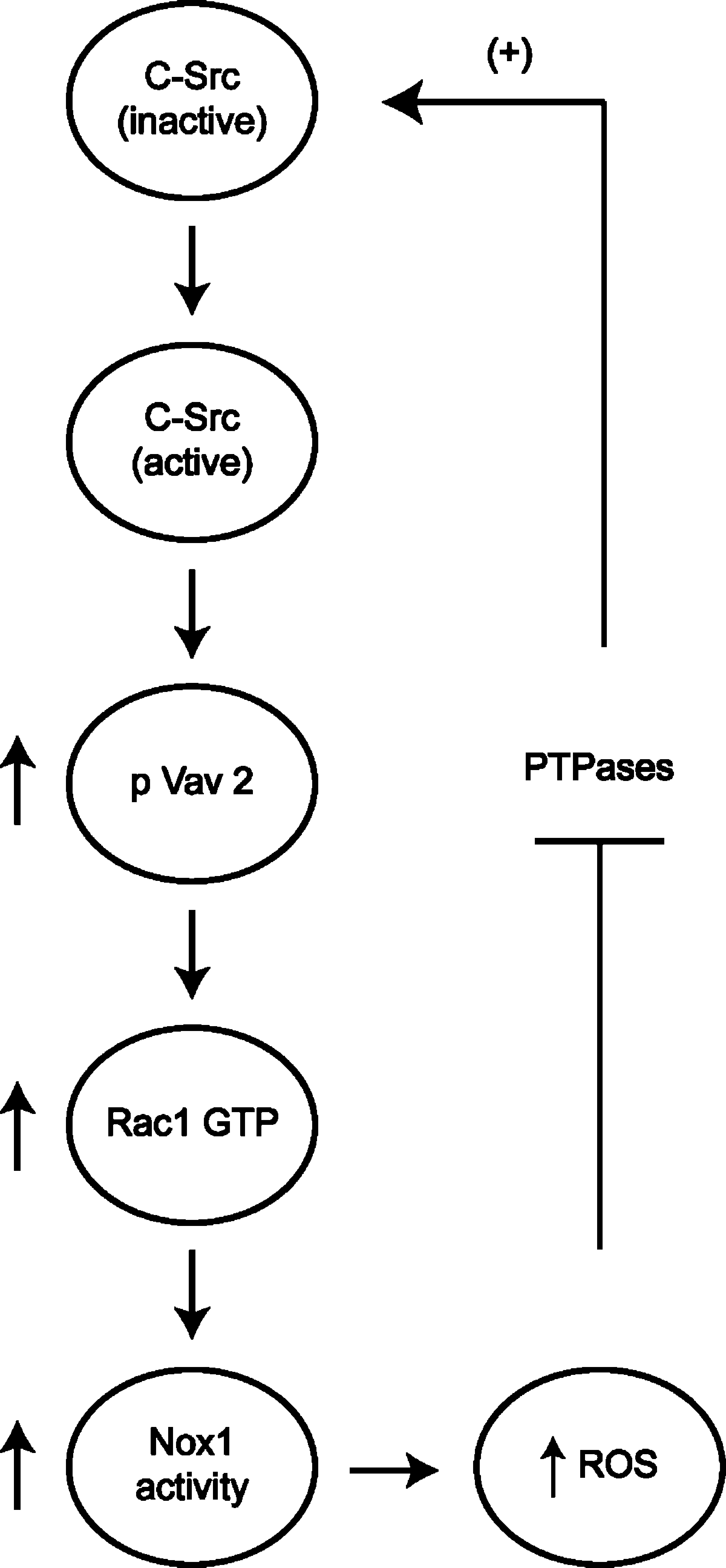
The ability of c-Src to increase Nox1 activity was found to be sensitive to inhibition by dominant negative Rac1T17N, and was not seen in an HEK293 co-expression system in which either a non-Rac-binding NoxA1 protein or a non-Rac-binding Nox1 was used (48). c-Src was shown to increase endogenous Rac1 activity in HT29 cells, and the increase in Rac1 activity was inhibited when the Rac exchange factor Vav2 was depleted by using siRNA. Consistent with this observation, c-Src induced the tyrosine phosphorylation of Vav2 on Tyr174, a site that is indicative of Vav2 activation. Other exchange factors did not appear to be involved. Thus, at least one major pathway by which c-Src regulates ROS formation in colon cancer cells is through the activation of Rac1 via the exchange factor Vav2 to regulate Nox1 activity.
c-Src was previously shown to be involved in signaling events stimulated by ROS production (1). c-Src itself undergoes activation on cellular ROS formation through multiple mechanisms (4). Src activity can be regulated through the control of its phosphorylation status by protein tyrosine phosphatases (PTPases) (79). The PTPases, which contain sulfhydryl (93) groups on cysteine residues in the catalytic domain that undergo oxidative modification to impair catalytic activity, are well-known targets of ROS (97). Two major regulatory phosphorylation sites are present in human c-Src: Tyr527 and Tyr418. The phosphorylation of Tyr418 is responsible for c-Src activation, whereas phosphorylation of Tyr527 maintains Src in an inactive state. The inability to dephosphorylate Tyr418 by ROS-inactivated PTPases on Src-induced ROS generation in colon cancer cells could explain the higher level of Src activation in such cells. More important, c-Src–induced ROS formation by Nox1 could provide an elegant positive feed-forward mechanism through which c-Src induces its autoactivation by increasing ROS production via Nox1 (Fig. 6). However, whether such a mechanism contributes to formation and progression of colon cancers remains to be clarified.
