
Abstract
The endoplasmic reticulum (ER) is the principal cellular organelle in which correct folding and maturation of transmembrane, secretory, and ER-resident proteins occur. Research over the past decade has demonstrated that mutations in proteins or agents/conditions that disrupt protein folding adversely affect ER homeostasis, leading to ER stress. This in turn initiates the unfolded protein response (UPR), an integrated intracellular signalling pathway that responds to ER stress by increasing the expression of ER-resident molecular chaperones, attenuating global protein translation and degrading unfolded proteins. Failure to relieve prolonged or acute ER stress causes the cell to undergo apoptotic cell death. Recent groundbreaking studies have provided compelling evidence that ER stress and UPR activation contribute to the development and progression of human disease, including neurodegenerative disorders, diabetes, obesity, cancer, and cardiovascular disease. Furthermore, the ability of the UPR to modulate oxidative stress, inflammation, and apoptosis provides important cellular clues as to how this evolutionarily conserved cellular-stress pathway maintains and responds to both normal physiologic and pathologic processes. In this Forum issue, many aspects of the UPR are reviewed in the context of how ER stress and UPR activation influence human disease. This current information provides a solid foundation for future investigations aimed at targeting the UPR in an attempt to reduce the risk of human disease. Antioxid. Redox Signal. 11, 2279–2287.
Overview of the Endoplasmic Reticulum and Its Function
The ER is also the major intracellular Ca2+ store with a resting concentration of Ca2+ of ∼400 μM (46, 57). These elevated ER Ca2+ levels facilitate interactions between Ca2+-dependent chaperones such as calreticulin and calnexin and nascent polypeptides (71). Depletion of the ER Ca2+ stores impairs chaperone activity and inhibits the secretion of specific proteins, demonstrating that proper protein processing requires high levels of Ca2+ within the ER lumen (38). The ER lumen is also the cellular site for the addition of sugar moieties to asparagine residues, a process termed N-linked glycosylation. Given that glycosylation is an important component of correct protein folding, underglycosylated proteins have increased association with ER-resident chaperones, leading to decreased cell-surface expression or secretion (16).
