
Abstract
Reactive oxygen species (ROS) were seen as destructive molecules, but recently, they have been shown also to act as second messengers in varying intracellular signaling pathways. This review concentrates on hydrogen peroxide (H2O2), as it is a more stable ROS, and delineates its role as a survival molecule. In the first part, the production of H2O2 through the NADPH oxidase (Nox) family is investigated. Through careful examination of Nox proteins and their regulation, it is determined how they respond to stress and how this can be prosurvival rather than prodeath. The pathways on which H2O2 acts to enable its prosurvival function are then examined in greater detail. The main survival pathways are kinase driven, and oxidation of cysteines in the active sites of various phosphatases can thus regulate those survival pathways. Regulation of transcription factors such as p53, NF-κB, and AP-1 also are reviewed. Finally, prodeath proteins such as caspases could be directly inhibited through their cysteine residues. A better understanding of the prosurvival role of H2O2 in cells, from the why and how it is generated to the various molecules it can affect, will allow more precise targeting of therapeutics to this pathway. Antioxid. Redox Signal. 11, 2655–2671.
Introduction
It is not just the source of H2O2 that is important when considering its role as a survival molecule; it also is important to understand how it interacts with its targets molecules. The main way that H2O2 affects cell-signaling pathways is through oxidation of specific target molecules. If the concentration of H2O2 is high, then these oxidation processes may lead to irreversible damage, followed by cell death. However, this is not always the case, and H2O2 at low concentrations is capable of reversible inhibition of many enzymes, including phosphatases, which are the second focus of this review. We examine Nox proteins as a source of H2O2 and the oxidation of phosphatases to demonstrate how H2O2 can act as a survival molecule.
The Nox family
Seven Nox family members in mammals are known with at least 101 orthologues across 25 species (71). The seven members are Nox1-5, and dual oxidases (Duoxs) 1 and 2. Nox2 is the prototype member of this family and originally was called gp91 phox . It is highly expressed in neutrophils and macrophages, in which it produces an oxidative burst to destroy pathogens. This burst occurs when these cells engulf microbes, containing them in a phagosome, and was characterized first in the early 1960s (61). It was not until the mid-1980s that the gene responsible for the burst was cloned (128, 150). Activated Nox2 in the membrane of the phagosome produces high levels of O2 •−, which is converted to H2O2, either through the action of SOD, or spontaneously at the low pH of this organelle (75). The O2 •−/H2O2 can be in the micromolar concentration range in the phagosome (57), which is lethal for the engulfed microbe. Understandably, the production of this high level of H2O2 is strictly regulated to avoid damaging the cell itself, and this high activity level allowed the regulatory mechanisms of Nox2 to be studied comprehensively.
Nox1 was the next member to be discovered and was first cloned in 1999 (143). It is highly expressed in the colon (21), with no expression in Nox2-rich phagocytes (143). In the following 5–6 years, genetic database mining and subsequent cloning led to the discovery of the other family members. Nox3 is expressed predominantly in the octonia of the inner ear (9), and it also is found in some fetal tissues (21). Nox4 is found in many systems, with high expression in the kidney (47, 138). It can have different subcellular localizations, which can affect its function (19). Nox5, highly expressed in testes, spleen, and lymph nodes, stands apart from Nox1 to 4, as it is calcium dependent (7). Duox1 and Duox2 are found mainly in the thyroid and in airway epithelium (32), have an extracellular peroxidase-like domain, and also are calcium dependent.
Nox proteins are integral membrane proteins, with six transmembrane domains, which together are thought to form a channel to allow the successive transfer of electrons (for a review on the structure of Nox proteins, see ref. 146) (Fig. 1). They operate by transferring electrons from NADPH (converting it to NADP−) to FAD to heme and finally to oxygen to make O2 •−. This transfer is dependent on the presence of two heme groups associated with the inner and outer cellular membrane envelope. Two His residues, in two of the transmembrane domains, are responsible for binding the hemes, and these are the most conserved regions of the Nox family. Nox1, - 2, - 3, and - 4 are similar in sequence and only differ in length by 14 amino acids, the shortest being Nox1, which is 564 amino acids in length (143), and the longest, Nox4 of 578 amino acids (138). Their amino acid homology ranges from 56% (Nox1 and Nox2), and 58% (Nox2 and Nox3), to 39% (Nox2 and Nox4) (145). Nox5 is significantly different because of its four EF domains, which confer its dependence on calcium for activation (7, 10) (Fig. 1). Duox1 and 2 both have a peroxidase-like domain on their N-terminal (36), which means that these two members of the family produce H2O2 and not O2 •− as their final product (48, 106, 142). They also have two EF domains and so are also calcium dependent (28, 33) (Fig. 1). With the discovery of the other members of the Nox family, it became apparent that they do not always produce the lethal levels of O2 •−/H2O2 characteristic of Nox2 in neutrophils. Because of their different locations, both within cells and within tissues, this group of proteins are now known to affect many cellular functions including growth, motility, and cell survival.

