
Abstract
The discovery of some additional properties and functions of reactive oxygen species (ROS), beyond their toxic effects, provides a novel scenario for the molecular basis and cell regulation of several pathophysiologic processes. ROS are generated by redox-sensitive, prosurvival signaling pathways and function as second messengers in the transduction of several extracellular signals. A complex intracellular redox buffering network has developed to adapt and protect cells against the dangerous effects of oxidative stress. However, pathways involved in ROS-adaptive response may also play a critical role in protecting cells against cytotoxic effects of anticancer agents, thus supporting the hypothesis of a correlation between adaptation/resistance to oxidative stress and resistance to anticancer drugs. This review summarizes the main systems involved in the adaptive responses: an overview on the pathophysiologic relevance of mitochondria on redox-sensitive transcription factors and genes and main antioxidant networks in tumor cells is provided. One of the major aims is to highlight the adaptive mechanisms and their interplay in the intricate connection between oncogenic signaling, oxidative stress, and chemoresistance. Clarification of these mechanisms has tremendous application potential, in terms of developing novel molecular-targeted anticancer therapies and innovative strategies for rational combination of these agents with chemotherapeutic or tumor-specific biologic drugs. Antioxid. Redox Signal. 11, 2701–2716.
Introduction
Since 1980, it has been known that, at high concentrations, free radicals and radical-derived species are hazardous for living organisms because they damage all major cellular components (2, 11, 22, 90, 102). However, because a comprehensive description of the chemical and physical properties of ROS is beyond the purpose of this review, a few examples of review articles in which these properties are broadly discussed are provided (81, 98, 173).
Protection against the noxious effects of ROS is achieved in aerobic organisms by a wide spectrum of defensive natural antioxidant systems, consisting of antioxidant enzymes and numerous endogenous and dietary antioxidant compounds that react with and inactivate ROS (65). The primary antioxidant enzymes include superoxide dismutases (SODs), which catalyze the dismutation of O2 •− to O2 and the less reactive H2O2 (54, 97). Other powerful scavenging enzymes include catalase and glutathione peroxidase (GPX). Catalase is a tetrameric peroxidase enzyme that converts H2O2 to water and molecular oxygen. Unlike O2 •−, which remains near the site of production, H2O2 can diffuse across membranes and through the cytosol (5). For these reasons, the main focus in the study of ROS as physiological regulators in intra- and intercellular signaling (as is seen in this review) has been on H2O2 as the most efficient signal messenger. GPX, conversely, is a selenium-containing tetrameric enzyme that reduces H2O2, lipoperoxides, and other organic hydroperoxides to their corresponding hydroxylated compounds by using glutathione (GSH) as a hydrogen donor (148, 149). Finally, because of their recently characterized roles in cellular response, peroxiredoxins (PRXs) and thioredoxin (Trx) systems are described later in this review. Meanwhile, the nonenzymatic antioxidants include, among others, vitamin C, vitamin E, β-carotene, reduced GSH, and numerous phytochemicals. GSH, a ubiquitous thiol, plays an important role in maintaining intracellular redox equilibrium and has evolved into one of the major detoxifying antioxidants and most abundant thiols in almost all mammalian cells (146).
Adaptation to Oxidative Stress and Redox Signaling
A fundamental scientific crossroad in the history of ROS came from the elegant studies of Finkel and co-workers (157). In 1995, they proposed new important functions for these toxic, “undesirable” molecules as subcellular messengers in growth-factor signal transduction. This finding opened up a multiplicity of still relevant new concepts, in such complex cellular processes as mitogenic signal transduction (40, 58), gene expression, regulation of cell proliferation, replicative senescence, apoptosis, and cancer (39). In the last decade, it has emerged that intracellular generation of ROS serves to maintain homeostasis and that the activation of several pathways by ROS could be part of a defensive mechanism against oxidative stress, an adaptive response serving to counteract redox perturbations.
To study adaptive responses to oxidative stress, our group set up an experimental system based on transient adaptation to mild oxidative stress of human osteosarcoma cells grown in the presence of subtoxic concentrations of diethylmaleate (DEM), a GSH-depleting agent. The adapted cells, compared with untreated cells, contain increased concentrations of GSH (four- to sixfold), which, on DEM removal from the culture medium, return to normal values and are more resistant to subsequent oxidizing stress induced either by toxic concentrations of the same agent or by H2O2 treatment (Fig. 1). To investigate the molecular mechanisms involved in the adaptive response to oxidative stress, we analyzed the gene-expression profiles of DEM-adapted cells by differential display. The expression of some known genes involved in energy metabolism, protein folding, and membrane traffic is upregulated in adapted cells (Table 1). Two genes were further characterized by our group for their involvement in oxidative stress adaptive mechanisms: the mitochondrial heat-shock protein TRAP1 (TNF receptor–associated protein 1) and the adaptive response to oxidative stress (AROS)-29 gene, coding for a transmembrane protein of unknown function. The increased resistance to DNA damage and apoptosis, in cells stably overexpressing either AROS-29 (105) or TRAP1 (106), confirmed their functional roles in the adaptation to oxidative stress. For these features, a further study of their role in tumor cell survival has been performed, and the results are presented later.