Once a protein is successfully modified and folded in its native state, it is transported out of the ER in coatomer protein (COP) II–coated vesicles to the cis-face of the Golgi complex (3). Proteins that are irreversibly damaged and cannot be correctly folded are eliminated via the ER-associated degradation (ERAD) pathway. ERAD involves the retrotranslocation of damaged proteins out of the ER into the cytosol via the Sec61 translocon (74). Once in the cytosol, these proteins are ubiquitinated and targeted for proteosome-mediated degradation (44). A growing body of evidence demonstrates that the accumulation of mutant proteins or conditions/agents that disturb protein folding/processing cause ER stress and contribute to a wide range of human diseases, including diabetes, cancer, neurodegenerative disease, and heart disease. Whether this impairment in the cellular secretory pathway is the primary cause or a secondary event in disease progression is yet to be determined.
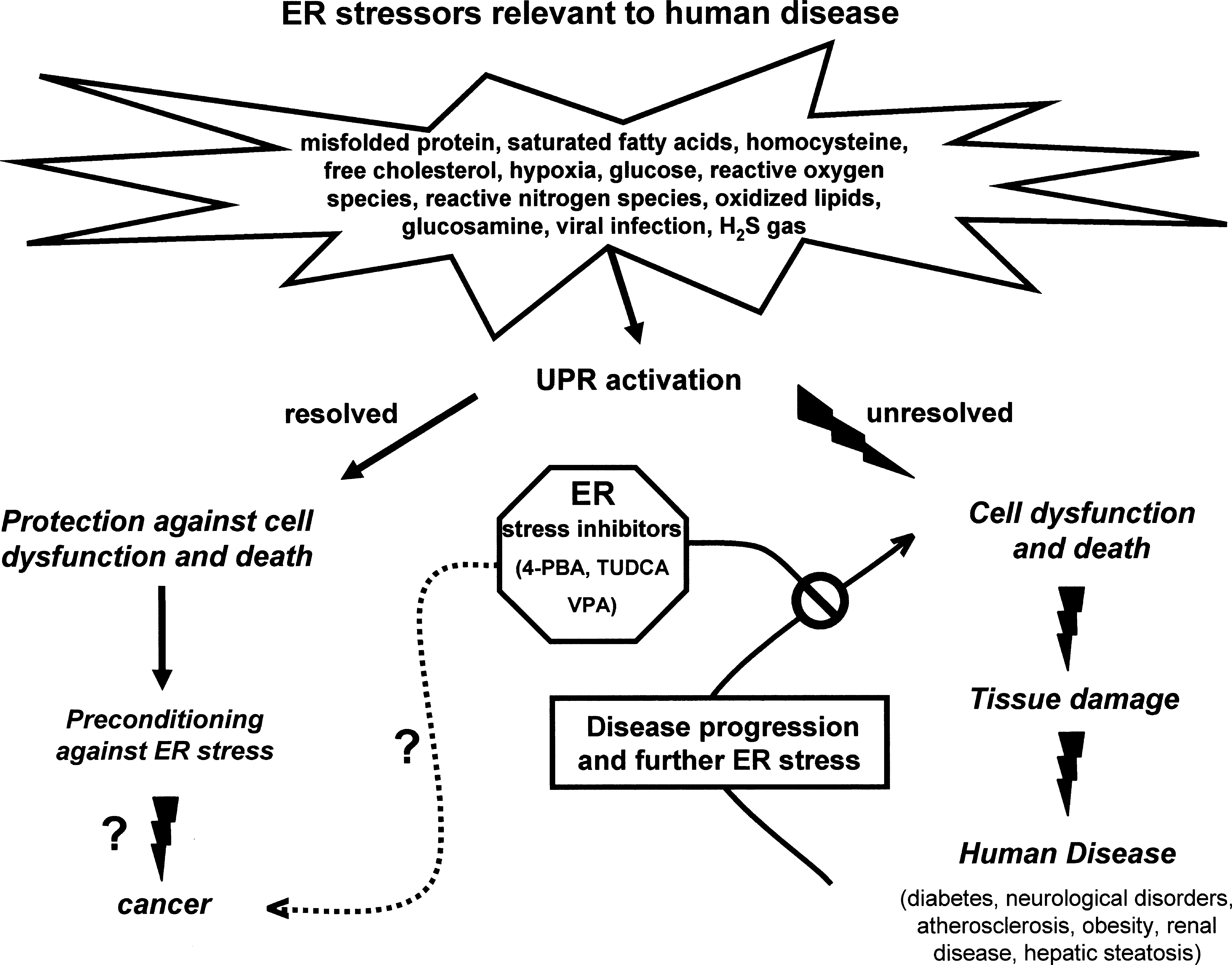
This Forum issue focuses on the physiologic roles of the unfolded protein response (UPR) pathway and its connection to reactive oxygen species (ROS) production and inflammation, as well as its relation to the development and progression of certain human diseases, such as cancer, atherosclerosis, renal disease, and diabetes (Fig. 1). In addition, unique properties of the ER-resident chaperone, GRP78, are highlighted with respect to its role as a multifunctional cell-surface receptor and as a modulator of early embryonic development.
ER-resident chaperone proteins
To assist and regulate the folding process, the ER lumen contains a family of proteins known as ER chaperones that act as foldases and stress sensors in the ER quality-control system. These chaperones include protein disulfide isomerase (PDI), glucose-regulated protein-78/immunoglobulin-binding protein (GRP78/BiP), glucose-regulated protein-94 (GRP94), ERp72, calreticulin, and calnexin (59). ER chaperones target unfolded or misfolded proteins that enter the ER, thereby simultaneously preventing inappropriate aggregate formation and maintaining the proteins in a conformation compatible with correct folding. Several chaperones, including GRP78, PDI, and GRP94, have been identified in a large complex, together with incompletely folded immunoglobulins, suggesting that chaperones form a multimeric network that binds unfolded proteins (45). Neither calreticulin nor calnexin was identified in this multimeric complex, providing evidence of a separation of chaperone systems by function or temporal action (45). Within the ER lumen, chaperones bind to and interact with unfolded proteins through their inappropriately exposed hydrophobic domains or underglycosylated residues (59, 64).
