
Abstract
Tumor necrosis factor-α (TNF) is a key cytokine that has been shown to play important physiologic (e.g., inflammation) and pathophysiologic (e.g., various liver pathologies) roles. In liver and other tissues, TNF treatment results in the simultaneous activation of an apoptotic pathway (i.e., TRADD, RIP, JNK) and a survival pathway mediated by NF-κB transcription of survival genes (i.e., GADD45β, Mn-SOD, cFLIP). The cellular response (e.g., proliferation versus apoptosis) to TNF is determined by the balance between the apoptotic signaling pathway and the NF-κB survival pathway stimulated by TNF. Reactive oxygen species (ROS) are important modulators of signaling pathways and can regulate both apoptotic signaling and NF-κB transcription triggered by TNF. ROS are important in mediating the sustained activation of JNK, to help mediate apoptosis after TNF treatment. In some cells, ROS are second messengers that mediate apoptosis after TNF stimulation. Conversely, ROS can cause redox modifications that inhibit NF-κB activation, which can lead to cell death triggered by TNF. Consequently, the redox status of cells can determine the biologic response that TNF will induce in cells. In many liver pathologies, ROS generated extrinsically (e.g., inflammation) or intrinsically (i.e., drugs, toxins) may act in concert with TNF to promote hepatocyte death and liver injury through redox inhibition of NF-κB. Antioxid. Redox Signal. 11, 2245–2263.
Introduction

Key regulators of TNF signaling pathways are reactive oxygen species [ROS; e.g., superoxide (
In this review, we examine how ROS and redox changes are important in modulating both apoptotic signaling and NF-κB signaling in response to TNF. Because TNF and NF-κB signaling pathways vary greatly in cells, a great deal of our discussion focuses on TNF signaling in hepatocytes and liver pathologies mediated by TNF.
TNF Signaling Pathways
TNF and TNF receptor
TNF is a 17-kDa cytokine that is released during inflammation by macrophages and other immune cells (14, 151). TNF is the founding member of the TNF superfamily that includes some 19 proteins such as Fas ligand and tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) (87). Two different TNF receptors have been identified in cells: TNF receptor-1 (TNF-R1) and TNF receptor-2 (TNF-R2). A soluble form of TNF receptor (sTNF) also is found in vivo and can reduce free circulating levels of TNF. TNF-R1 is expressed in virtually all cells, whereas TNF-R2 is found primarily in immune cells, where it regulates inflammation (151). TNF-R2 may enhance TNF-R1–induced signaling and apoptotic signaling, but does not directly trigger apoptosis in cells. Consequently, this review focuses on TNF-R1–mediated signaling important in regulating cell differentiation and death in liver and other tissues.
Apoptotic signaling pathway activated by TNF
As previously discussed, the opposing biologic responses (differential and death) that TNF can elicit in tissues, such as liver, are due to TNF activation of both apoptotic and survival signaling pathways in cells (14, 37, 126). The binding of TNF to TNF-R1 will induce an apoptotic response mediated by JNK and caspase-8 and a survival response mediated by NF-κB. However, it must be noted that TNF signaling is very cell specific, and a great degree of variability in signaling pathways activated by TNF can be found in the literature. Consequently, when possible, we focus on TNF signaling in primary hepatocytes or hepatocyte cell lines or both. In most cells, the binding of TNF to TNF-R1 causes a conformation change that recruits TRADD, receptor-interacting kinase (RIP), and TNF-receptor–associated factor 2 (TRAF2) to form a complex, referred to as complex I (Fig. 1) (95). Complex I is believed to be important in triggering both NF-κB activation and JNK activation. Complex I eventually dissociates from TNF-R1 and becomes internalized to the cytoplasm, where it integrates Fas-associated death domain (FADD) and procaspase-8 to form a complex referred to as complex II (95). The formation of complex II is believed to be important in activating apoptotic signaling pathways through activation of caspase-8, which promotes the cleavage of Bid to tBid (truncated Bid) (160). tBid was shown to translocate to mitochondria and permeabilizes the mitochondrial outer membrane to allow cytochrome c release, which can further activate other caspases, leading to a positive-feedback loop resulting in extensive caspase activation and ultimately apoptosis (160, 169). In many cells, ROS have been suggested to increase during or after complex I and II formation (1‱6 h after TNF treatment) to mediate or help potentiate apoptosis after TNF stimulation (24, 36, 121).
TRAF-2 in complex I also is believed to activate the MAP kinase cascade [apoptosis signal-regulating kinase 1 (ASK-1), mitogen-activated protein kinase kinase 4/7 (MKK4/7) that leads to the activation of JNK, a member of the mitogen-activated protein kinase (MAPK) family important in stress response] (104, 160). JNK is important in the stress response and is activated by a wide range of environmental stresses (i.e., ROS, heat) and various cytokines (130). Whereas JNK is important in the stress response, when its activation is prolonged, JNK is believed to mediate both apoptotic and necrotic cell death (51, 85, 130). Sustained JNK activity is a key component of hepatocyte injury during acetaminophen-induced liver injury and ischemia/reperfusion injury in liver (51, 59). JNK activation by complex I after TNF treatment was shown to be transient (30‱60 min), because NF-κB activation is believed to shut down JNK (104, 131). In many cells, including hepatocyte cell lines (e.g., RALA255-10G), prolonged JNK activity (>120 min) is essential for TNF-induced apoptosis to occur (85). In these cells, inhibition of JNK, through chemical or genetic means, prevented TNF-induced apoptosis. However, the requirement of JNK in mediating TNF-induced cell death may be cell specific. JNK 1−/− and JNK 2−/− embryonic fibroblasts were found to have more apoptosis when treated with TNF than did wild-type cells (67). In addition, a recent study showed that concanavalin A (Con A)-induced hepatitis (liver injury mediated through TNF) does not require JNK activation in hepatocytes (29). Thus, JNK activation may be integral for TNF-stimulated apoptosis in some cells, but may not be necessary for apoptosis in all cells.
Survival pathways activated by TNF–NF-κB activation
NF-κB is an essential transcription factor that regulates inflammation, stress response, and survival. NF-κB is a dimer (homo or hetero) composed of members of the NF-κB subfamily (i.e., p50, p52) and Rel subfamily [i.e., p65 (Rel A), c-Rel, RelB]. In most cells, the most prominent form of NF-κB is the p50/p65 heterodimer, which binds to the κB sites in DNA. Normally NF-κB is found anchored in the cytoplasm with IκB-α. When TNF binds TNF receptor, complex I forms and will activate IκB kinase (IKK) (47, 151). IKK is a complex composed of two catalytic subunits (i.e., IKK-α, IKK-β) and a regulatory subunit, IKK-γ. Once activated, IKK will phosphorylate IκB-α, which triggers ubiquitination and degradation of IκB-α. The degradation of IκB-α releases NF-κB, allowing NF-κB to translocate to the nucleus and to promote transcription of survival genes. NF-κB is essential in survival, and mice lacking the p65 subunit of NF-κB or various IKK subunits will die during embryogenesis because of hepatocyte apoptosis (47). NF-κB upregulates antiapoptotic proteins such as bcl-xL, c-FLIP, and XIAP, which can inhibit proapoptotic bcl-2 family members and caspases (151, 160). For example, c-FLIP will inhibit caspase-8, central in mediating TNF-induced apoptosis (94). XIAP, A20, and GADD45β (105, 160) are NF-κB–transcribed genes that have been suggested to inhibit JNK activated by complex I after TNF stimulation. The importance of GADD45β in regulating JNK (through MKK7, the MAPK kinase upstream of JNK) in liver was recently observed in GADD45β(−/−) mice (105). GADD45β(−/−) mice were found to have increased sustained JNK activity, decreased hepatocyte proliferation, and increased cell death during liver regeneration (a process mediated by TNF) than did wild-type mice (106). In addition to GADD45β, iNOS, A20, and bcl-xl transcribed by NF-κB contribute in protecting liver from cell death induced by TNF (8, 62). NF-κB also upregulates antioxidant enzymes such as Mn-SOD (SOD1‱mitochondria isoform) and ferritin, which are essential in inhibiting TNF-induced apoptosis in some cancer cells (65, 108, 157). NF-κB was suggested to inhibit ROS generated after TNF treatment in many cells (121), although this mechanism remains unclear. TNF can induce both apoptosis and necrosis in cells, with FADD/caspase 8 important in mediating apoptosis and RIP important in mediating necrosis (104). For this review, we focus mainly on TNF-induced apoptosis, the most common consequence after TNF treatment when NF-κB is inhibited. Although NF-κB activation primarily activates prosurvival pathways in cells, in some examples, NF-κB activation induces apoptosis in cells (104).