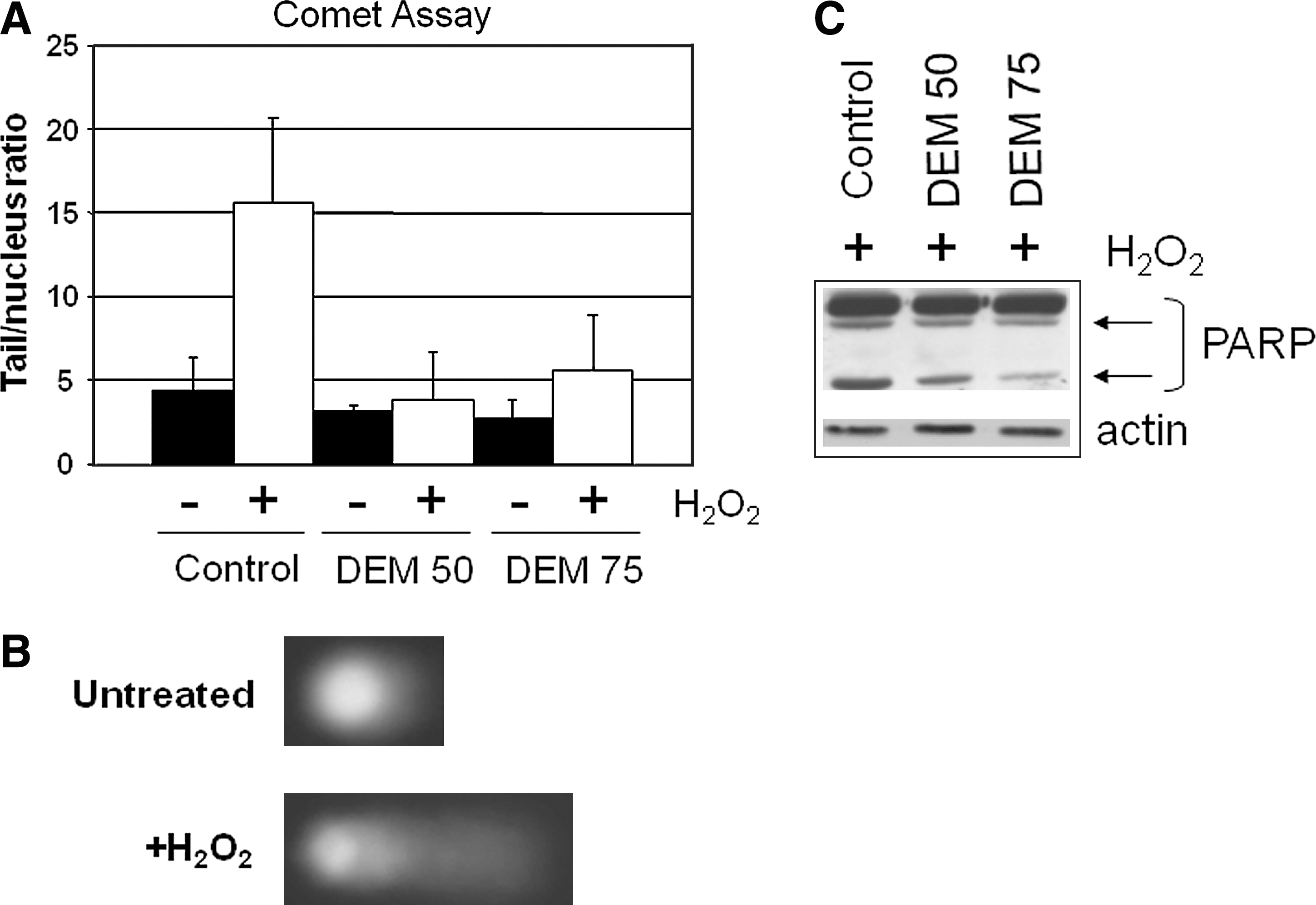
Meanwhile, some major “end points” involved in the regulation of cellular adaptation to oxidative stress are presented.
Role of mitochondria
Adaptive response to agents that challenge cellular homeostasis involves important changes in mitochondrial function. Given their crucial role in cell physiology, mitochondria are among the first responders to various stressors. Adaptive response of mitochondria to stress includes mainly the enhanced expression and activity of oxidative phosphorylation subunits, generation of ROS for signaling or defense, and induction of the apoptotic cascade, as reviewed in Manoli et al. (96).
One of the initial regulatory steps in mitochondria-mediated adaptive responses occurs through the opening of the permeability transition pore (mtPTP) located in the inner mitochondrial membrane (17). In recent years, the mtPTP has received considerable attention as a potential mechanism for the execution of cell death, and findings that link the mtPTP to key cellular signaling pathways are emerging, thus increasing interest in the pore as a pharmacologic target (133). Mitochondrial fission and fusion are also part of the adaptive response of the organelle to various environmental challenges. “Mitochondria-shaping” proteins such as the GTPases Mitofusin (Mfn) 1 and 2, Opa1, as well as the cytosolic dynamin-related protein 1 (Drp1) and its receptor on the outer mitochondrial membrane Fis1, influence not only the shape of mitochondria but also the function of the organelle and eventually integrate cellular signaling cascades, including apoptosis (32, 145).
Key genes involved in mitochondrial adaptive response to oxidative stress are the heat-shock proteins (HSPs), a highly conserved and functionally interactive network of intracellular “chaperones” that disaggregate and refold, misfolded proteins resulting from various environmental, physical, and chemical stressors. HSP gene expression and other cellular responses limit the damage caused by stress and, thus, facilitate cellular recovery through interactions of the major HSPs with components of the apoptotic pathways. HSPs promote cell survival by preventing mitochondrial outer membrane permeabilization and subsequent cytochrome c release, caspase activation, and apoptosome assembly (14).
However, even though they function primarily in cell-survival pathways, these chaperones can also inhibit or even promote cell-death pathways at multiple levels to ensure homeostasis at cell or organism level or both. Therefore, the heat-shock genes do much more than simply help cells survive stress (8). Further roles of HSPs in the regulation of cell survival in drug resistance are described later.
Transcription factors
Modulation of gene expression is a common end point exerted by cells to defend themselves against ROS insults. In this context, we have chosen not to discuss the influence of ROS or redox metabolites on the activity of enzymes responsible for initiating and perpetuating epigenetic control of gene expression, because this topic is considered too broad and requires a dedicated review.
The regulation of gene expression is a control mechanism that arises from a complex series of signaling pathways, starting from the cellular “periphery” and reaching the nucleus, where redox-sensitive transcription factors are the downstream targets that can regulate the expression of specific genes, such as those involved in cellular proliferation and apoptosis, DNA repair systems, control of protein synthesis, enhanced detoxification of free radicals, and antioxidant production. These mechanisms are simultaneously activated by the cells to cope with and defend themselves against oxidative stress. Transcription factors could be considered immediate-response genes that respond to a wide variety of environmental agents inducing oxidative stress (64, 83). Redox modulation of transcription factor activity includes oxidative modification of the DNA-binding motif of the transcriptional factor itself by ROS or posttranslational modifications or both, mainly a phosphorylation/dephosphorylation switch consequent to the effects of the redox-regulated intracellular signaling pathway (39). Our group studied the redox regulation of some Zn-finger transcription factors that are extremely sensitive to oxidative stress, such as Sp1, glucocorticoid receptor, and Egr1 both in vitro (4) and in vivo (3, 38, 41). Another Zn-finger–like and redox-sensitive transcriptional factor is the p53 tumor suppressor involved in the regulation of cell-cycle arrest and apoptosis. We demonstrated that the oxidative stress induced by DEM inhibits site-specific p53 binding and p53 transactivation ability, but enhances p21wafl expression in intact cells (138) through a p53-independent, Ras-ERK–dependent pathway (42). Other observations suggest that in the absence of stress or after mild stress, low levels of p53 drive the expression of several antioxidant genes that decrease ROS levels and protect cells against DNA damage. Stronger activation of p53 after severe or extended stress results in elevated ROS levels and cell death, probably through the loss of p53 function, which ultimately damage DNA and increase the mutation rate. The inability to activate p53-induced apoptosis may result in the development of cancer (15). The studies on p53 redox regulation and adaptive responses are still very active. It was recently demonstrated that p73, a member of the same family as p53, is involved in the control of apoptotic pathways in response to DNA damage (119).