The regulation of Nox proteins
Nox subunits
The activity of the best known Nox, Nox2, is dependent on its regulatory subunits, reviewed in depth by (84, 146) and summarized here. Normally, Nox2 is found at the cytoplasmic membrane with its essential partner p22 phox , and together, they form a complex called cytochrome b558 . In the resting cell, p47 phox , p67 phox , and p40 phox form a large complex in the cytoplasm (62, 163), and it is only when the cell is stimulated that they are recruited to cytochrome b558 at the membrane (26) (Fig. 2). During cellular stimulation, p47 phox is phosphorylated, removing its autoinhibition (60). This phosphorylation is correlated with Rac (Rac2 in myeloid cells or Rac1 in other cell types) recruitment to this complex, and together these four proteins translocate to cytochrome b558 in the membrane, resulting in the production of O2 •− and H2O2. The interactions between the complex members are determined by their structures (Figs. 2 and 3). p47 phox binds p22 phox (90) and p67 phox (62). In turn, p67 phox also binds Rac (78, 85). Some doubt exists over the necessity of p40 phox in vitro and in vivo (41, 79), but in vivo, p47 phox remains essential for Nox2 activation (95). In the Nox1 system, homologues exist for two of these subunits. NOXO1 is homologous to p47 phox , and both are seen as Nox-organizing proteins. NOXA1 is homologous to p67 phox , and both are the activating proteins. To date, no homologues to p40 phox or p22 phox have been discovered. In different cell systems, these regulatory proteins were found to be interchangeable between Nox1 and Nox2 (8, 149). It remains unknown what actually happens to the Nox protein when all the subunits arrive and interact with it. It is assumed that it leads to a conformational change in the protein, opening up fully the channel formed by the six transmembrane domains, and so allowing the flow of electrons and the generation of O2 •−. None of the seven Nox family members have been crystallized yet, so their actual structures have yet to be solved, but models of their structures support this hypothesis (146).
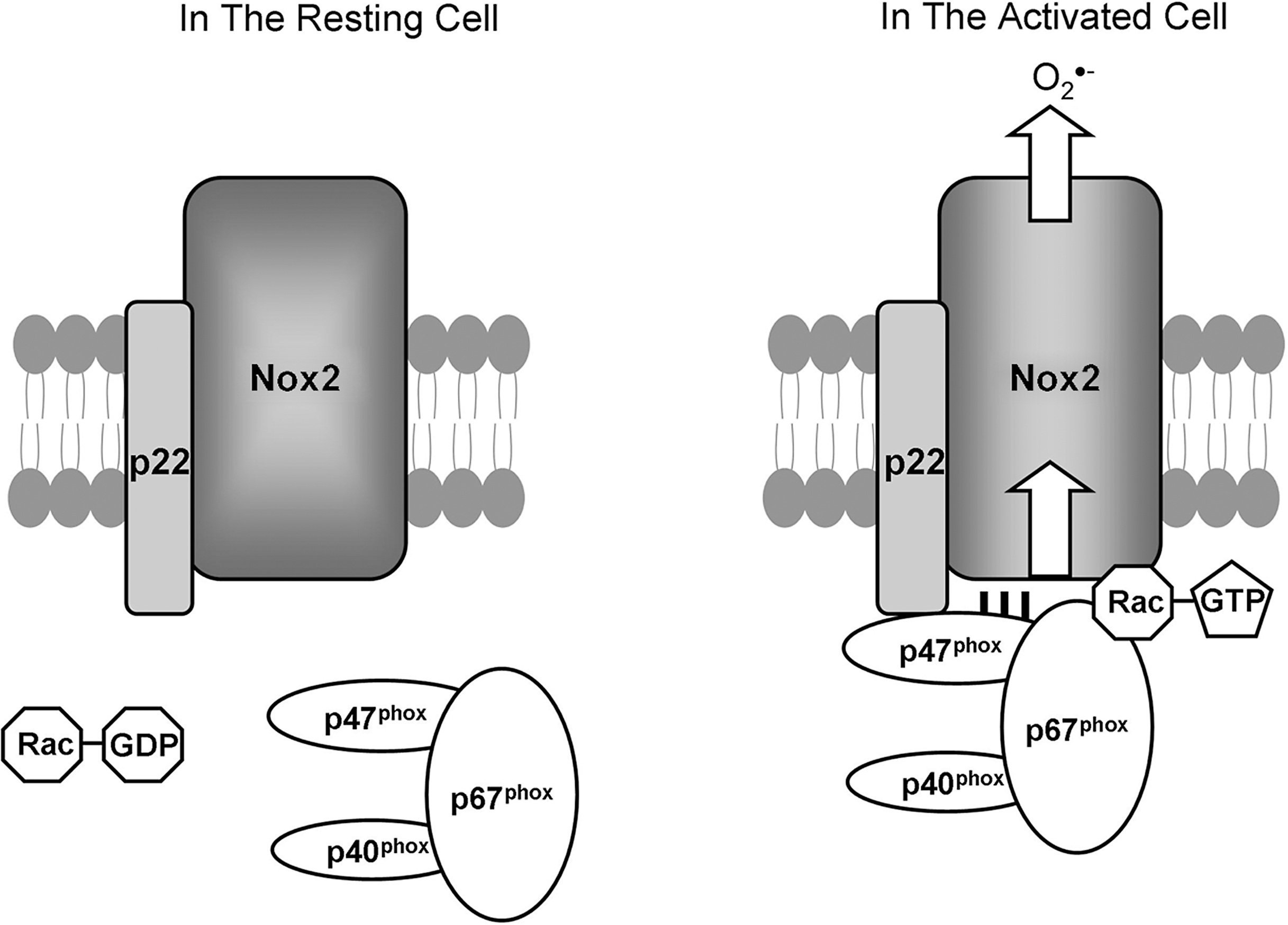