The activation of NF-κB and transcription of NF-κB genes is a major response to TNF stimulation in cells (151, 160). In immune cells, NF-κB transcribes many cytokines and proteins, such as iNOS, important in inflammation. In liver and other tissue, NF-κB–dependent genes inhibit apoptotic pathways and promote survival and the stress response (73, 126). Liver regeneration requires NF-κB activation to prevent apoptosis and help normal cell progression after injury (16). Because NF-κB is activated, most primary cells are highly resistant to TNF, and very little cell death ensues in culture (36, 41, 55). Consequently, to study apoptotic signaling pathways induced by TNF, NF-κB activation or transcription (or both) of NF-κB–dependent survival genes must be inhibited in most cells (85, 160). NF-κB transcription of survival genes is generally inhibited chemically (i.e., actinomycin, RNA synthesis inhibitor, or cycloheximide, a protein synthesis inhibitor), or through genetic modulation (e.g., knocking down IKK). Co-treatment of hepatocytes with cycloheximide or actinomycin D renders hepatocytes and hepatocyte cell lines susceptible to the cytotoxic effects of TNF by inhibiting expression of NF-κB–transcribed survival genes (36, 85). However, the application of findings from cell-culture studies that use chemical inhibition of NF-κB to situations in vivo is somewhat problematic, because the mechanism of NF-κB inhibition in vivo is likely through very different mechanisms. Thus, some findings from cell-culture studies may not completely reflect the TNF signaling pathways that occur in vivo in TNF-mediated pathologies such as alcohol-induced liver injury.
Another uncertainty in studying TNF signaling in cell culture is the dose of TNF administered to cells. TNF doses used in cell culture can vary greatly, with 5 ng/ml to up to 100 ng/ml of TNF being reported to be used in studies involving cultured primary hepatocytes. Because of NF-κB activation, primary hepatocytes can withstand extremely high doses of TNF (370 ng/ml) without any significant apoptosis occurring (32). However, increasing doses of TNF can cause more apoptosis in hepatocytes sensitized to TNF-induced cell death by treatments such as alcohol, suggesting that varying doses of TNF may modulate the extent of TNF signaling pathways (23). The levels of TNF that cells encounter in vivo during various pathologies are difficult to ascertain. During septic shock episodes in vivo, plasma levels of TNF have been measured to be ∼3 ng/ml (28), suggesting that many doses used in cell culture studies may be on the high side. However, during inflammation, cells such as hepatocytes may encounter highly localized levels of TNF secreted by neighboring macrophages and therefore exposed to higher levels of TNF than seen in plasma. More studies using varying doses of TNF in cell-culture studies must be examined, because in vivo doses of TNF remain uncertain.
Regulation of TNF Signaling by ROS
ROS have been suggested to be important regulators of signaling pathways stimulated by TNF (55, 56, 73, 131). In the past two decades, a great deal of work has shown that both

In cells, most thiols are in the reduced form with the GSH/GSSG ratio being >100:1 and most proteins being in the reduced thiol form (75, 123). The protein redox status in cells is regulated by GSH along with the proteins, glutaredoxin and thioredoxin (Trx) (56). Glutaredoxin catalyzes the deglutathionylation of proteins by using the reducing power of GSH, whereas Trx (Trx-1 cytoplasmic isoform; Trx-2 mitochondria isoform) contains vicinal thiols that react with and reduce disulfide crosslinks. GSH, along with these redox-regulating proteins, help keep most of the protein in the intracellular environment in the reduced thiol redox state. ROS, conversely, can alter the redox state of cells and oxidize protein thiols. The oxidation of thiols by ROS have been shown to alter the activity of many proteins (56, 123). H2O2 can oxidize cysteines in proteins to disulfides, sulfenic, sulfinic, and sulfonic acids, depending on the concentrations and reactivity of the thiol (Fig. 2). Sulfenic acids (--SOH) can react with GSH and become converted to a glutathionylation bond (--SSG). Glutathionylation of proteins also can occur when GSSG levels increase in response to increased H2O2 levels (due to increased GSH peroxidase activity, which uses the reducing power of GSH to form GSSG during H2O2 detoxification). Not all protein thiols have the same reactivity, and the localized environment (e.g., pKa of neighboring amino acids) will greatly affect thiol reactivity. Thiolates (--S−, deprotonated thiols), being strong nucleophiles, are generally more reactive than thiol groups. It is believed that H2O2 modifies signaling pathways through posttranslational modification of cysteine or other amino acids (i.e., methionine, proline) on proteins (139).
Many pathways activated by TNF, such as JNK and NF-κB, have clearly been shown to be modulated by ROS through redox changes to various regulatory proteins. Proteins, such as Trx-1, involved in JNK activation (through ASK-1 binding), and IKK, involved in NF-κB activation, contain critical cysteines that regulate function (82, 120). Consequently, ROS have been shown to modulate both the apoptotic signaling pathway and NF-κB survival pathways stimulated by TNF, through redox changes to signaling proteins. This has been supported by studies that showed the addition of exogenous ROS or agents that trigger increase in endogenous ROS generation (mitochondria inhibitors that increase ROS) sensitize many cells, including primary hepatocytes, to TNF-induced apoptosis (without actinomycin or cycloheximide treatment) (24, 55, 125, 167). ROS have been shown to sensitize hepatocytes to TNF-induced apoptosis, through inhibition of NF-κB transcription of survival genes (55). The notion that ROS inhibits NF-κB through redox changes to proteins is supported by the observations that redox-modulating agents or GSH-depleting agents [i.e., diamide, diethylmaleate (DEM)] also sensitize hepatocytes to the cytotoxic effects of TNF (55, 88, 92, 99). The sensitization of hepatocytes to TNF by ROS was attributed mainly to redox inhibition of NF-κB, but changes to other redox-sensitive pathways, such as JNK and Akt, also may contribute to increased sensitization to TNF. Redox modulation of TNF signaling may be an important mechanism of sensitizing hepatocytes to the cytotoxic effects of TNF, which may be important in many liver pathologies that are discussed later in the review.