Finally, TP53INP1, a major actor of the p53-driven oxidative-stress response with antioxidant functions, was recently identified: it was shown that this factor elicits a p53-independent intracellular ROS regulatory function and a p53-dependent transcription regulatory function (21).
Another redox-sensitive factor is NF-E2–related factor 2 (Nrf2) (104). In response to oxidative stress, Nrf2 controls the fate of cells through transcriptional upregulation of antioxidant response element (ARE)-bearing genes, including those encoding for endogenous antioxidants, phase II detoxifying enzymes, and transporters. Expression of the Nrf2-dependent proteins is critical to maintain cellular redox homeostasis through elimination of toxicants/carcinogens. Nrf2 activity is regulated in part by the association with Keap1 protein: a disruption of this association in response to stress signals causes the release of Nrf2 and its translocation into the nucleus, to effect its transcriptional activity (72). Antioxidant response by Nrf2 activation can also involve interaction with p53. It has been demonstrated that if oxidative stress provokes DNA damage, a second response of the cell takes place, based on the activation of p53, which induces cell-cycle arrest or apoptosis or both. The cross-talk between these two regulatory mechanisms has been explored. The results show that p53 counteracts the Nrf2-induced transcription of three ARE-containing promoter genes (46). However, the regulatory mechanisms involved in mediating Nrf2 activation, the nature of the Keap1 pathway, or the mechanisms by which Keap1 acts to repress Nrf2 activity remain to be fully characterized (124). Further observations linking Nrf2 to chemoresistance are discussed later.
One of the most-characterized signaling pathways, highly sensitive to changes in the intracellular redox environment and activated by oxidative stress, is regulated by NF-κB. Numerous authors have reviewed NF-κB–activating mechanisms and signals (for a review, see 18) and have highlighted the role of the proinflammatory cytokines TNF-α or IL-6, irradiation, endotoxins and ROS, as well as the steps involved in initiating the transcription of several antioxidant genes, such as MnSOD and enzymes involved in GSH biosynthesis. For the purposes of this review, it is interesting to note that NF-κB and its inhibitor IκBα are also localized in the mitochondria and can negatively regulate mitochondrial gene expression, thus reinforcing the role of mitochondria in redox-adaptive responses (29).
Other protective mechanisms against ROS-derived cell death involve local activation of AP-1 and c-Jun N-terminal kinase (JNK) in the mitochondria, a particularly significant pathway in tumor cell progression (93). However, even though many other relevant examples of redox-sensitive transcription factors could still be provided, we chose to focus on a few proteins involved in the regulation of genes, whose expression strongly contributes to the redox adaptive response and, in this context, defends cells against ROS-damaging effects. Although the adaptive response to stress could represent a mechanism of cell protection against a hostile environment, this review focuses on the correlations between adaptation/resistance to oxidative stress, chemoresistance, and cell survival, and on the hypothesis that these mechanisms may also be used by tumor cells to evade and defend against the cytotoxic effects of anticancer agents. Thus, we present the most recent findings on the redox-regulatory mechanisms in chemoresistance, giving examples of specific genes—mainly antioxidants—altered in tumors.
Correlations Between Oxidative Stress and Drug Resistance
Most malignancies are resistant to therapies or become resistant during anticancer therapy. Drug resistance can be described as a lack of meaningful response or a partial response to therapy, or regrowth of tumor after an initial response. Tumor cell population is characterized by subclones resistant to anticancer drugs, which are responsible for the regrowth of the tumor mass after or during therapy. A wide variety of mechanisms have been identified as being responsible for drug resistance, but these insights have resulted only in limited improvements in patient survival. Drug resistance is a multifactor phenomenon involving multiple interrelated or independent pathways, including changes in cellular responses, such as increased cell ability to repair DNA damage or tolerate stress conditions and acquiring mechanisms for escaping apoptosis (134). A better knowledge of the molecular mechanisms underlying drug resistance is fundamental for the design of novel treatments that have a chance to improve tumor response and patient survival.
Overwhelming evidence supports the association of nonfunctional apoptotic pathways or the activation of survival mechanisms with resistance to chemotherapy. This topic was recently reviewed by Rodriguez-Nieto et al. (137) and de Bruin et al. (33). The activation of tyrosine kinase (TK)-receptor signaling results in the induction of survival mechanisms and in the downregulation of proapoptotic pathways responsible for resistance to anticancer drugs (33, 137). Furthermore, several lines of evidence suggest that adaptation to oxidative stress and drug resistance may share common mechanisms (122) (Fig. 2). Tumor cells produce increased amounts of ROS because of the altered metabolic demand, and this induces an antioxidant adaptive response, which, in turn, favors a redox imbalance, an altered redox regulation of cellular signaling, and the activation of prosurvival mechanisms. These signaling factors/proteins are involved in the transmission of inter- or intracellular information through multiple transduction pathways and are critical for supporting cell proliferation and establishing cell fate. Furthermore, it has been suggested that these pathways involved in the ROS-adaptive response may also play a critical role in protecting cells against the damaging and cytotoxic effects of anticancer agents (122). Many anticancer drugs induce oxidative stress either as a direct mechanism of cell death, such as ionizing radiation, or as an indirect effect of exposure, as observed with several chemotherapeutic agents (158). Recently, it was reported that breast-cancer stem cells contain lower ROS levels and enhanced ROS-scavenging systems compared with their normal counterparts. Interestingly, lower ROS levels in breast cancer stem cells are associated with less DNA damage with ionizing irradiation and with radiosensitization after depletion of ROS scavengers (35). Thus, the antioxidant network of cancer cells may be relevant in maintaining intracellular homeostasis and in favoring cell survival. With such a perspective, many redox-dependent mechanisms of adaptation to stress have been explored in terms of their potential for inducing drug resistance, with those proposed including the induction of DNA repair systems, the reprogramming of cell-cycle–regulation systems, and the upregulation of nonenzymatic and enzymatic antioxidant defenses, and also of molecular chaperones and of stress-responsive proteins (120, 122).