The regulation of Nox3, Nox4, Nox5, Duox1, and Duox2 has been less comprehensively investigated. Nox3 requires co-expression of p22 phox alone to generate H2O2, and this can be enhanced by both p47 phox and p67 phox , but Rac is not required in either of these situations (156), which is somewhat at odds with data showing that Nox3 has a Rac-binding site (67). Nox3 appears to use NOXO1 as a signaling partner, as mice knockouts of NOXO1 have the same phenotype as that of Nox3 knockouts (74). Nox4 generates O2 •−/H2O2 constitutively when it is stabilized by the coexpression of p22 phox (4). None of the other known regulatory subunits appears to play a role in Nox4 generation of O2 •−/H2O2 (99). Nox5, in contrast to the first four family members, does not require any of the known regulatory subunits, including p22 phox (70), and its activity is dependent on calcium (7, 9). Duox1 and Duox2 also are activated by calcium and have yet to show any reliance on the known regulatory subunits. Two maturation factors are associated with Duox proteins, DUOXA1 and DUOXA2, and they are necessary for the full maturation and stabilization of the two Duox proteins (53), but not their activity per se. It remains unknown whether other homologous and nonhomologous Nox regulatory subunits could play a role in the activation of these newer family members.
Upstream regulation of Nox proteins
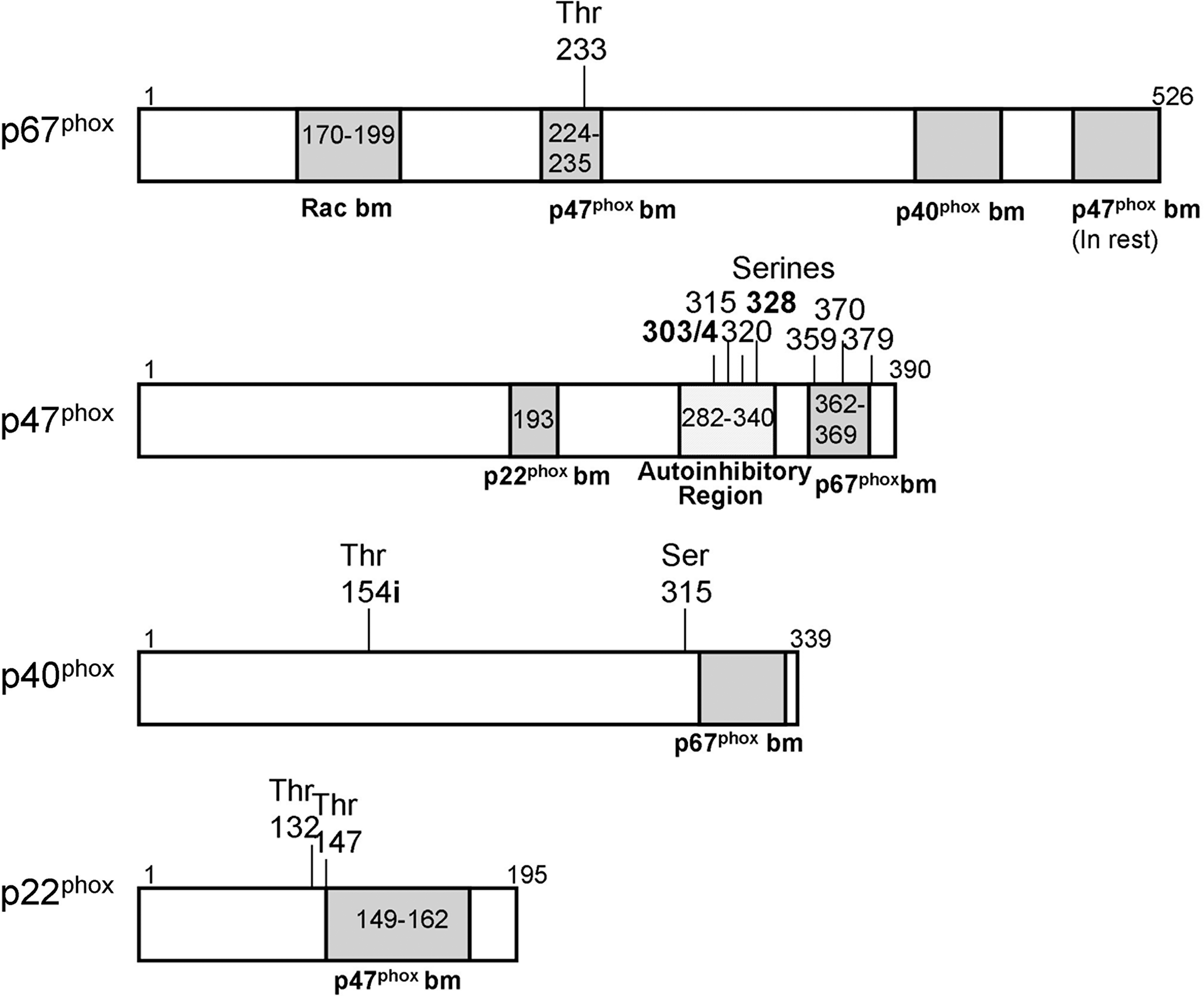
Although the preceding paragraphs detailed the immediate regulation of the Nox family members, a wider view must be taken to understand how H2O2 can operate as a survival signal through Nox proteins. For H2O2 to act as a survival signal, it must be generated when a cell is stressed. Decoursey and Ligeti (29) summarized the known upstream regulation of Nox2, but they focused on this particular family member, and as this is a developing area of Nox biochemistry, more work has been published since then on it and the other family members. They considered the phosphorylation status of p47 phox and the activity of Rac to be the key determining factors, and although p22 phox (124, 125), p40 phox (15, 42), and p67 phox (34, 38) also can be phosphorylated (Fig. 3), their phosphorylation statuses are not thought to play a pivotal role in Nox activity.