Calreticulin and calnexin specifically assist in glycoprotein folding by recognizing underglycosylated residues within the nascent polypeptide chain. As the polypeptide chains enter the ER lumen, they are modified by the addition of glucose moieties that provide binding sites for calreticulin and calnexin, thus retaining these proteins in the ER (65, 71). The glucose moiety is removed by glucosidase I and II, releasing the protein from calreticulin/calnexin and allowing the newly synthesized protein the opportunity to fold correctly (46). During this period, the oxidoreductase PDI catalyzes the correct formation of disulfide bonds by reshuffling incorrect disulfide bonds and ensuring flexibility of the disulfide bonds until all folding is completed (7). Once correctly folded and modified, the protein continues onto the Golgi; however, if folding is impaired, the glycoprotein is reglycosylated by UDP-glucose:glycoproteins glucosyltransferase (UGGT), recreating the calreticulin/calnexin binding site (46). This cycle of addition/removal of glucose moieties and binding and release from calreticulin/calnexin continues until proper protein folding occurs.
Chaperones from the heat-shock 70 family, including GRP78 and GRP94, recognize unfolded proteins by their inappropriately exposed hydrophobic domains (19). The chaperone GRP78 undergoes cycles of protein binding and release mediated by its N-terminal ATPase activity until the protein is folded correctly and no further GRP78 binding motifs are exposed (20). When GRP78 is bound to ATP, the conformation of GRP78 is modified to allow the binding of unfolded proteins (via their hydrophobic domains) (20). ERdj1 through 5 are members of the DnaJ family of proteins and co-chaperones of GRP78 that bind to the GRP78/protein complex and stimulate the ATPase domain of GRP78 to hydrolyze ATP to ADP (8, 10, 61, 62). The conversion of ATP into ADP causes a conformational change in GRP78, which increases the binding affinity of GRP78 to the unfolded protein (20). The nucleotide exchange factor BiP-associated protein (BAP) then mediates the exchange of ADP to ATP, thereby weakening the GRP78/protein interaction and releasing the protein from GRP78 (9). These cycles of binding and release between GRP78 and the unfolded protein, mediated by ATP hydrolysis, continue until the protein is correctly folded, and no further hydrophobic residues remain exposed.
GRP78 and other ER chaperones assist in maintaining the intracellular Ca2+ balance by sequestering Ca2+ within the ER (34). Ca2+ does not bind to ER chaperones at specific Ca2+-binding motifs but rather to paired anionic amino acids present in their structure (40). Although GRP78 contains the greatest number of anionic residues of any chaperone, it has a relatively low number of paired anionic residues and thus has a low capacity for Ca2+ (1‱2 mole Ca2+/mole), compared with calreticulin (20 mole Ca2+/mole) or PDI (23 mole Ca2+/mole) (34, 40). Nevertheless, overexpression of GRP78 has been shown effectively to increase the Ca2+ storage capacity of the ER (34), attenuate oxidant-induced alterations in intracellular Ca2+ levels, and decrease apoptotic cell death (36). Conversely, reducing GRP78 expression increases intracellular Ca2+ levels in response to the agonists glutamate (79) or H2O2 (1). These findings demonstrate that ER-resident chaperones have the capacity to bind Ca2+ and play a major role in maintaining cellular Ca+ homeostasis.
ER stress and the UPR
ER stress is defined as a disruption in ER homeostasis that leads to the accumulation of unfolded or misfolded proteins in the ER (58). Mutations in secretory proteins or receptors that alter their proper folding induce ER stress and cause human disease (81). Furthermore, a large number of pharmacologic agents or physiologic conditions can alter ER homeostasis, thereby inducing ER stress. For example, depletion of ER Ca2+ stores by Ca2+ ionophores, such as A23187, thapsigargin, or ionomycin, induces ER stress by impairing Ca2+-dependent chaperone activity (38). The addition of reducing agents such as dithiothreitol (DTT) or homocysteine induces ER stress by impairing disulfide bond formation, leading to protein misfolding (39). Tunicamycin or 2-deoxyglucose induces ER stress in vitro by antagonizing N-linked glycosylation, an important posttranslational modification required for proper protein folding (17).
ER stress also is induced by many physiologic conditions or insults that overwhelm the folding/secretory capacity of the ER. Viruses exploit the host cell's translational machinery to synthesize their viral proteins, leading to increased folding pressure on the ER and chaperone upregulation (66). Differentiation of B cells to antibody-producing plasma cells activates the UPR to accommodate the dramatic increase in the expression of IgG (30).