Several studies evaluate the reprogramming of gene expression in human tumors after treatment with antiblastic agents. The major aim of these analyses was to identify a gene-expression signature associated with drug resistance that can be used in daily clinical practice to select patients who are likely to be sensitive to a specific drug regimen. These studies suggest that oxidative stress induced by antiblastic agents (e.g., taxanes, anthracyclins) may play a direct or indirect role in tumor cell death. Interestingly, the gene-expression profile of nonresponder breast carcinomas treated with docetaxel was characterized by elevated expression of genes controlling the cellular redox environment, such as Trx, GST, and PRX (73).
The following paragraphs focus on the redox factors that have been described as critical for the protection of tumor cells from the damaging activity of anticancer agents and discuss the hypothesis that such molecules may be or become potential molecular targets to improve the cytotoxic effects of drug therapies and overcoming drug resistance. A list of the most-studied pharmacologic agents modulating some antioxidant systems is provided in Table 2.
Antioxidant Responses and Redox Signaling in Drug Resistance
Antioxidants and antioxidant enzymes are the front-line intracellular defenses against oxidative stress (65). Collectively, these systems constitute a complex intracellular “redox buffer,” a network of molecules whose function is to maintain a slightly reduced environment. Interestingly, normal and transformed cells can induce this intracellular network when the levels of ROS exceed the intracellular antioxidant capacity. Several studies link the upregulation of antioxidants and antioxidant enzymes to the onset of drug resistance in human malignancies (122).
Glutathione
GSH is a major antioxidant that removes ROS directly and acts as a substrate for several peroxidases (36, 150). This tripeptide also is involved in the conjugation of foreign molecules catalyzed by GST. Even though GSH synthesis occurs in the cytoplasm, the mitochondrial pool is more important than the cytoplasmic in maintaining cell viability and protecting against toxic agents. Several studies demonstrated that GSH depletion in mitochondria is associated with dysfunction and loss of cell viability in response to oxidative events (30, 47, 174). The GSH content of cancer cells is particularly relevant in regulating mutagenic mechanisms, DNA synthesis, growth, and multidrug and radiation resistance. Owing to its reactivity and high intracellular concentrations, GSH has been implicated in resistance to several chemotherapeutic agents. With such a perspective, several approaches designed to deplete intracellular GSH levels have been pursued, including the use of buthionine-(S, R)-sulfoxime (BSO), a potent and specific inhibitor of γ-glutamyl cysteine synthetase (γ-GCS), the rate-limiting step in the synthesis of GSH, as well as a hammerhead ribozyme against γ-GCS mRNA aiming specifically to downregulate GSH levels and to target c-Jun expression to reduce GSH levels. Interestingly, GSH-depleting strategies have clearly been shown to improve the capacity of antiblastic agents to induce apoptosis in cancer cells both in vitro and in murine models (44).
Furthermore, several studies link the pleiotropic effects of GSH in promoting cell growth and broad resistance to therapy with the upregulation of Bcl-2, which inhibits the activation of apoptosis and contributes to the elevation of GSH (55, 100, 117). This issue was clearly demonstrated in highly metastatic murine B16 melanoma cells that are characterized by elevated levels of both GSH and Bcl-2. Interestingly, the reduction of Bcl-2 and GSH—combined with treatment with paclitaxel, radiation, and cytokines—eliminated melanoma cells from liver and all other systemic disease, leading to long-term survival without recurrence in melanoma-injected mice (100). Similarly, breast carcinoma MCF-7 cells overexpressing Bcl-2 protein are resistant to cisplatin and doxorubicin (Adriamycin), and this resistance was clearly correlated with the inhibition of an early GSH efflux (117). GSH was also found to play a critical role in regulating apoptotic pathways triggered by APO-1/Fas or anticancer drugs in leukemia T cells. CD95-resistant or Bcl-x(L) overexpressing leukemia cells are resistant to apoptosis and exhibit high intracellular GSH levels after treatment with anticancer drugs or CD95 triggering. Downregulation of GSH, by BSO, reversed the deficiencies in activation of apoptotic pathways induced by anticancer drugs or CD95 (55). These lines of evidence suggest that the resistance to apoptosis observed in human tumor cells may depend, at least in part, on intracellular GSH levels, which may affect apoptotic signaling in different compartments (for example, the death receptor or mitochondria).
Manganese superoxide dismutase
MnSOD is an important antioxidant enzyme that has received growing attention as a negative modulator of cellular apoptosis and as a survival factor for cancer cells. Several studies describe increased levels of MnSOD in human neoplasms, in a fashion that often correlates with the degree of their malignancy (28, 88, 114, 136, 162). We previously showed that the overexpression of MnSOD in human brain gliomas significantly correlates with high tumor grade (88, 136), and much evidence suggests that even small amounts of this enzyme may be crucial for cell resistance to inflammatory stimuli and anticancer drugs and may prevent the oncogene-induced apoptosis triggered by the tumor-suppressor protein p53 (56).
Great interest was aroused by the recent observation that MnSOD and mitochondria-generated ROS-signaling (superoxide and hydrogen peroxide) could regulate radiation-induced G2 checkpoint activation and radioresistance in human pancreatic cancer cells, as well as mechanisms that increase resistance to apoptosis in ovarian cancer cells (50, 176). In such a perspective, knockdown of MnSOD by siRNA leads to sensitization of ovarian cancer cells to the two front-line anticancer agents doxorubicin and paclitaxel, whose action involves the generation of free radicals. Furthermore, MnSOD downregulation enhanced the drug-induced phosphorylation of extracellular signal-regulated kinase 1/2, suggesting that a combination of drugs capable of suppressing MnSOD with conventional chemotherapeutic agents may provide a novel strategy with a superior therapeutic index and advantages for the treatment of refractory ovarian cancer (176).