Many works have also suggested that ROS act as second messengers and mediate TNF-induced apoptosis in various cells, when NF-κB transcription of survival genes is inhibited [i.e., actinomyin, cycloheximide, or through genetic means, as reviewed in (104, 131, 160)]. The evidence that ROS are essential in TNF signaling is based on the following observations: (a) an increase in ROS generation [by using fluorescent probes such as dichlorodihydrofluorescein (DCFH)] is detected after TNF treatment to cells, generally before apoptosis (36, 72, 121, 149); (b) antioxidant treatment can prevent TNF-induced cell death in many cells (36, 121); (c) redox changes in proteins (increased disulfide formation) have been observed after TNF treatment to cells (72, 141). Taken together, many researchers have suggested ROS are essential mediators of apoptosis induced by TNF in many cells, including primary hepatocytes. However, the evidence that ROS are second messengers or are necessary during TNF-induced apoptosis has had some methodologic problems. One problem is that the measurement of ROS, because of their reactivity, is extremely difficult (57). Most studies have relied on fluorescence probes such as DCFH to detect ROS, which are nonspecific and prone to artifacts that can generate false-positive signals (153). In addition, studies using antioxidants to demonstrate involvement of ROS are not always conclusive, because antioxidants can have secondary effects, and inhibition of signaling pathways by antioxidants does not necessarily prove ROS involvement (15, 64). Thus, the role of ROS in TNF-stimulated apoptosis may be overstated in some cells, although, in other cells, ROS appear to play an essential role. In the next section, we examine (a) the experimental data implicating ROS as second messengers in TNF signaling, and (b) the proteins involved in TNF signaling that are regulated by redox changes.
ROS as Second Messengers in TNF-Induced Apoptosis
Methodologic problems
Because of their reactivity and consequent short half-life, ROS are very difficult to detect intracellularly. ROS cannot be directly detected; instead, exogenous probes that react with ROS to form a unique measurable product are commonly used (57, 153). In most, if not all TNF studies, researchers have relied on fluorescent probes that react with ROS and become oxidized to a molecule with a different fluorescent characteristic than the parent compound, which can be readily measured. Two probes, DCFH and hydroethidine (HE), are probably the most widely used probes to measure ROS in cells and have been used in the majority, if not all, of studies involving TNF (24, 36, 72, 121, 149). Although these two probes are convenient and easy to use, they have many inherent problems that make problematic the assessment of ROS in TNF signaling. Both probes can generate many false-positive signals during apoptosis, making results obtained by using these probes very difficult to interrupt (153). In the next section, we review the use of DCFH and HE in measuring ROS in TNF signaling and problems in interpretation that arise from using these probes.
Dichlorodihydrofluorescein (DCFH)
DCFH is probably the most widely used probe to measure H2O2 in cells. Most researchers use DCFH-DA (DCFH diacetate ester form), which is believed to enter cells and become cleaved by esterases to trap DCFH inside cells. DCFH, which is nonfluorescent, is believed to be oxidized by H2O2 to DCF (dichlorofluorescein), which can be measured fluorometrically in cells. DCF fluorescence is usually observed to increase after TNF stimulation before apoptosis (about 3 to 6 h when NF-κB is inhibited), in a wide range of cells, including primary hepatocytes (24, 49, 60, 121, 133). When NF-κB is not inhibited and apoptosis is inhibited, DCF fluorescence is usually not detected or is detected at much lower levels than during TNF-induced apoptosis. Antioxidants such as trolox (water-soluble analogue) and butylated hydroxyanisole (BHA) have been shown to prevent apoptosis and DCF fluorescence, supporting the role of ROS in TNF-induced apoptosis in cells (36, 121). However, many inherent problems exist in using DCFH, and it was widely criticized as a specific measure of H2O2. First, DCFH retention (conversion from DCFH-DA) and diffusion inside cells is dependent on esterase activity and cellular pH. Cellular pH levels frequently change during apoptosis; these are likely to affect DCFH uptake and retention (110, 153). Second, DCFH is not oxidized to DCF by H2O2, but rather by more-reactive oxidants (HO·, ONOO−) and labile iron (117, 142). DCFH is also oxidized by various oxidases and peroxidases through a catalytic process, which does not necessarily depend on changes in H2O2 levels (13, 153). Third, antioxidants, especially exogenously added antioxidants, greatly affect DCF fluorescence independent of H2O2 levels (153). Most problematic in the use of DCFH for TNF studies is that cytochrome c can act as a peroxidase and oxidize DCFH to DCF, independent of changes to H2O2 levels (18, 84). Because cytochrome c is released during TNF-induced apoptosis (when DCF fluorescence is the greatest), DCF oxidation observed after TNF treatment may be a consequence of cytochrome c release that preceded apoptosis rather than changes in H2O2 levels. Free iron and GSH redox, which have been shown to change after TNF stimulation in some cells, have also been shown to affect DCF fluorescence (84, 153). DCFH oxidation may reflect free cytochrome c levels or other redox changes rather than changes in H2O2 or
Hydroethidine
HE (sometimes referred to as dihydroethidine) and its mitochondrial counterpart, MitoSOX (hydroethidine linked to hexy-triphenylphosphonium cation to target mitochondria) are commonly used to assess
Overall, TNF studies using DCFH and HE do indicate a redox change in cells, but whether the redox change is due to increased
Antioxidants and TNF-Induced Apoptosis
The protective effects of exogenous antioxidants or genetic modulation of antioxidant enzymes against TNF-induced apoptosis have lent support to the notion that ROS are second messengers or mediate apoptosis after TNF. Both the addition of low-molecular-weight antioxidants and the genetic modulation of antioxidant enzymes have been shown to decrease ROS (measured by using DCFH and HE) and in some cases partially prevent TNF-induced apoptosis (15, 37, 38, 121). However, antioxidant treatment, especially at the high doses used in most cell-culture studies, can affect many cellular pathways, independent of antioxidant activity. For example, pyrrolidine dithiocarbamate (PDTC), a metal chelator, was used in many cell-signaling studies to inhibit ROS. However, it was observed that PDTC caused an increase in oxidized glutathione, suggesting that it acts as an oxidizing agent to inhibit NF-κB activation in Jurkat cells (15). Similarly, another commonly used antioxidant, N-acetylcysteine (NAC), was found to inhibit TNF-induced NF-κB activation by reducing the affinity of the TNF receptor (64). Antioxidants also were shown directly to modulate DCFH and HE fluorescence (153). Thus, decreases in the ROS signal observed with antioxidant treatment with DCFH and HE does not necessarily reflect changes in ROS levels in cells. Given that antioxidants work through other mechanisms besides antioxidant action, some caution is required in the interpretation of data that demonstrate the protective effects of antioxidants against TNF-induced apoptosis. These data obtained with antioxidants still have some merit, but additional experiments are needed to confirm that TNF leads to increased ROS generation in cells. In hepatocytes, the antioxidants Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid, a water-soluble form of vitamin E) and Mn-TAP (a chemical mimic of superoxide dismutase) prevent apoptosis induced by TNF and cycloheximide (to inhibit synthesis of NF-κB proteins) (37). Because Mn-TAP detoxifies
In some cells, NF-κB is involved in the transcription of several key antioxidant enzymes, including Mn-SOD, ferritin, and thioredoxin, which play an essential role in preventing apoptosis after TNF treatment. The upregulation of antioxidant enzymes by NF-κB suggests that TNF triggers an increase in ROS generation that is important in mediating apoptosis. Genetic studies modulating antioxidant enzymes, such as Mn-SOD in cells, also support the involvement of ROS in TNF-induced apoptosis in some cells. In several cell lines (i.e., 293 human embryonic cell line, pulmonary adenocarcinoma cells, breast cancer cell lines), Mn-SOD appears to be an essential defense against TNF-induced apoptosis (65, 97, 157). In these cells, NF-κB activation by TNF treatment causes an upregulation of Mn-SOD, which is essential in preventing apoptosis stimulated by TNF. This is supported by the observations that Mn-SOD confers increased resistance to apoptosis induced by TNF-plus-cycloheximide treatment. Conversely, antisense to Mn-SOD made these cells sensitive to TNF, even in the absence of cycloheximide (65, 157).