To relieve the accumulation of misfolded/aggregated proteins and reduce ER stress, the cell initiates the activation of a series of integrated stress pathways collectively known as the unfolded protein response (UPR). The UPR consists of three mechanistically distinct but integrated pathways that include the IRE1, ATF6, and PERK pathways. One of the first responses to ER stress involves activation of the PERK pathway. PERK is an ER transmembrane sensor bound to GRP78 under nonstressed conditions (5). With ER stress, GRP78 dissociates from PERK to bind the accumulating unfolded proteins in the ER lumen (5). After its dissociation from GRP78, PERK phosphorylates the α subunit of the eukaryotic translation-initiation factor 2 (eIF2α), thereby leading to the attenuation of general protein translation (60). This prevents any further increase in ER protein load during the initial stages of ER stress. Although eIF2α phosphorylation attenuates translation in a global manner, transcription and translation of ER chaperones such as GRP78 and GADD153/CHOP can occur through translation of the transcription factor ATF4 (23, 60). Similar to PERK, ATF6 is an ER transmembrane-bound transcription factor with its ER luminal domain bound to GRP78 under nonstressed conditions (26, 41). Dissociation of GRP78 under ER-stress conditions releases ATF6 from the ER, allowing it to transit to the Golgi, where it is cleaved by site-1 and site-2 proteases (S1P and S2P) (76). Cleavage of the transcriptionally active domain of ATF6 from the Golgi membrane allows it to migrate to the nucleus, where it binds to the promoter of UPR-inducible genes, specifically ER chaperones and XBP-1 (33, 78).
The final pathway activated with ER stress is the IRE1 pathway. Similar to the PERK and ATF6 arms of the UPR, IRE1 is an ER transmembrane sensor protein that is bound to GRP78 under nonstressed conditions (5). After its dissociation from GRP78, activated IRE1 exhibits endoribonuclease activity, resulting in unconventional splicing and subsequent translation of the mRNA encoding X-box–binding protein (XBP1). The removal of a 26-bp intron from the XBP1 message by IRE1 produces an active XBP1 transcription factor that drives the expression of several ER chaperones, including GRP78.
Although the UPR is orchestrated to alleviate ER stress, severe or prolonged ER stress can lead to apoptotic cell death. One mechanism by which ER stress can induce cell death is through activated IRE1, which recruits TRAF2 to the ER membrane (70). TRAF2 activates apoptosis-signaling kinase 1 (ASK-1), which in turn activates JNK and caspases in a mitochondria-mediated manner (70). Recruitment of TRAF2 to the ER membrane by IRE1 also releases TRAF2 from pro-caspase 12 in the cytosol, thus allowing the active form of caspase 12 to activate downstream executioner caspases (77). Furthermore, release of ER Ca2+ stores can activate calpain, which in turn activates procaspase 12 and other caspases, leading to cell death (51).
ER stress also can induce death through the PERK- and IRE1-mediated activation of proapoptotic proteins, specifically the growth arrest–DNA damage gene (GADD153/CHOP), which can induce apoptosis by decreasing Bcl-2 expression (41, 58). Recent studies by our group have shown that ER stress can induce the expression of TDAG51 (27), a member of the pleckstrin homology–related domain family having proapoptotic characteristics. Overexpression of TDAG51 leads to dramatic alterations in cell shape and increases detachment of cells from their appropriate matrix (27), supporting a mechanism involving detachment-induced PCD or anoikis. The observation that TDAG51-induced shape changes are independent of caspase activation and occur prior to apoptosis is consistent with this mechanism. Therefore, both caspase-dependent and -independent pathways of cell death are activated in response to prolonged or severe ER stress.
Cytoprotective effects of the ER chaperone GRP78
Although GRP78 plays a critical role in ER protein quality control and regulation of the activation of the ER transmembrane signaling molecules, it also confers protection against cytotoxicity and apoptosis (36, 55, 72). GRP78 overexpression protects cells from injury induced by increases in intracellular Ca2+ or oxidative stress by sequestering Ca2+ and in turn inhibiting oxidant-induced increases in intracellular Ca2+ (1, 36). GRP78 has also been shown to be protective against apoptotic cell death induced by topoisomerase inhibitors such as etoposide, doxorubicin (Adriamycin), and camptothecin (55).
A proportion of GRP78 was found to be expressed on the cytosolic side of the ER membrane in a complex with procaspase 7, suggesting that GRP78 exerts its antiapoptotic effects through the sequestration of caspase 7 and thus the prevention of further caspase activation and caspase-mediated cell death (55). Caspase 12 also may be involved, because it has been identified in complex with GRP78 and caspase 7 (54). That deletion of the ATP-binding domain of GRP78 prevents complex formation between procaspase 7 and GRP78 suggests that binding ATP is essential for its antiapoptotic properties (55).