When p47 phox is phosphorylated, its autoinhibition is removed, resulting in its being able to bind to the cytoplasmic tail of p22 phox and allowing activation of the entire Nox complex (54, 115). Phosphorylation is generally under the control of kinases, and protein kinase C (PKC) has been shown to be critical for Nox2 activity in neutrophils (27, 123) and also can activate Nox1 (20, 22) and Nox3 (156) (Fig. 4). Two other kinases have been shown to phosphorylate p47 phox : p38 mitogen-activated protein kinase (89) and p21-activated kinase-1 (127). But what activates these kinases in the case of Nox proteins? Phospholipase A2 (PLA2) activation causes the release of arachidonic acid, which is known to activate PKC directly (72), and the inhibition of PLA2 inhibits Nox activity (2, 152). Recently, Kambayashi et al. (65) showed that diacylglycerol, produced through the action of phospholipase C, can activate Nox through its well-known activation of PKC. Oxidative stress can cause a direct activation of some types of PKC (11), but this has not yet been linked to Nox activity. Many different ways can be used to stimulate PKC and the other kinases capable of activating Nox, so it is highly unlikely that one single pathway exists between stress stimuli and Nox activation, which would lead to H2O2 production. This also shows that many ways exist to activate Nox proteins, and so illustrates that possibly many different types of stresses exist to which Nox could respond.
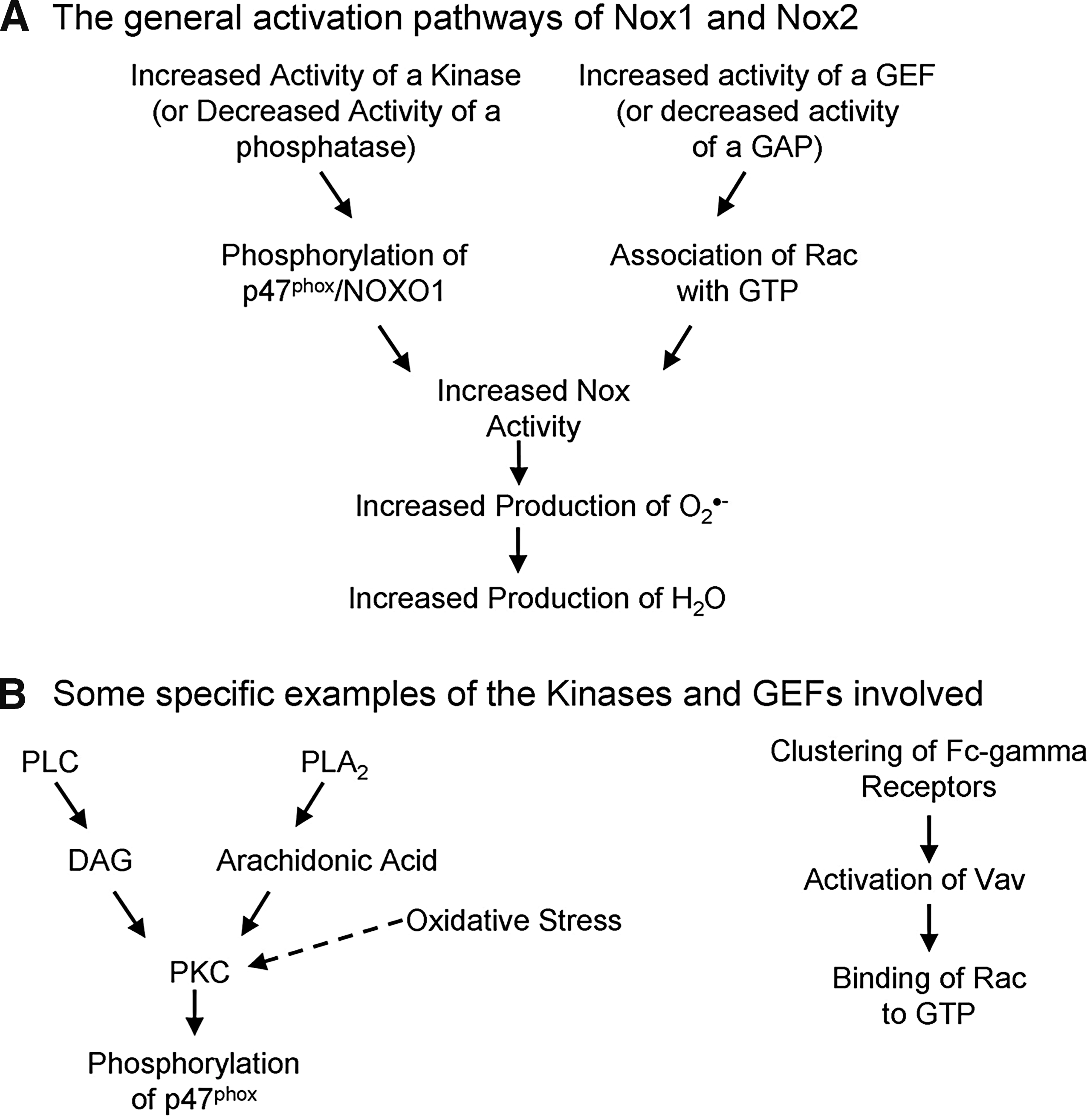
The second key determinant of Nox activity, according to Decoursey and Ligeti (29), is the status of the Rac associated with the complex (Fig. 4), but so far this is proved for only Nox1 and Nox2. Rac is a small RhoGTPase and can be bound to guanosine diphosphate (GDP), making it inactive, or to guanosine triphosphate (GTP), making it active. Two groups of molecules control which of these is bound to Rac, guanine nucleotide exchange factors (GEFs), which promote the exchange from GDP to GTP, increasing the activity of Rac, whereas guanine-activating proteins (GAPs) do the opposite and switch it off. Seventy different GEFs and 80 GAPs are expressed in various cells (45), and so these might be quite cell-type and stimulus-type specific in their control of Nox activity. For example, Vav1 is upstream of Nox2 signaling in a murine microglial cell line (127), whereas Tiam1 plays a role in Nox-mediated signaling in keratinocytes (129), and βPix1 can bind to Nox1 and is involved in its activation (113, 114). Recently, Umoto et al. (157) showed that the clustering of Fc-γ-receptors on neutrophils resulted in the activation of Vav, and therefore also in Nox2 activation, illustrating nicely the role of GEFs in Nox activation.