Mn-SOD resides in the mitochondrial matrix, where it catalyzes the dismutation of
In other cells, ferritin, a protein important in binding and neutralizing labile iron, appears to be important in preventing cell death induced by TNF. In 3DO cells, NF-κB was found to upregulate the ferritin heavy chain (FHC), but not Mn-SOD or thioredoxin (108). Upregulation of FHC by NF-κB was found to be important in preventing ROS generation (measured by using DCFH), JNK activation, and apoptosis caused by TNF (108). In another cell line (human prostate cancer cells, DU145), TNF treatment caused a decrease in levels of the ferritin light chain and increased the levels of labile iron pool (LIP), through a process mediated by JNK (2). Similarly, in L929 cells, an increase in LIP was observed after TNF treatment (161). Ferritin is not normally induced by NF-κB in L929 cells, but the introduction of TNF-inducible FHC prevented TNF-induced ROS generation (measured by using DCFH and HE) and apoptosis (161). In mice, the iron chelator, deferoxamine, increased survival after various TNF-mediated stresses (i.e., LPS, TNF plus galactosamine), suggesting a possible role of iron in mediating TNF-induced liver injury (150).
Taken together, these studies suggest that TNF may increase the LIP to mediate ROS and apoptosis in certain cell lines. LIP can induce the Fenton reaction and cause the formation of the hydroxyl radical, the most reactive and damaging ROS (Fig. 2).
Protein Redox Changes After TNF Treatment
Additional evidence of increased ROS production during apoptosis comes from studies that observed cellular redox changes and protein redox changes before apoptosis induced by TNF. Several studies reported that a decrease in GSH precedes apoptosis after TNF stimulation (109, 121). As previously mentioned, H2O2 and possibly
Sources of ROS During TNF-Induced Apoptosis
It is clear that ROS, particularly
Mitochondria are major sources of ROS in cells, and it stands to reason that mitochondria are the major source of ROS (both
Mitochondria have been reported to swell, aggregate, and undergo other morphologic changes that precede cytochrome c release and loss of membrane potential during TNF-induced apoptosis in cells (31, 125, 145). This suggests that mitochondria are an early target of signaling pathways during TNF-induced apoptosis. Many potential signaling pathways are activated during TNF signaling that can potentially affect mitochondrial respiration and consequently mitochondrial ROS generation. Three potential pathways activated by TNF that could affect mitochondrial bioenergetics and ROS generation are (a) ceramides, (b) kinases such JNK, and (c) caspases and proapoptotic bcl-2 family members (Fig. 3).
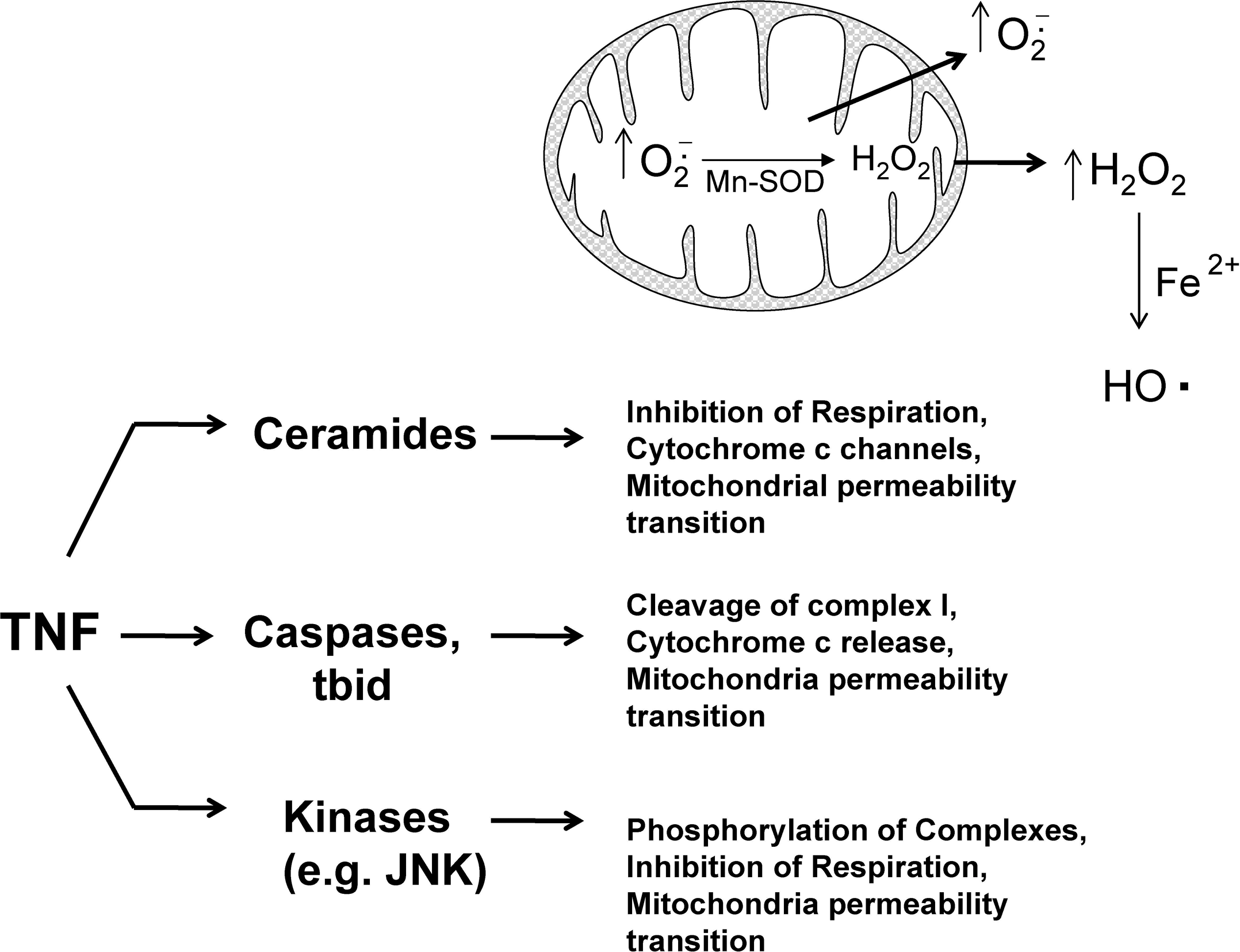
Ceramides
Ceramides are signaling lipids that have been shown to modulate many signaling pathways, including apoptosis in cells (7, 124). The binding of TNF to TNF receptor was shown to activate sphingomyelinase (SMase), which liberates ceramides from sphingomyelin in the plasma membrane (42). A neutral SMase (NSMase, optimum pH of 7.5) and acidic SMase (ASMase, optimum pH of 4.8) have been identified in cells. The activation of SMase and generation of ceramide have been suggested to mediate, at least in part, ROS generation and apoptosis stimulated by TNF (50). Ceramides have been shown to target mitochondria and form channels in mitochondrial membranes that allow passage of cytochrome c. Ceramides cannot form channels in the plasma membranes (137). The addition of ceramides to isolated heart mitochondria was shown to inhibit respiration and increased ROS generation (measuring DCFH plus horseradish peroxidase, which adds specificity for H2O2) (33). In HUVECs (human umbilical vein endothelial cells), TNF was shown to increase ROS generation from mitochondria (measured by using DCFH) that was inhibited by despiramine (SMase inhibitor), suggesting that ceramides mediate increased ROS generation from mitochondria after TNF treatment (24). In primary hepatocytes, ASMase was suggested to mediate TNF-induced cell death (45). Hepatocytes from ASMase(−/−) mice were resistant to apoptosis and increased ROS generation (measured by using DCFH) caused by TNF and mitochondrial GSH depletion (a redox change that sensitizes hepatocytes to TNF-induced apoptosis).