The thioredoxin reductase system
Several studies suggest that thioredoxin reductase (TrxR) is a selenoprotein involved in the regulation of the redox status of cells and in many cellular functions, including redox control of transcription factors, protection against oxidative stress, and cell growth (19, 112). TrxR, Trx, and NADPH compose a highly conserved, ubiquitous system (108) that plays an important role in the redox regulation of multiple intracellular processes, such as DNA synthesis, regulation of gene expression, cell proliferation, and resistance to chemotherapeutic drugs that induce oxidative stress and apoptosis (12, 126, 128). After ROS production, TrxR initiates a signaling cascade of events, which begins with the activation of Trx-1 (67, 68, 170). Trx-1 is regulated by the ability of TrxR to reduce oxidized Trx-1 at cysteines 32 and 35. However, posttranslational modifications, including glutathionylation, thiol-oxidation, and S-nitros(yl)ation, at the nonactive cysteines, importantly contribute to the regulation and functions of Trx-1 (63). The activated Trx-1 translocates to the nucleus and turns on the nuclear component of the redox-sensitive signaling pathway Ref-1, which then activates transcription factors, such as AP-1 and NF-κB (82, 152). As described earlier, these transcription factors are involved in regulating the expression of specific target genes responsible for the protective or reparative cellular response to the damaging effects of oxidative stress induced by exogenous cytotoxic agents (6, 127). Altogether, the key biologic functions of Trx-1 are antioxidative, antiapoptotic, and pro-proliferative properties.
TrxR expression is upregulated in several human cancers, such as breast, thyroid, liver, prostate carcinoma, and melanoma; human colorectal carcinomas are characterized by either upregulation of TrxR expression or increased TrxR activity (6, 16, 75, 127, 159). TrxR-1 upregulation in human gastrointestinal cancer correlates with resistance to cisplatin (6). Trx also is upregulated in a variety of cancers, including lung, colorectal, cervical, hepatic, and pancreatic cancer (121), and its upregulation seems to correlate with aggressive tumor behavior, poorer prognosis, and decreased patient survival (76, 129). These findings are in agreement with other evidence suggesting that the inhibition of TrxR prevents the activation of prosurvival signaling pathways in response to cytotoxic agents and can sensitize tumor cells to agents that induce cell death (121).
The glutathione reductase/glutaredoxin system
The glutathione reductase (GR) and glutaredoxin (GRX) system represents another redox-sensitive signaling pathway that has been suggested to play a role in how cells respond to oxidative stress (57). Similar to TrxR and Trx, GRX also contains a common active-site motif and a fold known as a Trx-fold that is critical for its function. However, unlike Trx, which is reduced by its own reductase, GRX is reduced by GSH, which in turn is reduced by GR. Both GR and GRX are upregulated in tumor cells and tumor cell lines (37) and are involved in the control of several redox-sensitive signaling pathways that likely play a role in cellular response to oxidative stress (13, 67). To address the issue that upregulation of GR and GRX gene expression may represent a mechanism used by tumor cells to escape the cytotoxic effects of some anticancer agents, several studies have been performed by using siRNA technology (92). In such a perspective, siRNA-interfering technologies against GRX in HeLa cells resulted in an increased sensitivity to doxorubicin and phenylarsine oxide (92). Furthermore, it has been proposed that the mitochondrial isoform, GRX-2, may play a central role in the glutathionylation and deglutathionylation of proteins in multiple signaling pathways, suggesting that it may be involved in regulating both ROS production and scavenging in mitochondria (13). Finally, it has been proposed that the GR/GRX system plays an important role in controlling programmed cell death, because the upregulation of GRX-2 results in a significant survival effect in cells exposed to antiblastic agents, likely by preventing cytochrome c release (37).
Peroxiredoxins
PRXs reduce alkyl hydroperoxides and H2O2 to the corresponding alcohol or water, although, unlike other peroxidases that generally contain heme or selenocysteine in their active site, PRXs contain one or two cysteines at their active site. PRXs are ubiquitous enzymes with specific isoforms located in different subcellular compartments, and importantly also in mitochondria (PRXs III and V) (85). Several observations underline the role of PRXs as the regulators of physiologic (135) and pathologic processes, including cancer (111). Today, PRXs are considered one of the most important cell redox state–regulating enzymes. To our knowledge, the most interesting feature about these enzymes is that cancer-specific PRX overexpression appears to be present in various human primary tumors, including esophageal squamous cell, lung, and oral carcinoma, follicular thyroid neoplasms, malignant mesothelioma, and pancreatic adenocarcinoma (52, 91, 147). Preliminary data also suggest that all PRX isoforms are overexpressed in breast carcinomas compared with healthy controls (80). Interestingly, PRX II seems to have a role in the resistance to ionizing radiation in breast carcinoma cells, and PRX II antisenses could therefore theoretically be useful as radiosensitizers (167).
Redox Regulation of Gene Expression in Drug Resistance
Several lines of evidence suggest that a complex network of redox regulatory mechanisms of gene expression occur in adaptive responses to chemotherapy by tumor cells (122). Among several others, Nrf2 is certainly one of the most general and heterogeneous transcription factors involved in these processes. It has been recently proposed that the Nrf2 system exerts a dual role in carcinogenesis: it seems to promote the survival of malignant cells exposed to a hostile environment, besides also being involved in protecting cells from transformation (89). Nrf2 and its downstream gene, GSTP1, are upregulated during hepatocarcinogenesis (70); several other findings support a role for Nrf2 in cancer chemoresistance. For instance, many Keap1 mutations or loss of heterozygosity in the Keap1 locus have been identified in lung cancer cell lines, as well as in lung cancer patients, especially in those with lung adenocarcinoma (115, 118, 151). Furthermore, a recent report indicates that hypermethylation of Keap1 promoter may account for the downregulation of Keap1 in lung cancer cell lines and tissues (166). Interestingly, it has been suggested that Nrf2 may be involved in protecting cancer cells from ROS, because in a hypoxia/reoxygenation condition, which mimics a tumor microenvironment, Keap1 expression decreased, whereas Nrf2 was upregulated (84). Collectively, these results suggest that loss of function of Keap1 and a parallel activation of Nrf2 provide cancer cells with a growth advantage because of the upregulation of Nrf2 downstream genes.
Whereas the Nrf2-dependent adaptive response to counteract environmental insults has emerged as a promising strategy for cancer prevention, recent data revealed that Nrf2 is upregulated in resistant cancer cells and is probably involved in chemoresistance and, conversely, inhibition of the Nrf2 pathway during chemotherapy should be required (89). It has been reported that the pharmacologic activation of Nfr2 induces mechanisms of cell survival in response to doxorubicin, cisplatin, and etoposide (169), whereas the downregulation of Nfr2 expression by using Nfr2-siRNA restores sensitivity to several chemotherapeutic and biologic agents (123).