Rac is known to play a role controlling actin through the formation of membrane ruffles (108); therefore, it might be that the activation of Rac binds it to p67 phox and also results in actin changes, thereby moving the entire complex to the membrane and inducing Nox activation. Rac activity is not necessarily the main or only way in which the complex could translocate, as both p47 phox and p40 phox bind moesin, which is an actin-binding protein, and so could also be responsible for the translocation (164). It has been shown that the phosphorylation of p47 phox and the activation of Rac are required in tandem for the maximal production of H2O2 through Nox2 (1) and Nox1 (20), and one publication even linked Vav to PKC(theta) activation (161), implying that it could have a dual role in Nox activation. Because of the large number of GEFs and GAPs, and the variability in their structures, it is difficult to pick out any common traits that might show which of them respond to specific stress stimuli and to determine how they would do so. This large number could also allow specific targeting of certain Nox proteins under certain conditions by therapeutics.
As mentioned earlier, little evidence exists of p47 phox or Rac playing a role in regulating Nox4 or Nox5 or Duox1/2 (40). This is reinforced by the recent data of Kao et al. (67), which showed that a conserved Rac binding site is found on Nox1, Nox2, and Nox3, but not on the others. For Nox5, Duox1 and Duox2 calcium triggers their generation of O2 •− (7, 28, 33). In the case of Nox5, when calcium binds to the protein, the N-terminal undergoes a large conformational change, presumably then allowing the transfer of electrons (10). Calcium concentrations in the low micromolar range were enough to trigger activation of Nox5 (10) and Duox1 and Duox2 (5, 105). Therefore, any stimulus that increases intracellular calcium concentrations is likely to result in increased O2 •−/H2O2 production. Nox5 may also be phosphorylated, during which it becomes more sensitive to calcium (63), and this phosphorylation may be under the control of PKC (135) or of c-Abl (37). When calmodulin binds to the opposite end of the molecule from the calcium-binding domains, sensitivity to calcium increases (151). A recent publication showed an interaction between Duox1 and NOXA1 and that this interaction is inhibitory of activity (111). Nox4 stands out as being constitutively active when expressed with p22 phox (47, 99) and capable of stimulation to produce extra O2 •−/H2O2 (51, 81, 112), but as yet very little is known about its regulation and how it could be activated by a stress stimulus, even though it has been linked to several different signaling pathways in cells involving different stresses. So although many structural similarities are found between the Nox family members, the differences in their regulation make it highly unlikely that some overriding controlling factor exists for all Nox proteins and hence O2 •−/H2O2. Conversely, it probably means that they can be stimulated by a variety of stressors and respond in subtle ways to varying therapeutic treatments.