Phosphorylation by kinases such as JNK
The phosphorylation or glutathionylation of mitochondrial complex I in the respiratory chain was shown to regulate ROS generation from that complex (111, 144). In addition, phosphorylation was shown to regulate mitochondrial complexes such as cytochrome c oxidase (COX), which could theoretically modulate ROS production. TNF treatment of bovine and murine liver homogenates results in the phosphorylation (tyrosine 304, subunit I) and inhibition of COX (60%) (122). Whether this phosphorylation of COX results in an increased generation of ROS from the respiratory chain remains to be determined, but based on other works, inhibition of respiratory complexes often leads to increased ROS generation. The kinase(s) responsible for COX phosphorylation after TNF treatment has not been identified; however, many cytoplasm kinases such JNK have been shown to modulate mitochondria bioenergetics. Recent studies suggested that many kinases (i.e., JNK, PKC) found in cytoplasm translocate to mitochondria after activation to phosphorylate mitochondrial protein and regulate mitochondria bioenergetics (6, 21, 59). For example, after activation, JNK translocates to mitochondria and initiates a signal cascade that leads to pyruvate dehydrogenase phosphorylation and inactivation, loss of membrane potential, and cytochrome c release (6, 59, 170). JNK activated by TRAF-2 after TNF treatment may therefore be important in mediating mitochondrial dysfunction and ROS generation during TNF-induced apoptosis. However, whether JNK is directly responsible for increased mitochondrial ROS production remains to be confirmed experimentally. Initial studies using DCFH observed JNK(−/−) knockout mice have lower ROS generation than do wild-type mice after TNF treatment (149).
Caspases and proapoptotic bcl2 family members
Caspases have been suggested to modulate mitochondria bioenergetics and ROS generation. Caspases have been shown to cleave the NDUFS1 subunit in mitochondrial complex I of the respiratory chain and to inhibit respiration (114). This inhibition of mitochondrial complex I may potentially increase
Modulation of TNF Signaling by ROS: Redox Targets in TNF Signaling
ROS are important second-messenger molecules in some cells, but whether this is a universal mechanism remains to be determined. However, it is clear that many proteins involved in TNF signaling are redox regulated. This has been demonstrated by many studies demonstrating that addition of sublethal levels of exogenous ROS or causing increased endogenous ROS generation (e.g., mitochondria inhibitors) sensitizes cells to TNF-induced cell death (no actinomycin or other NF-κB inhibition treatment is necessary) (55, 125, 159, 167). In primary hepatocytes, we similarly observed that sublethal doses of H2O2 and mitochondrial inhibitors (e.g., antimycin) caused apoptosis when simultaneously treated with TNF (55). The notion that ROS sensitize hepatocytes to TNF-induced apoptosis through redox changes is supported by the fact that redox-modulating agents also sensitized hepatocytes to apoptosis stimulated by TNF. Agents that deplete GSH (i.e., DEM, phorone, or acetaminophen) or oxidizing agents such as diamide at sublethal doses also sensitized hepatocytes to TNF-induced apoptosis (55, 88, 92, 99). In addition, GSH depletion in the liver of mice increased liver injury and mortality caused by galactosamine and lipopolysaccharide (LPS) treatment (a liver-injury model mediated by TNF) (163). These observations do not necessarily support the notion that ROS are second messengers in TNF signaling; rather, these findings demonstrate that many proteins involved in TNF signaling can be redox regulated to promote apoptosis, suggesting that redox changes potentiate proapoptotic signaling pathways or inhibit NF-κB expression of survival genes or both. These redox modulations that activate apoptotic signaling and inhibit NF-κB may be important mechanisms by which TNF induces liver injury in vivo.
Studies from our and other laboratories suggested that redox alteration affects many signaling pathways to modulate survival after TNF treatment. Although many pathways that are redox regulated may be regulated by TNF, we focus on redox regulation of JNK signaling and NF-κB transcription. The redox regulation of JNK and NF-κB may underlie the sensitization of hepatocytes to TNF-induced apoptosis caused by ROS and redox-modulating agents.
JNK
As previously stated, JNK was shown to be activated by various stresses including oxidative stress. Exogenous H2O2 was shown to activate JNK in primary neurons, primary hepatocytes, and various cells lines (59, 130, 170). Similarly, high doses of acetaminophen, which depletes GSH, causes sustained JNK activation, important in mediating hepatocyte death and liver injury (51, 59). Several mechanisms underlie the activation of JNK by ROS (Fig. 4). JNK is phosphorylated primarily by MKK (a MAPK kinase), which in turn is phosphorylated by ASK-1 (a MAPK kinase kinase) in liver. ASK-1 was shown to be inhibited in cytoplasm by an association with thioredoxin, which contains critical redox-sensitive thiols (cysteine 32 or 35) (86, 120, 168). The oxidation of these critical thiols on thioredoxin by H2O2 or other oxidants will cause thioredoxin to disassociate from ASK-1, which subsequently self-activates. Once activated, ASK-1 will phosphorylate MKK, which then activates JNK. Trx-1 association with ASK-1 has also been shown to be important in ubiquitination and degradation of ASK-1 (86). However, other studies showed that JNK is inhibited through association with redox-regulated proteins such as glutaredoxin in human cancer cells (MCF-7/ADR and DU-145) (138), and glutathione S-transferase in the NIH3T3 cell line and mouse embryo fibroblasts (1, 152). ROS was shown to cause the detachment of JNK from these redox-inhibitor proteins, which subsequently become activated by MKK or through autophosphorylation. In addition, sustained JNK activity may be due to redox inhibition of MAP kinase phosphatase, important in dephosphorylating and turning off JNK (72). Whether these redox mechanisms of JNK activation are cell specific and which pathway(s) work in hepatocytes remain to be elucidated. Overall, ROS can alter the redox status of many proteins involved in JNK regulation. Consequently, JNK activation is a common response to ROS in most cells.