Based on their ability to function as antioxidants and detoxifying enzymes, many Nrf2 downstream genes have been shown to contribute to the observed Nrf2-dependent chemoresistance. HMOX-1 is an enzyme responsible for the degradation of prooxidant heme into ferrous iron, carbon monoxide, and biliverdin, which is converted into bilirubin; its end-products have antioxidant activities that are able to defend cells from oxidative stress. Therefore, activation of HMOX-1 could be involved in the antiinflammatory response and cell survival (74). The expression of HMOX-1 is up-regulated in several human malignancies, and it has been suggested that HMOX-1 may facilitate cancer cell growth and survival in many aspects, such as stimulating rapid growth, enhancing cell resistance to stress and apoptosis, promoting angiogenesis, and facilitating the metastatic process (74).
Several other Nrf2-downstream genes, such as PRX-1, GPX, TrxR, and GST, act as antioxidant or detoxifying enzymes, are upregulated in human cancer cells or tissues, and may contribute to chemoresistance (19, 84, 86, 99). GST is a phase II conjugating enzyme that detoxifies reactive electrophilic metabolites, whose expression is increased in cancer cell lines and tumors with a multidrug-resistant phenotype (99). It has been shown that the transfection of GST Pi subclass in human carcinoma cells results in the induction of resistance to doxorubicin, cisplatin, etoposide, and melphalan (9), whereas inhibition of GSH synthesis by pharmacologic agents induces a phenotype sensitive to antiblastic drugs (1). This suggests that redox-sensitive detoxifying enzymes may be critical in protecting tumor cells from exogenous molecules, as demonstrated for the MDR transporters. From such a perspective, several studies also suggest that redox signaling is critically involved in the regulation of the expression and activity of MDR transporters by multiple mechanisms, as reviewed by Kuo et al. (87). Several redox-regulated signaling pathways and transcription factors, such as the PI3K/Akt pathway, p53, Nrf2, and NF-κB, play a central role in the regulation of the expression of these transporters (87).
Heat-Shock Proteins in Tumor Drug Resistance
Most HSPs have strong cytoprotective effects and have been characterized as molecular chaperones, proteins with the property of modifying the structures and interactions of other proteins (53, 110). Inappropriate activation of signaling pathways could occur during acute or chronic stress as a result of protein misfolding, protein aggregation, or disruption of regulatory complexes. Levels of HSPs are elevated in many cancers, and HSP overexpression results in a poor prognosis in terms of survival and response to therapy in specific cancer types (26, 163). Elevated HSP expression in malignant cells plays a key role in protecting from the spontaneous apoptosis associated with malignancy as well as the apoptosis generated by therapy, mechanisms that may underlie the role of HSPs in tumor progression and resistance to treatment (26, 61).
Several indications suggest a direct correlation between HSP27 overexpression and resistance to chemotherapy in ovarian carcinoma, head and neck cancer, esophageal squamous cell carcinoma, and leukemia (26). HSP70, like HSP27, is also emerging as a predictor of resistance to chemotherapy and shorter disease-free survival in breast cancer (26, 164).
The molecular mechanisms involving HSPs in resistance to cancer therapies can be explained in several ways: (a) as molecular chaperones, they can confer cytoprotection by repairing more efficiently the damaged proteins resulting from cytotoxic drug administration; (b) protecting cancer cells from apoptosis (7); (c) protecting the microvasculature inside tumors, because HSP27 is found in endothelial cells (27); and (d) enhancing DNA repair (101).
Neutralizing HSPs is therefore an attractive strategy for anticancer therapy. The only inhibitors to have been developed are against HSP90, and they are now under clinical evaluation (Table 2). However, inhibitors of HSP70 or other HSPs would be very useful in cancer therapy alone and in combination with the inhibitors of HSP90. Several reports suggest that HSP70 or HSP27 antisense constructs have chemosensitizing properties and may even kill cancer cell lines in the absence of additional stimuli (60, 178). Interestingly, the cytotoxic effect of HSP70 downmodulation is particularly strong in transformed cells, yet is undetectable in normal, nontransformed cell lines or primary cells (142). This evidence suggests that targeting HSPs is one of the most promising approaches to anticancer therapy.
Because of our specific scientific interest in the functional characterization of the mitochondrial HSP TRAP1, especially its roles in protection against apoptosis and in chemoresistance, we hereby conclude our discussion by presenting a recently identified role of the TRAP1/HSP90 pathway in cell survival and drug resistance (77). A major advance in the knowledge of the mitochondrial HSP/TRAP1 function in chemoresistance was achieved by recent observations suggesting that TRAP1 and HSP90 are components of a mitochondrial survival pathway that is selectively activated in tumor cells and is responsible for antagonizing the proapoptotic activity of cyclophilin D (CypD), thus favoring mitochondria integrity and cell survival (77). The disruption of TRAP1/HSP90 interaction, by using the mitochondria-directed ATP-binding peptidomimetic shepherdin, induces CypD release and the consequent opening of the mtPTP (Fig. 3). This finding correlates with several other observations that suggest that HSP90 is involved in favoring resistance to antiblastic agents in human tumors. It has been proposed that, because of its restricted repertoire of client proteins, mainly kinases and signaling molecules, HSP90 chaperones occupy a critical role in cellular homeostasis (172, 177). HSP90 chaperones are required for the activity of several key regulators of apoptosis and, through these associations, may confer survival advantages to tumor cells (172). A derivative of geldanamycin, 17AAG, has been proposed for clinical use in cancer patients because of its ability to block the HSP90 pathway, by binding the regulatory pocket in the N-terminal domain of HSP90 (103). 17AAG has shown a significant antitumor activity in vitro and in animal models and, thus, has entered clinical testing in cancer patients (25). Downregulation of client proteins (i.e., Raf-1, cdk4, and Akt) has been reported in lymphocytes at well-tolerated doses of 17AAG (10), and early evidence of therapeutic activity has been described in human solid tumors by using 17AAG alone or in combination with paclitaxel (131, 153). However, recent reports suggest that 17AAG is unable to accumulate in mitochondria (77, 125): hence, HSP90 inhibition as well as cytochrome c release cannot occur, and therefore, apoptosis is not activated. The advantage of using shepherdin to block ATP binding and ATPase activities, compared with the widely used pharmacologic ATPase inhibitors 17AAG, geldanamycin, and radicicol (62, 77, 125), depends on the specificity of effects. Shepherdin is directed to mitochondria because of the Antennapedia sequence, in which TRAP1 and HSP90 are active in protecting from apoptosis (Fig. 3). Recently, a novel class of small molecules, known as gamitrinibs, were described. These agents are designed to target HSP90 selectively in human tumor mitochondria and were shown to accumulate in the mitochondria of human tumor cell lines and to inhibit HSP90 activity by acting as ATPase antagonists (78).