When discussing H2O2 as a survival molecule, it is important to know how quickly the Nox signaling complex can be deactivated, as this determines the amount of H2O2 produced. The deactivation depends mainly on the dephosphorylation of p47 phox , typically accomplished by phosphatases and the binding of Rac to GDP, through the activity of GAPs. Although much work has shown that phosphatases are important (for review, see ref. 29), little progress has occurred in identifying which specific phosphatase(s) is involved, as most of the studies to date have used general phosphatase inhibitors. It is likely that protein phosphatase 1 and protein phosphatase 2B or both are involved (14). The second step is the nature of the nucleotide bound to Rac, which is controlled by GAPs, but little is known about how these interact with Nox2 (59, 148), and nothing with Nox1. Other regulatory mechanisms of Nox proteins are being discovered. A recent publication demonstrated that the binding of NOXA1 to 14-3-3 proteins results in the inhibition of Nox1 activity (73), and it is likely with time that more of these types of interactions will be revealed. It is assumed that once the calcium levels in cells decrease, the activity of Nox5, Duox1 and Duox2 also decreases. Again very little is known about the deactivation of Nox3 and Nox4. Overall the deactivation of the Nox signaling complex remains an underinvestigated area in the Nox field, but it could prove to be very important when trying to control the concentration of H2O2, and its possible effects, in cells.
In comparing studies on the prodeath against the prosurvival effects of Nox proteins, it becomes apparent that the final outcome on the cell is probably due to several factors (e.g., the amount of O2 •−/H2O2 produced, the location of the O2 •−/H2O2 production, and the duration of O2 •−/H2O2 production). It is widely believed that a shorter/smaller production of H2O2 is prosurvival, whereas longer or larger amounts (or both) would be prodeath, and this was proved in a p53 system (130), but not yet for Nox proteins. One way in which H2O2 probably acts as a survival factor is through preconditioning, which relies on a system receiving a smaller stimulus before it experiences a major stress. A well-known example of this is ischemic preconditioning, in which the heart is stopped for a few times of short duration before it is stopped for a number of hours as a person goes onto a bypass machine during heart surgery (104). Nox activity has been shown to act as a preconditioning factor in a model of ischemic preconditioning (133). For a preconditioning stimulus to work, it must be of a similar nature to the main one, so it is likely that this prosurvival effect of H2O2 generated by Nox proteins is true only when the cell is under oxidative stress. We recently showed that this is the case in retinal cells, as a prosurvival burst of H2O2 occurs only when these cells are treated with an oxidative stress, and not with other types of stressors (96).
Other sources of ROS
As mentioned in the opening paragraph of this review, Nox proteins are not the only enzymes capable of forming ROS. The mitochondria are the organelles with the largest oxygen-consumption rate within cells and hence the largest producers of ROS, but because of their many redox systems, they contribute little to the overall redox status of cells (141). Only if mitochondria malfunction do they tend to play a role in the oxidation status of proteins elsewhere in the cell. Nonmitochondrial sources of ROS other than Nox proteins are known, but their biochemistry is less well studied, and so their relevance to survival signaling remains unknown. Overall, they tend to be expressed by specific cell types only. For example, xanthine oxidase is expressed mainly in the liver and epithelia and can produce O2 •− and uric acid from xanthine and oxygen (25). Lipoxygenases, cyclooxygenases, and nitric oxide synthases tend to produce ROS indirectly, through the formation of reactive intermediates, when they form their main respective products of leukotrienes (80), prostaglandins (144), and nitric oxide (117). These intermediates can then interact with oxygen to form ROS. Nitric oxide synthase, in certain circumstances, may be uncoupled, and then it generates O2 •− directly (160). The smaller space allocation in this review to these many other enzymes capable of generating ROS reflects simply the paucity of knowledge about them, particularly in relation to cell-survival signaling, but does not reflect their possible importance to this key cellular pathway.
H2O2-driven cell-survival pathways
Up to this point, the main sources of H2O2 in cells have been outlined, with particular focus on Nox proteins. To understand how H2O2 can act as a survival molecule, it is important to understand how it can interact with its target molecules, and this is the focus of the remaining sections. The scientific evidence to date is in two separate halves, reflected in the dichotomy of this review, with one half showing that Nox proteins can produce ROS, and the other demonstrating that ROS can promote cell-survival signaling. A limited number of articles bridge this gap and show precisely how Nox proteins can promote cell survival, and these are highlighted in the next section. However, all possible ways that H2O2 can interact with prosurvival downstream targets are mentioned, as it is likely that Nox-derived H2O2 can play a role in these events.