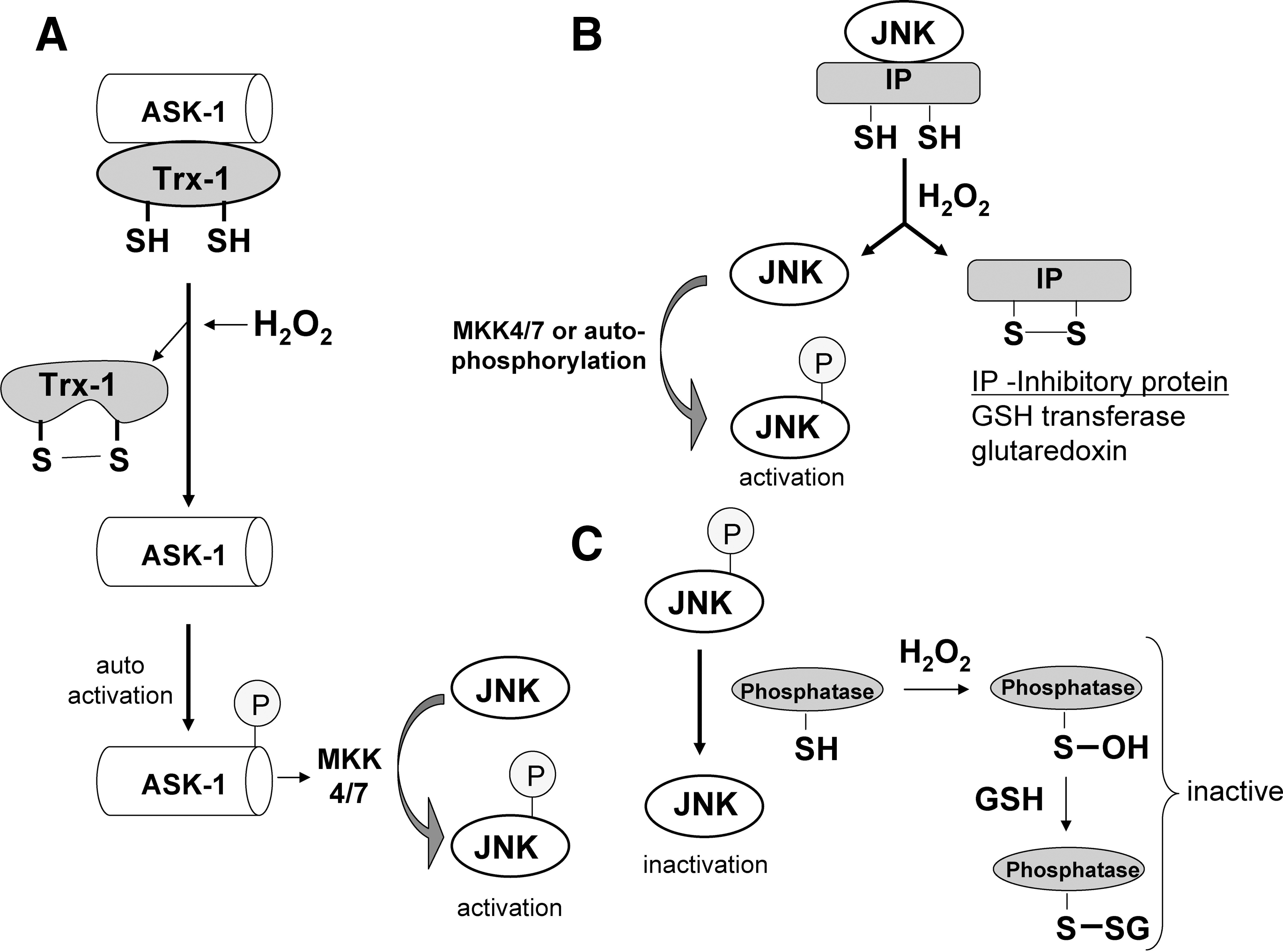
In most cells after TNF treatment, JNK is transiently activated by TRAF2, probably through ASK-1 (104, 160). It was shown that ASK-1(−/−) embryonic fibroblasts have a severely inhibited (but not complete) JNK activity in response to TNF or H2O2 (146). As previously mentioned, several NF-κB–transcribed proteins (i.e., GAPDD45β, XIAP, A20) may be responsible for turning off JNK activity (105, 143, 160). However, during TNF apoptosis [when NF-κB is inhibited, chemically (e.g., actinomycin D) or genetically (e.g., IKK knockout)], ROS are believed to be important in sustaining JNK activation (121), probably through increased ASK-1 activity (146). A feed-forward loop may exist in which activated JNK translocates to mitochondria and promotes increased ROS generation from the respiratory chain, which in turn will trigger more JNK activation (131, 149). Thus, although TRAF-2 in complex I (TNF-R1, RIP, TRAF2 complex) may be responsible for initial activation of JNK, ROS are believed to be responsible for mediating sustained JNK activity through redox alterations. The increased sensitization of primary hepatocytes to TNF-induced apoptosis by H2O2 and other oxidants may be due in part to the increased sustained JNK activity these agents trigger. Once activated, JNK is believed to target mitochondria and promote cytochrome c release, the mitochondrial permeability transition, and to phosphorylate and inactivate the antiapoptotic bcl-xl protein (6, 39, 59). JNK also was shown to accelerate the degradation of the NF-κB–regulated c-FLIP, an inhibitor of caspase 8, to promote apoptosis in the liver (20). The JNK promotion of c-FLIP degradation shows that prolonged JNK activity may counteract some protective effects of NF-κB, to promote apoptosis stimulated by TNF. In mouse lung epithelial cells, H2O2 treatment inhibited TNF-induced NF-κB activation, but apoptosis still proceeded because of sustained JNK activation (103). Whether an increase in JNK activity caused by ROS alone is enough to sensitize hepatocytes to TNF-stimulated apoptosis, or whether some concurrent redox inhibition of NF-κB also is necessary remains to be determined.
NF-κB signaling
As described by many reviews in this forum, a great deal of cell specificity exists regarding the redox modulation of NF-κB. H2O2 was suggested to activate NF-κB in some cells and to inhibit NF-κB in others cells (48). The dose of H2O2 and delivery method may a play a role in the activation or inhibition of NF-κB in cells (30). Similarly, TNF induction of NF-κB activation was shown to require ROS in some cells (60, 134), whereas ROS were shown to inhibit TNF-stimulated NF-κB activity in other cells (159, 167). In most cells, high doses of ROS are likely to inhibit NF-κB, because of redox inhibition of many proteins involved in NF-κB activation, such as IKK. In hepatocytes, ROS or redox-modulating agents primarily inhibit NF-κB activation after TNF stimulation, leading to a sensitization to TNF-induced apoptosis (55, 88, 92, 99). However, redox changes caused by mitochondrial GSH depletion, by using 3-hydroxy-4-pentenoate (HP), was shown to sensitized hepatocytes to TNF-induced apoptosis, even though NF-κB activation still occurred [high doses of TNF (100 ng/ml) were used in the study] (91). In general, redox changes inhibit NF-κB activity in hepatocytes, and many proteins involved in NF-κB signaling were shown to be regulated by redox changes. Because little evidence exists of activation of NF-κB by ROS after TNF stimulation in hepatocytes and other primary cells, we focus on NF-κB inhibition by ROS after TNF treatment, which may play a pathophysiologic role in many liver diseases.
Many potential sites involved in NF-κB transcription of survival genes that are regulated by redox changes are shown in Fig. 5. NF-κB, like many transcription factors, contains critical cysteine residues that are needed for binding to DNA. The modification of thiols of cysteine residues can potentially affect binding to DNA and thus transcriptional activity. NF-κB was shown to be S-glutathionylated and S-nitrosylated, both modifications causing a decrease in transcriptional activity because of decreased binding to DNA (81). The NF-κB p50 subunit has a critical cysteine-62 thiol that is surrounded by a cationic environment, making the thiol ionized, and highly reactive to thiol-disulfide exchange (93, 100). As previously mentioned, protein redox status is regulated by GSH along with the proteins, glutaredoxin and Trx. In the nucleus, Trx1 serves as a positive regulator of NF-κB and AP-1 transcription, either directly reducing critical cysteines of these proteins, or acting via reduction of GRX and Ref-1 (66). In HeLa cells, TNF was shown to induce a nuclear translocation of Trx1, which directly associates with p50 as well as Ref-1 (61).