GRP78 also has been shown in a cell-free system to inhibit cytochrome c–mediated caspase activation and subsequent cell death (54). An interaction between GRP78 and cytochrome c is postulated to occur in the cytosol, based on observations that after prolonged ER stress, GRP78 is redistributed from the ER lumen to the ER transmembrane and the cytosol.
The cytoprotective property of GRP78 also has been demonstrated to contribute to tumor survival and chemotherapeutic drug resistance (15, 72). The tumor microenvironment of hypoxia and glucose starvation induces UPR activation and the subsequent upregulation of GRP78. This provides an antiapoptotic benefit to tumor cells, which contributes to resistance to chemotherapeutic drugs such as etoposide (15).
ER luminal versus cell-surface GRP78
GRP78 is sequestered in the ER lumen by its C-terminal KDEL (lys-asp-glu-leu) sequence, an ER-retention signal that binds to the KDEL receptor. However, GRP78 as well as other KDEL-containing chaperones, including PDI and calreticulin, have been identified on the surface of several human cancers, including prostate and breast cancer (2, 22, 72). GRP78 also associates with the cytosolic surface of the ER membrane, suggesting that the structure of GRP78 is consistent with that of a transmembrane protein (55). However, it is unclear as to how GRP78 is orientated in the plasma membrane; whether it is a true integral membrane protein or whether its cell-surface expression is dependent on its association with other cell-surface proteins.
It also remains unclear as to how GRP78 (and other ER chaperones) escapes ER retrieval mechanisms mediated by the KDEL sequence, especially given that at least one study has demonstrated that GRP78 retains its KDEL sequence when expressed on the cell surface (55). Two possible explanations are that the KDEL receptors on the ER membrane are saturated (possible during ER stress and GRP78 upregulation) or that the KDEL signal on GRP78 is masked by its interaction with other proteins such as MTJ-1 (49).
Physiologic functions of cell-surface GRP78
Although the chaperone function of GRP78 is well characterized, current studies indicate that the physiologic function of cell surface GRP78 appears to be that of a receptor (22). GRP78 can associate with MHC class I molecules on the cell surface and acts as a co-receptor for virus internalization (69). Absence of MHC class I molecules increases GRP78 expression, thereby compensating for the loss of MHC molecules and suggesting that GRP78 may represent a unique antigen-presentation system (69). Cell-surface GRP78 also is the receptor for α2-macroglobulin (α2M*), mediating its downstream signaling and regulating cell-survival/cell-death pathways (47 –49). Binding of α2M* to cell-surface GRP78 regulates the Ras-dependent MAPK and PI3 kinase pathways, leading to increases in cell proliferation, IP3 synthesis, intracellular Ca2+ levels, and expression of the docking proteins Grb2, Shc, and Sos (48). Binding of α2M* to GRP78 also mediates macrophage motility through the recruitment of p21-activated protein kinase-2 (PAK-2) to the plasma membrane via the adaptor protein NCK. PAK-2 is activated by tyrosine phosphorylation (49). PAK-2 activation leads to LIM kinase activation, which in turn phosphorylates cofilin, an actin-binding protein, and inhibits its depolymerization, thus regulating actin assembly and motility (49). Cell-surface GRP78 also was shown to act as a receptor for kringle 5 of plasminogen and to mediate its antiangiogenic properties by inducing apoptosis of proliferating endothelial cells (12). Cell-surface GRP78 is also a major autoantigen in several forms of cancer, including prostate, ovarian, and gastric cancer (22). Recent studies suggested that the effects of anti-GRP78 IgG on tumor-cell proliferation, survival, or apoptosis is dependent on the epitope binding domain. In general, autoantibodies directed against the N-terminal region of GRP78 enhance cell survival and proliferation, whereas C-terminal–specific antibodies inhibit cellular proliferation and induce apoptosis (21). In addition to its effects as a receptor, a role for cell-surface GRP78 in thrombosis has been reported (6, 73). This study identifies GRP78 on the cell surface and demonstrates that anti-GRP78 antibodies can increase TF procoagulant activity (6). Although no specific mechanism was addressed, a direct interaction between GRP78 and TF exists on the cell surface was proposed (6). These results support previous findings that GRP78 plays an important role in the regulation of TF PCA (73) and provides evidence that cell-surface GRP78 may affect TF directly. Together these findings demonstrate that cell-surface GRP78 displays unique topology that allows it to interact with and regulate a diverse group of signaling pathways and surface proteins that control the balance between cell proliferation and cell death.