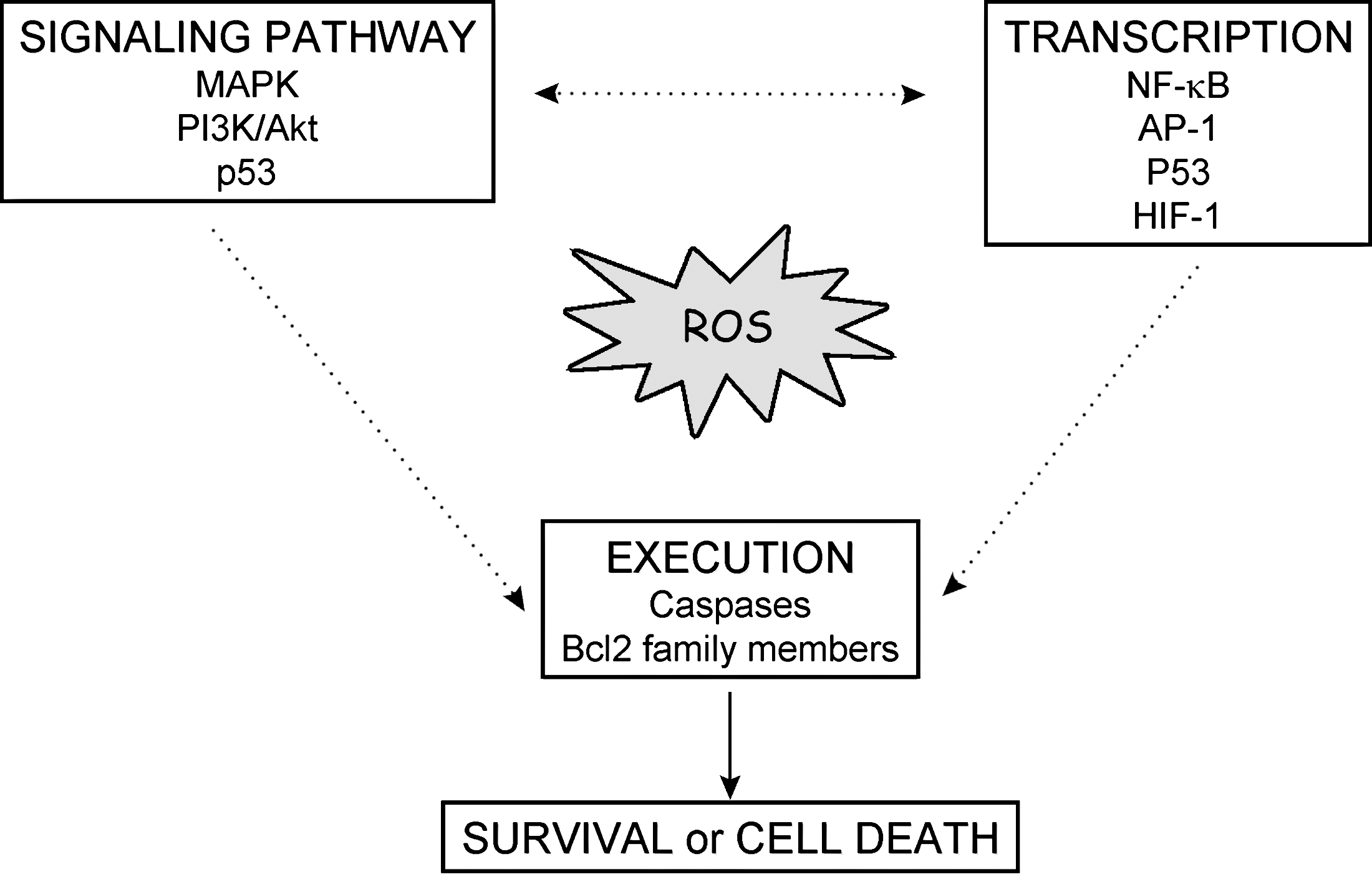
H2O2 affects cell-signaling pathways mainly through oxidation of specific molecules. If the concentration of H2O2 is high, then these oxidation processes may lead to irreversible damage, followed by cell death. However, this is not always the case, and H2O2 is capable of reversible inhibition of many proteins (i.e., phosphatases) along the main activation cascade of survival pathways (Fig. 5). First we provide a quick overview of the different survival pathways and how they can be redox regulated by phosphatases.
Key survival pathways
Mitogen-activated protein kinases (MAPK) constitute a large kinase network that regulates physiologic processes such as cell growth, differentiation, and apoptosis. Three MAPK pathways have been characterized: the extracellular signal-regulated kinase (ERK) pathway, the c-Jun N-terminal kinase (JNK)/stress-activated protein kinase, and the p38 pathway. MAPK operates in a cascade fashion with MAPKKK phosphorylating and activating MAPKK, which then activates MAPK. Generally ERK1/2 signaling promotes cell survival under mild oxidative stress, whereas JNK and p38 can induce apoptosis by involving direct phosphorylation of pro-/antiapoptotic Bcl-2 family members, transactivation of transcription factor AP-1, and stabilization of p53.