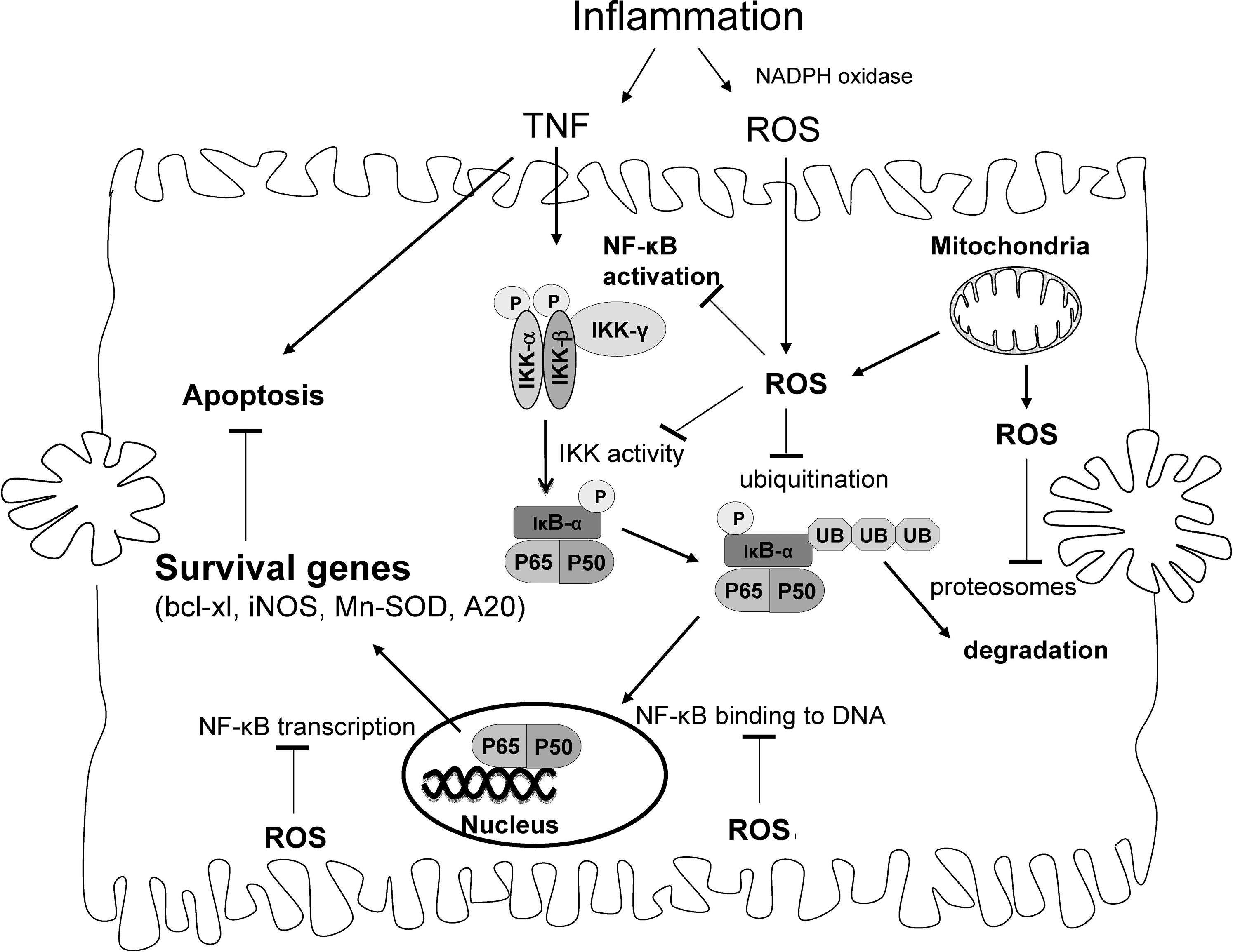
Other potential sites of redox regulation of NF-κB activity after TNF stimulation include IKK activity, NF-κB transcriptional activity, ubiquitination of IκB-α, and proteasome activity necessary for IκB-α degradation. The proteins involved in NF-κB signaling that are modulated by redox changes may depend on the redox potential of cells. Work with DEM showed that at different doses, which cause different degrees of redox changes in cells, different sites involved in NF-κB activation were inhibited after TNF simulation. A high dose of DEM (0.5 mM), which depletes cytoplasm and mitochondrial GSH (the latter could potentially increase H2O2 release from mitochondria), inhibited TNF-stimulated NF-κB activation through an IKK-dependent pathway involving decreased IKK activity, IκB-α phosphorylation, and ubiquitination in primary hepatocytes (88). IKK appears to be redox regulated, and H2O2 was shown to inhibit IKK activity through oxidation of cysteine-179 on the IKK-α or IKK-β subunit. (82, 102, 112). Inhibition of IKK by redox changes caused inhibition of IκB-α phosphorylation and degradation, leading to an inhibition of NF-κB translocation to the nucleus (82, 112). RIP in complex I, important in activating IKK after TNF stimulation, also was shown to be downregulated by H2O2 (103). Ubiquitination of IκB-α and proteasomes, necessary for IκB-α degradation after ubiquitination, also appear to be regulated by redox changes (70, 101, 159, 173). IκB-α also was shown to be glutathionylated, which reduces phosphorylation and ubiquitination of the protein (78). Conversely, lower doses of DEM (0.10 mM), which depletes cytoplasmic but not mitochondrial GSH and causes a less-significant redox change in cells, inhibited NF-κB transcriptional activity without inhibiting IKK activity or NF-κB translocation to the nucleus in primary hepatocytes (88). NF-κB was observed bound to DNA, as assessed by the Chip (chromatin immunoprecipitation) assay, suggesting that NF-κB binding to DNA was not inhibited by low-DEM treatment, but rather the NF-κB transcriptional machinery was being affected by redox changes to prevent NF-κB transcription of survival genes (88). The mechanism of how low doses of DEM inhibit the NF-κB transcriptional machinery to prevent transcription of survival genes is being investigated. Buthionine sulfoximine (BSO; a GSH-synthesis inhibitor), which slowly depletes cytoplasmic GSH, inhibited NF-κB transcription triggered by TNF in primary hepatocytes, without affecting NF-κB translocation or binding to DNA, similar to low-DEM treatment (88). In liver, NF-κB also is regulated by Akt (protein kinase B), which is required for maximal NF-κB activation in mouse hepatocytes (63), and Akt signaling was suggested to be redox regulated (83). Taken together, the data suggest that many potential sites exist where redox changes by ROS could inhibit NF-κB transcription of survival genes to promote apoptosis after TNF stimulation. The extent of redox change may be a key determinant of which sites in NF-κB signaling may be redox regulated.
Is redox inhibition of NF-κB a key factor in sensitizing cells to TNF in many diseases?
TNF was shown to play a pathophysiologic role in many diseases, including rheumatoid arthritis, psoriasis, myocardial infarction, and atherosclerosis. In liver, TNF was suggested to mediate various liver pathologies including liver ischemia reperfusion injury, alcoholic liver disease, viral hepatitis, and liver toxicity caused by toxins such as carbon tetrachloride (118, 136, 166). TNF, released during inflammation, is believed to be important in mediating hepatocyte death and tissue injury in these liver pathologies. Because the activation of NF-κB normally confers resistance in hepatocytes and other primary cells to TNF, the mechanism by which TNF is mediating liver injury in these pathologies is not completely known. Based on cell-culture studies, the presumption is that signaling changes occur in hepatocytes that result in sensitization to apoptosis stimulated by TNF. Because NF-κB is the essential pathway preventing TNF-induced apoptosis, inhibition of NF-κB may be an important factor in sensitizing hepatocytes to apoptosis stimulated by TNF. Most of these liver pathologies involving TNF are associated with increased ROS generation from external sources during inflammation (i.e., macrophages and immune cells) or internally (e.g., increased mitochondrial ROS by the herpes virus), that likely causes redox changes in cells. Consequently, redox changes that occur in these pathologies may be an important mechanism of inhibition of NF-κB signaling, leading to sensitization of hepatocytes to TNF-induced cell death.
We examine the possibility that redox inhibition of NF-κB is important in alcoholic liver disease and drug-induced liver injury. Our focus is on NF-κB; however, many important signaling pathways that modulate TNF signaling are also regulated by redox changes (i.e., Akt, PTEN, JNK) and therefore may contribute to sensitization of hepatocytes to apoptosis stimulated by TNF.