ROS Production, Inflammation, and the UPR
Unlike the cytosol, the ER contains a unique oxidizing environment that allows the formation of intra- and interchain disulfide bonds critical for the correct folding and assembly of nascent polypeptides. The formation or rearrangement of these disulfide bonds involves a cascade of disulfide-bond transfers that are catalyzed by protein disulfide isomerase (PDI) and ER oxidoreductase 1 (ERO1), with molecular oxygen serving as the terminal electron acceptor (63). Recent studies suggested that ROS generated as a byproduct of disulfide bond formation in the ER cause oxidative stress and contribute to apoptotic cell death (24, 42, 43). As a response to the increase in oxidative stress, a UPR-mediated integrated stress response involving the Perk pathway is activated to preserve cell function and survival (24). Activation of the Perk pathway by the accumulation of misfolded proteins in the ER, amino acid starvation, or oxidants leads to the phosphorylation of eIF2α. This inhibits general protein synthesis while promoting translation and expression of ATF4, a transcription factor that regulates amino acid import, glutathione biosynthesis, and resistance to oxidative stress. Thus, lack of ATF4 results in reduced GSH levels, accumulation of ROS, and apoptotic cell death (24).
Furthermore, ATF4-deficiency significantly reduces cystathionine γ-lyase (CGL) protein expression, a critical enzyme in the transsulfuration pathway that regulates cysteine synthesis (Austin and Dickhout, unpublished data). Supplementation of the media with high-dose amino acids necessary for GSH synthesis showed cytoprotection by decreasing ROS levels. Consistent with these studies, Malhotra and colleagues (43) demonstrated that antioxidant treatment in CHO cells overexpressing factor VIII (coagulation factor sensitive to misfolding) reduces UPR activation, oxidative stress, and apoptosis, as well as increasing factor VIII secretion in vitro and in vivo. These studies imply that persistent oxidative stress and protein misfolding act synergistically to increase apoptotic cell death and tissue damage that occurs in several human diseases such as diabetes, neurologic disorders, and atherosclerosis. Thus, antioxidant treatment may improve the clinical outcome in patients with congenital diseases associated with protein misfolding (81).
In addition to ROS production, ER stress can cause activation of nuclear factor-κB (NF-κB), a key transcription factor that regulates inflammation (32). Recent studies also demonstrated that ER stress causes a biphasic, bidirectional regulation of NF-κB that can modulate the expression of inflammation-associated genes encoding cytokines, chemokines, adhesion receptors, and various enzymes, leading to initiation, amplification, and perpetuation of inflammatory responses. Given the importance of the inflammatory process in a host of human disease, such as atherosclerosis and cancer, future experimentation will likely focus on the bidirectional roles of ER stress in the inflammatory processes so that approaches to suppress the proinflammatory response can be developed.
UPR and Human Disease
Many human diseases, including cancer (72), atherosclerosis (67), diabetes (31), neurologic disorders (72), and renal disease (14), are associated with ER stress and UPR activation (Fig. 1), prompting the hypothesis that ER stress contributes to the pathology of these diseases. This is particularly relevant in the development and progression of atherosclerosis, which is highlighted below.