Nox5: phosphorylation-mediated regulation of activation and calcium sensitivity
The ∼75- to 85-kDa Nox5 is distinguished from the Nox1 through 4 enzymes by the presence of a long intracellular NH2 terminus containing four Ca2+-binding EF-hand domains (6). Nox5 does not require p22 phox for activity, nor is it regulated by any of the known cytosolic Nox regulatory proteins (Fig. 2) (7). Instead, Nox5 undergoes Ca2+-induced conformational changes that lead to enzyme activation (7).
The median effective concentration (EC50) of Nox5 for Ca2+(∼1 μM) is rather high and is unlikely to be reached in most stimulated cells. Jagnandan et al. (57) showed that the phorbol ester PMA (phorbol 12-myristate 13-acetate) increased Nox5 activity, accompanied by a potentiation of Nox5 sensitivity to calcium. PMA stimulates the phosphorylation of Nox5 at Thr494 and Ser498, presumably through the activation of a protein kinase C (PKC) isoform. This appears to be a direct phosphorylation of Nox5, although this has not been conclusively demonstrated. Mutation of Thr494 and Ser498 to Ala abolished PMA-mediated Nox5 phosphorylation and Ca2+ sensitization. Conversely, mutation of these residues to gain-of-function glutamic acid mutations increased Nox5 activity and Ca2+ sensitivity to a more physiologic range. Although the exact PKC enzyme responsible for PMA-dependent Nox5 phosphorylation is unknown, phosphorylation and Ca2+ sensitization was blocked by the specific PKC-β inhibitor LY379196, suggesting that PKC-β or a similar isoform mediates this effect.
Clark and his colleagues (35) recently described the ability of H2O2 to stimulate Nox5-dependent ROS formation. This effect of H2O2 was not observed in a broken cell system, indicating it was unlikely to be due to a direct effect on Nox5 itself. However, H2O2-induced Nox5 activation was sensitive to general inhibitors of Src-family tyrosine kinases, as well as to the specific c-Abl inhibitor imatinib mesylate. The latter was verified by showing that the effects of H2O2 on Nox5 activity were also inhibited by kinase-dead versions of c-Abl. Co-expression of active c-Abl enhanced both basal- and H2O2-induced activity of Nox5. Such activation was associated with increases in Ca2+ influx, although the actual sensitivity of Nox5 to Ca2+ concentration was not examined. c-Abl was observed to relocalize into small internal vesicles when the K562 cells were treated with H2O2, and this redistribution was Ca2+ dependent.
Nox5 regulation by c-Abl was associated as well with c-Abl tyrosine phosphorylation and oligomerization (35). Further, Nox5 could be coimmunoprecipitated with c-Abl, and this interaction was enhanced by H2O2 treatment. However, whether c-Abl was directly phosphorylating Nox5 to modulate its activity was not investigated. Thus, although c-Abl activation appears to mediate a redox-dependent regulation of Nox5 that involves Nox5 complexation with c-Abl, the mechanism by which this regulation occurs remains to be clarified.
Summary and Conclusions
In addition to the examples discussed here of Nox1 regulation via c-Src tyrosine kinase and of Nox5 regulation by c-Abl tyrosine kinase, it is known that many growth-factor receptors can stimulate the activity of various Nox enzymes (39). This may be a result of the intrinsic tyrosine kinase activities of the receptors themselves, or of downstream receptor-associated tyrosine kinases. Although the tyrosine phosphorylation of Nox enzymes or their regulatory proteins directly has not yet been described, it is likely that this occurs, given the existence of predicted sites for tyrosine phosphorylation in these proteins. Protein tyrosine phosphorylation is known to induce the binding of regulatory proteins through phosphotyrosine-binding motifs present on such proteins (SH2 domains, PTP domains, etc.) (84). Thus, as with the phosphorylation-induced binding of 14-3-3 proteins to NoxA1, we speculate that tyrosine phosphorylation-induced regulatory protein interactions will also significantly influence Nox-enzyme function, affecting their localization and activity.
The kinase-mediated regulation of the phagocyte NADPH oxidase and nonphagocyte Nox increasingly appears to be an important means of controlling the formation of ROS in various systems. The disruption of regulatory feedback loops involving kinases, phosphatases, and redox state as a means of initiating pathologic conditions appears to be an attractive hypothesis. Given the accumulating evidence for Nox-mediated ROS production in various disease situations, increased efforts to identify and characterize such phosphorylation-dependent regulatory mechanisms are called for. The availability of effective kinase inhibitors that are well tolerated in vivo suggests the possibility of modulating Nox function indirectly by pharmacologic intervention in their kinase-mediated regulatory pathways.
Footnotes
Acknowledgments
We thank Ms. Emilie Broderick for editorial assistance and aid with the figures used in this review. We acknowledge support for this research from The National Institutes of Health. G.M.B. is supported by grant HL048008.