Our group previously demonstrated—in Saos-2 osteosarcoma cells permanently adapted to grow in the mild oxidizing conditions induced by the GSH-depleting agent DEM (43)—the upregulation of several genes and, among others, TRAP1 (105). Several recent findings obtained by our group on the protective role played by TRAP1 against apoptosis and in the induction of a drug-resistant phenotype, suggest that this chaperone seems to be involved in favoring a condition of resistance to apoptosis and antiblastic agents in human tumors (Fig. 4) and can be regarded as a suitable target for future therapeutic approaches to improve patient survival (31, 106). Saos-2 osteosarcoma cells expressing constitutively high TRAP1 levels (a) are more resistant to H2O2-induced DNA damage and cisplatin-triggered apoptosis, (b) do not release the apoptosis-inducing factor from mitochondria on cisplatin treatment, and (c) exhibit reduced activation of caspase 3 (106). In addition, we observed that TRAP1 and AROS-29, another DEM-induced gene (105), are overexpressed in human colorectal carcinomas (Table 3) and in drug-resistant colorectal carcinoma cells (31). Finally, we recently demonstrated that TRAP1 is responsible for a multidrug-resistant phenotype in human colorectal carcinoma cells (31). Accordingly, HT-29 colorectal carcinoma and Saos-2 osteosarcoma cells transfected with the TRAP1 gene exhibit a phenotype resistant to 5-fluorouracil, oxaliplatin, and irinotecan, three agents widely used in the treatment of human colorectal cancer (165), whereas both the transfection of a dominant negative N-terminal deletion mutant of TRAP1, and the inhibition of TRAP1 activity by shepherdin, increase the sensitivity to apoptosis induced by oxaliplatin and irinotecan in wild-type HT-29 colon cancer cells and in cells resistant to one or another of the agents (31). Taken together, these results suggest that TRAP1 may be regarded as a novel molecular target for overcoming drug resistance.
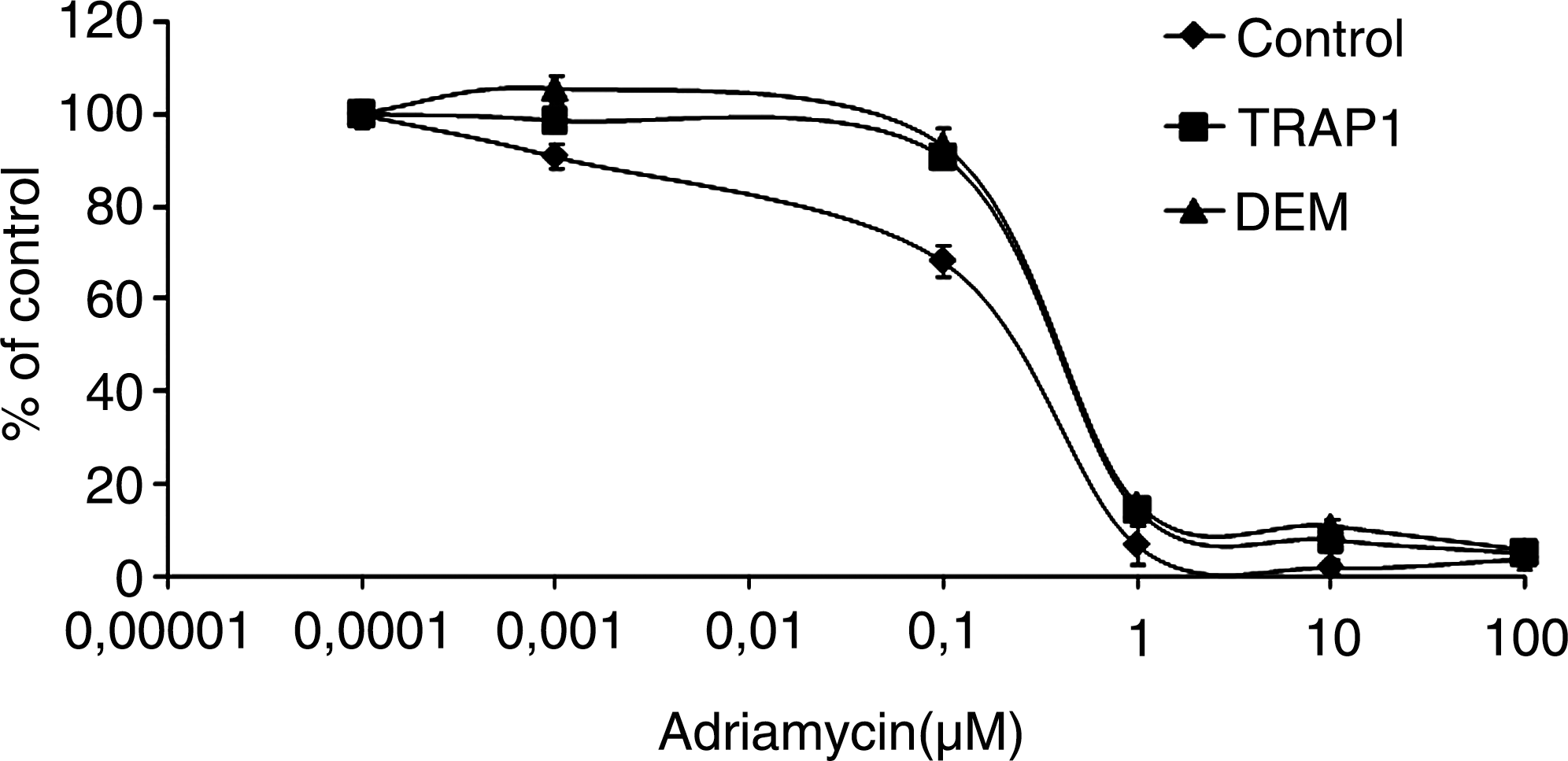
TRAP1
Statistical analysis: Pearson Chi-square test (by considering a significant cut-off value of gene expression of ± 3) p = 0.0106; Spearman Rank Correlation test (TRAP1 vs. AROS-29) p = 0.014.