UPR and atherosclerosis
UPR activation has been reported during all stages of atherosclerosis (82), and pathophysiologic conditions known to induce ER stress contribute to lesion development, plaque rupture, and thrombogenicity (67). Furthermore, a significant correlation between ER stress markers, plaque vulnerability, and lesion apoptosis was reported in human coronary artery plaques (50). Peroxynitrite, a byproduct of the reaction between nitric oxide (NO) and superoxide, elicits ER stress in cultured endothelial cells, resulting in cell dysfunction and apoptosis (13). Several cardiovascular risk factors, including hyperhomocysteinemia (HHcy), diabetes, and hyperlipidemia, are known to enhance the intracellular production of superoxide, which is rapidly scavenged by NO to form peroxynitrite. Recent studies demonstrated that HHcy increases superoxide production in atherosclerotic lesions from apoE−/− mice (75), thereby contributing to peroxynitrite formation. The fact that peroxynitrite induces ER stress and colocalizes with UPR markers in atherosclerotic lesions from apoE−/− mice (13) suggests a role for UPR-induced lesion development and plaque necrosis. In support of this concept, Tabas and colleagues (18) reported that the ER is the site of free cholesterol–induced cytotoxicity in cultured macrophages. The additional observations that cholesterol-induced apoptotic cell death was attenuated in CHOP/GADD153-deficient macrophages (18) and CHOP/GADD153 was markedly elevated in advanced atherosclerotic lesions from apoE−/− mice (82) suggests that activation of the CHOP/GADD153 arm of the UPR is a key signaling step in cholesterol-induced apoptosis in macrophages. This is supported by recent studies showing reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions from apoE-deficient or LDL receptor–deficient mice lacking CHOP/GADD153 (68). These findings suggest that therapeutic approaches that target the UPR may have beneficial effects on reducing atherosclerotic lesion progression and rupture.
Potential strategies aimed at reducing human diseases by blocking ER stress and UPR activation
Numerous reports support an important role for ER stress and UPR activation in the pathogenesis of a wide range of human diseases, including diabetes, obesity, renal disease, cancer, atherosclerosis, and neurologic disorders. This suggests that ER stress represents a unifying mechanism that contributes to a number of different human disease states and provides a unique opportunity for the development of novel therapeutic approaches aimed at reducing the effects of ER stress. Small chemical chaperones, such as 4-PBA and TUDCA, have been used successfully to reduce the effects of ER stress in mouse models of obesity (4), diabetes (52), and Alzheimer disease (56). Based on these findings, it is possible that the use of these chemical chaperones or their derivatives may provide a unique approach to treating congenital diseases resulting from the improper folding and processing of target proteins. This would provide several potential benefits: it could assist in proper folding, thereby rescuing protein function, and decrease the adverse effects of ER stress in the target tissue due to the accumulation of misfolded proteins.
Cell preconditioning with activators of the UPR has been examined as a potential protective approach against several diseases. Preconditioning with several ER stress-inducing agents/conditions, including ischemia, tunicamycin, or DTTox protects cultured renal epithelial cells, cardiomyocytes, or neurons from subsequent oxidative stress, ischemia/reperfusion, and nephrotoxins (25, 28, 37, 53, 80). In vivo studies have demonstrated that preconditioning with tunicamycin or thapsigargin ameliorates passive Heymann nephritis (11) and mesangioproliferative glomerulonephritis (29) in rats. Furthermore, induction of GRP78 by ischemic preconditioning was shown to reduce ER stress and delay neuronal cell death (25). Ischemic postconditioning also protects the heart from ischemia/reperfusion injury (37). Although it is still at an early stage for potential therapeutic use, alteration in UPR activation has emerged as a promising and novel approach for reducing disease progression.
Conclusions and Future Perspectives
Recent groundbreaking studies provide compelling evidence that ER stress and UPR activation contribute to a wide range of human diseases. Components of the UPR as well as cellular factors that regulate the UPR are expressed in a cell- and tissue-type–specific manner. It is well established that uncontrolled ER stress induces UPR-mediated apoptotic cell death and tissue damage. However, a detailed understanding of the cellular and molecular events controlled by each arm of the UPR and their contribution to the disease process is lacking. This is based on recent studies indicating that differential activation of the PERK and IRE1 pathways may determine life-or-death decisions after ER stress (35). Future studies will be designed to evaluate the contribution of these UPR pathways to other cellular stress responses and how they affect disease progression. Furthermore, the development of mouse models having tissue-specific deletions in specific components of the UPR should yield important information on disease progression that is more relevant to the human condition. The discovery of cell-surface GRP78 in cancer cells, as well as in cells from pathologic states, suggests additional novel functions distinct from chaperone activity that affect disease development and progression. Ultimately, future investigations will provide a solid foundation for the development of novel inhibitors of ER stress or activators of the UPR or both that modulate a variety of different human disease and their complications.
Footnotes
Acknowledgments
This work was supported in part by research grants to R.C.A. from the Heart and Stroke Foundation of Ontario and the Canadian Institutes of Health Research. Financial support from St. Joseph's Healthcare Hamilton is acknowledged. RCA is a Career Investigator of the Heart and Stroke Foundation of Ontario and holds the Amgen Canada Research Chair in Nephrology.