Alcoholic liver disease
TNF was shown to play an essential role in the pathogenesis of alcohol-induced liver injury. Alcoholic liver disease also involves extensive redox changes in hepatocytes and consequently may be a disease in which redox changes may lead to inhibition of NF-κB and sensitization to TNF-induced apoptosis. Alcoholic liver disease is associated with hepatocyte apoptosis, believed to be mediated by TNF. The role of TNF in alcoholic liver disease is supported by the observation that patients with alcoholic hepatitis have high serum plasma levels of TNF (12). In addition, mice lacking the TNF-R1 or injected with antibodies to neutralize TNF are more resistant to alcohol-induced liver damage, suggesting the importance of TNF in mediating liver injury (69, 71, 166). Consequently, antisera against TNF are used in the treatment of patients with severe alcoholic liver disease. Because hepatocytes are normally resistant to TNF, this suggests that prolonged alcohol intake causes TNF to have a direct lethal affect on hepatocytes. Long-term alcohol intake appears to cause biochemical changes in hepatocytes, leading to a sensitization to TNF-induced apoptosis and necrosis (14, 34). This is supported by the observation that hepatocytes isolated from rats treated with long-term alcohol are more sensitized to TNF-induced cell death (23, 43).
How does alcohol sensitize hepatocytes to the cytotoxic action of TNF? Alcohol intake is associated with increased ROS generation and redox changes that may lead to redox changes that inhibit NF-κB transcriptional activity and sensitize hepatocytes to TNF. Fernandez-Checa (44) first demonstrated that prolonged alcohol consumption causes a modest loss of cytoplasmic GSH (∼25%) while causing significant mitochondrial GSH depletion [up to 65% (44)], likely to decrease the thiol redox status of both cells and mitochondria. Mitochondrial GSH is important in detoxifying H2O2 formed in the matrix (4), and loss of mitochondrial GSH can cause increased H2O2 release from mitochondria (54). Mitochondria isolated from alcohol-treated mice generated more ROS [measured by using DCFH (9)]. Other sources of ROS during alcohol intake besides mitochondria include Kupffer cells and neutrophils, which generate ROS during inflammation in the liver (154). In addition, alcohol intake causes induction of the cytochrome p450 2E1 (CYP2E1), which is associated with ROS production and generation of reactive products from ethanol oxidation (32, 77, 158). Recent proteomic studies verified that increased ROS generation caused by alcohol intake leads to posttranslational redox changes to proteins (increased proteins disulfides and nitrosylation due to increased iNOS during inflammation) of the liver (96, 140, 148). These proteomic studies identified that many thiols in mitochondrial proteins, such as ATP synthase and 3-ketoacyl-CoA thiolase, become oxidized. Many of these posttranslational redox modifications were associated with a decrease in activity. Whether signaling proteins such as NF-κB undergo redox changes has not yet been determined, but remains a distinct possibility. Some evidence suggests that prolonged alcohol treatment causes the inhibition of NF-κB activation in liver. In alcohol-treated rats, NF-κB activation and translocation to the nucleus was found to be inhibited after partial hepatectomy (165). This supports the idea that alcohol promotes sensitization to TNF, possibly through redox inhibition of NF-κB signaling. Overall, because alcohol is associated with protein redox changes and because many proteins involved in NF-κB signaling are redox regulated, the redox inhibition of NF-κB may be an important mechanism of sensitizing hepatocytes to TNF during alcoholic liver injury, but further studies are needed to confirm this idea. Of course, in addition to redox changes, alcohol treatment affects many metabolic and signaling pathways (i.e., PTEN, Akt, ER stress pathways) that may contribute significantly to sensitization of hepatocytes to TNF-induced apoptosis (76, 135).
Drug-induced liver injury
In addition to alcohol, many other drugs that cause liver injury are believed to cause redox changes in liver. The redox changes caused by drugs may be important in causing hepatocyte apoptosis and liver injury during inflammation when TNF is secreted. Acetaminophen (Tylenol), a widely used analgesic, will cause redox changes in hepatocytes (GSH depletion, increased ROS generation, protein oxidation), even at sublethal doses. The redox changes by acetaminophen and can sensitize hepatocytes to TNF-induced apoptosis, probably through NF-κB inhibition (99). Whether acetaminophen can sensitize hepatocytes to TNF in vivo remains to be determined. Work from Roth's laboratory also shows the possibility of drugs sensitizing hepatocytes to TNF-induced apoptosis in vivo. Most drugs that cause idiosyncratic drug liver injury (rare drug injury that occurs at therapeutic doses) in humans do not cause liver injury in animals. However, work by Roth's group (17, 89) revealed that chlorpromazine (an antipsychotic drug), trovafloxacin (a fluoroquinolone antibiotic), and ranitidine (a histamine 2‱receptor antagonist), which do not normally cause liver injury in mice, cause liver injury when simultaneously injected with a sublethal dose of lipopolysaccharide (LPS; bacteria endotoxin). Sublethal levels of LPS will trigger inflammation and TNF secretion in the liver. Blocking TNF production or activity by using various TNF inhibitors prevented liver injury caused by co-treatment with LPS and trovafloxacin, suggesting an essential role of TNF in hepatocyte injury (128). The liver injury caused by LPS and drugs depended on TNF, suggesting that hepatocytes become sensitized to the cytotoxic effects of TNF. Many drugs have been shown to cause increased ROS generation and redox changes in liver (73, 74); consequently, drug-induced redox changes may inhibit NF-κB signaling to sensitize hepatocytes to the cytotoxic effects of TNF (whose secretion increases in response to LPS). Further studies are needed to investigate the synergistic actions of TNF, secreted during inflammation, and drugs, which can cause redox changes.
Redox regulation of NF-κB in Kupffer cells
Although our focus has been on NF-κB signaling in hepatocytes, NF-κB also is very important in other cells in the liver, including Kupffer cells (resident macrophages in liver). In Kupffer cells, NF-κB regulates TNF production and other inflammatory responses. Interestingly, the activation of NF-κB in Kupffer cells may be redox regulated; iron was shown to prime the activation of NF-κB in Kupffer cells (129, 162). Iron chelators were shown to decrease NF-κB activation caused by LPS in Kupffer cells, whereas addition of Fe2+, but not Fe3+, to media increased NF-κB translocation to the nucleus after LPS treatment (129). Thus, although redox changes alone do not activate NF-κB in Kupffer cells, they modulate the extent of NF-κB activation after LPS stimulation. This suggests that in liver, redox changes in Kupffer cells may help potentiate NF-κB activation that results in increased TNF generation, whereas in hepatocytes, redox changes may inhibit NF-κB activation, leading to increased sensitivity to TNF-induced cell death. Why redox changes potentiate NF-κB activation in Kupffer cells, but inhibit NF-κB activation in hepatocytes, remains uncertain. It would be of great interest to determine the underlying differences in hepatocytes and Kupffer cells responsible for the different NF-κB responses after oxidative stress. Overall, redox changes may have a dual affect in liver, potentiation of NF-κB activation in Kupffer cells and inhibition of NF-κB activation in hepatocytes, to help mediate the pathogenesis of alcohol- and drug-induced liver injury.
Perspective
TNF-induced cell death may be an important component of inflammatory injury in liver and other tissues, particularly when inflammation is prolonged. Because TNF and ROS are secreted simultaneous during inflammation, they may work synergistically to mediate liver injury by promoting hepatocyte apoptosis (Fig. 5). Hepatocytes may become sensitized to the lethal actions of TNF by redox changes induced by ROS that modulate NF-κB and other important signaling pathways. The sensitization of hepatocytes to TNF may be heightened by drugs, viruses, and other foreign agents that induce increased ROS generation and cause redox changes in hepatocytes (73). Overall, much circumstantial data suggest that redox changes in hepatocytes leads to their sensitization to the cytotoxic effects of TNF in many liver pathologies. However, because ROS and redox changes are very difficult to measure in vivo, direct proof has not been established, and further studies are needed.
Footnotes
Acknowledgments
This work is supported by the National Institutes of Health Grant DK067215.