Redox Signaling and Resistance to Molecular-Targeted Therapies
Molecular-targeted agents are new anticancer drugs characterized by a selectivity of action against cancer cells and a low-toxicity profile. These novel drugs, apart from their ability to block specific pathways that are selectively activated in tumor cells, are also able to synergize with traditional chemotherapeutic agents, and this synergism is largely dependent on their ability to block antiapoptotic pathways activated by TK-dependent growth factors (107). Thus, these novel molecular-targeted drugs act by enhancing the cytotoxic activity of standard chemotherapy or reversing drug resistance or both. This is confirmed by the evidence that these biologic agents are characterized by a modest single-agent anticancer activity in human malignancies, but show significant clinical activity when combined with chemotherapeutic regimens. Recent findings suggest a role for HSPs in resistance to these novel agents. This hypothesis is supported by studies showing that the activation of antiapoptotic pathways may represent potential mechanisms responsible for resistance to agents that inhibit ErbB signaling, such as trastuzumab, cetuximab, and panitumumab (34, 107). Furthermore, it has been reported that agents that block HSP90 function induce degradation of ErbB2, mutant epidermal growth-factor receptor (EGFR), and other client proteins. Interestingly, these effects correlate with inhibition of extracellular signal–regulated kinase and Akt activation, and with induction of a Rb-dependent G1 arrest with subsequent apoptosis in ErbB2-dependent cancer cell lines (23). Furthermore, inhibition of HSP90 by 17AAG induces degradation of EGFR and inhibits downstream signaling sensitizing lung carcinoma cells to paclitaxel (140) and induces the degradation of androgen receptor, ErbB2, and Akt in prostate carcinoma cells, inhibiting the growth of prostate tumors in mice (154). Recent findings also suggest a potential synergism of action between 17AAG and the anti-ErbB2 antibody trastuzumab, based on the ability of both agents to induce the recruitment of specific E3 ubiquitin ligases to ErbB2. This means that the 17AAG and trastuzumab combination could induce a higher level of ubiquitinylation and downregulation of ErbB2 as compared with single-drug treatments (130). This issue is even more relevant, considering that ROS act as second messengers, activating critical signaling pathways such as MAPK and Akt pathways, because of their ability to trigger intracellular TK and inhibit tyrosine phosphatases (24). Thus, because these signaling pathways are responsible for survival responses, it is likely that a condition of oxidative stress may contribute to deregulating key growth-factor signaling pathways, promoting cell survival, and inducing cancer cell resistance to apoptosis. It is intriguing to speculate that antioxidant agents may be synergic with TK inhibitors in reversing drug resistance.
Finally, great clinical interest has arisen from the finding that HSP90 inhibition can arrest tumor angiogenesis, the rationale being that the production of the angiogenic factor VEGF is regulated by HSP90 (175). The ability of tumor cells to produce angiogenic factors such as VEGF and to induce the formation of new blood vessels represents both a requirement for tumor growth and an important novel and valuable target for cancer therapy. The activity of HSP90 inhibitors to block tumor angiogenesis is likely caused by a decrease in protein levels and activity of the HSP90-dependent transcription factor HIF1α, which positively regulates VEGF expression (71). In addition, the protein stability of VEGF receptors requires HSP90 function, and HSP90 inhibitors induce the degradation of all three VEGF receptors (139). Thus, HSP90 function is involved in regulating the proliferation and differentiation of endothelial cells, and its inhibition may represent a valuable approach for blocking tumor neovascularization.
Concluding Remarks
Redox cellular homeostasis is a complex phenomenon, delicate and crucial for cellular well-being. A huge number of experimental results supporting the involvement of free radicals in disease development originated from numerous studies showing that an enhanced antioxidant status is associated with reduced risk of several diseases. However, the discovery of new properties and functions of ROS, beyond their toxic effects, has opened a new scenario that contributes to a different understanding of the molecular basis and the regulation of several pathophysiologic processes. In recent decades, studies have produced a wealth of information on the function of ROS as second messengers involved in the amplification and transduction of several extracellular signals. Increased ROS levels may modify the activity of several TKs, as well as inhibiting protein tyrosine phosphatase activity, further enhancing the function of the TK protein. ROS also influence gene and protein expression by activating/inhibiting transcription factors. Activation of these redox-sensitive pathways results in numerous cellular responses that, if uncontrolled, could contribute to different pathologic conditions. Therefore, given the crucial roles of most redox-regulated factors in mitogenic signals, it is plausible to assume that ROS could contribute to malignant cell growth and to cancer progression. In addition, an overwhelming number of studies support the correlations between adaptation/resistance to oxidative stress and drug resistance. A wide and overlapping variety of mechanisms have been identified as being responsible for drug resistance, such as changes in cellular responses leading to increased cell ability to tolerate stress conditions and acquiring mechanisms to escape apoptosis. The complex antioxidant network of cancer cells may be relevant in maintaining intracellular homeostasis and in favoring cell survival. In such a context, many redox-dependent mechanisms of drug resistance have been explored, mainly the upregulation of nonenzymatic and enzymatic antioxidant defenses, molecular chaperones, and stress responsive proteins, with the hypothesis that such molecules may be potential molecular targets for improving the cytotoxic effects of drug therapies and overcoming drug resistance.
It is tempting to speculate that elucidation of the exact molecular mechanisms that govern ROS-mediated regulation of cell signaling, as well as providing a better understanding of the molecular mechanisms underlying redox adaptive responses, may lead to an improvement in cancer treatment, risk assessment, and risk-management strategies, and will provide the basis for the development of new therapeutic strategies for cancer prevention and treatment. Based on the evidence that tumor cells activate multiple and interacting redox mechanisms of resistance to apoptosis, it is intriguing to speculate that therapeutic strategies aimed at blocking these redox-dependent pathways should take into account the complexity of such mechanisms and may need to modulate/inhibit multiple redox-dependent targets. Finally, great clinical interest is focused on novel molecular-targeted tumor-specific agents and on the role of redox signals in resistance to these molecular-targeted therapies. These studies, although still preliminary, provide critical information for the understanding of the molecular mechanisms involved in the resistance to molecular-targeted agents. Furthermore, an extremely innovative field is the design of novel multitarget strategies based on the simultaneous inhibition of redox-regulated signals and other tumor-specific pathways, with the aim of reversing drug resistance and improving antitumor activity.
Footnotes
Acknowledgments
This work was supported by a grant from MIUR-PS 35-126/Ind, grant 26/08 from Lega Italiana per la Lotta contro i Tumori, and grant 2009 from Fondazione Berlucchi. The authors thank Anthony Green for proofreading the manuscript and suggesting stylistic improvements.