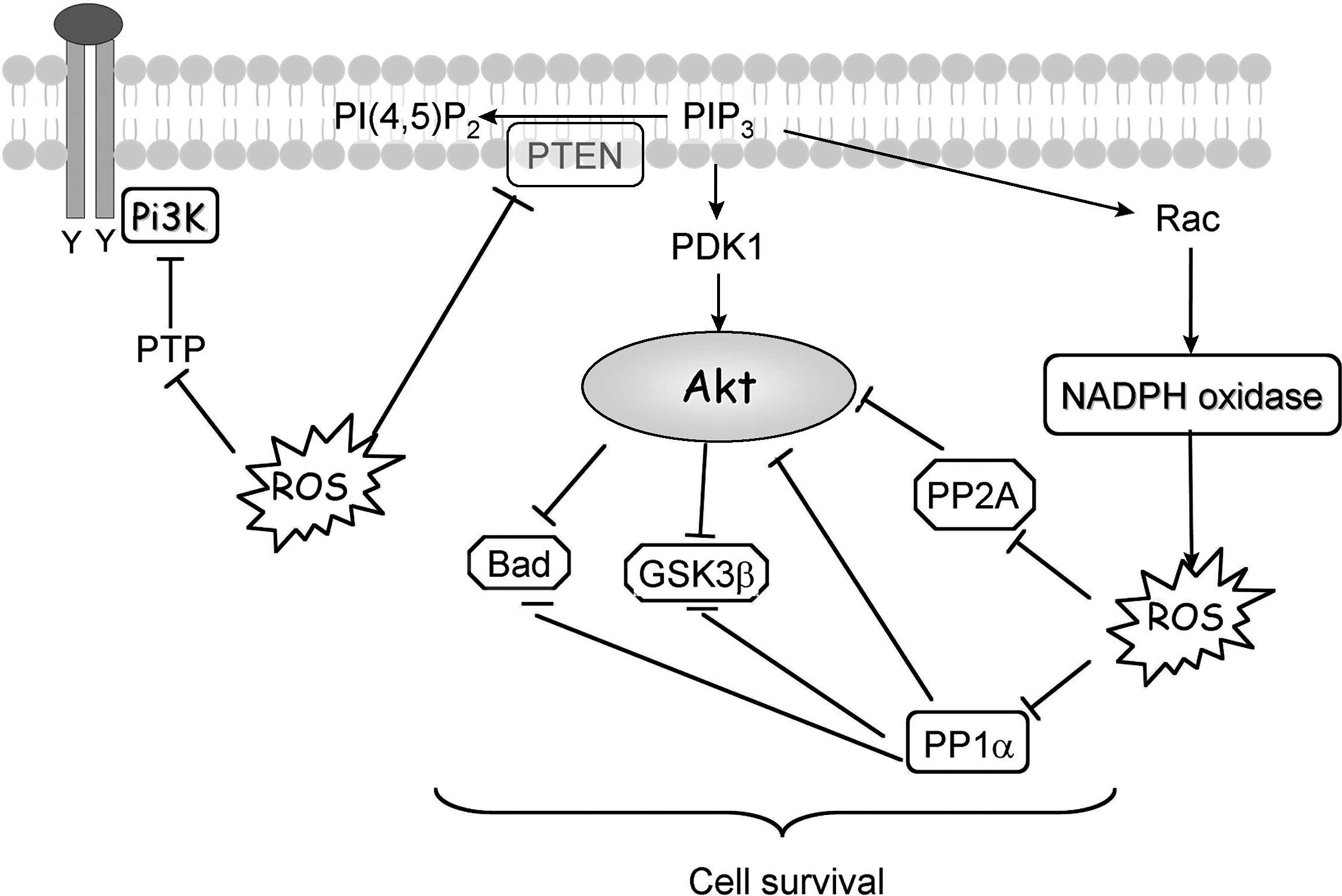
Signal transduction via phosphoinositide 3-kinase (PI3K) plays an important role in regulating proliferation, survival, and motility. A moderate level of ROS activates PI3K signaling and promotes cell survival. PI3K/protein kinase B (Akt) transduces the signal for cell survival mainly through phosphorylation of target molecules by Akt. This results in inactivation of proapoptotic proteins and activation of transcription factors that target the expression of antiapoptotic proteins.
Reliance on extracellular matrix (ECM) contact for cell survival is common in untransformed adherent cells; apoptosis induced by loss of ECM attachment is called anoikis and is a redox-regulated event (for a review, see ref. 24). Redox regulation of epidermal growth factor receptor (EGFR) signaling plays a crucial role in the protection from anoikis, as is shown in different cell types (49, 86, 119). During cell spreading, integrin activation through ECM-cell contact induces a significant increase in the intracellular level of ROS. This increase in ROS level is dependent on the activity of small GTPase Rac-1 (49), which could suggest that Nox1 or Nox2 is involved. The tyrosine kinase Src is the main target of the ROS and is consequently the main player in the crosstalk between ECM contact and generation of prosurvival signal. When Src is oxidized and fully activated, it allows the specific ligand-independent phosphorylation and activation of EGFR, downstream of which ERK and Akt signaling pathways are activated. They will eventually induce the phosphorylation of the proapoptotic protein Bim, which is then degraded, leading to the inhibition of apoptosis (116).
The survival pathways transduce their survival signal through phosphorylation of key proteins, as a consequence of which phosphatases are potent negative regulators, and, in particular, multiple steps along the PI3K/Akt survival pathway are negatively regulated by phosphatases.
Negative regulation by phosphatases
The phosphatase superfamily can be categorized into three smaller subfamilies based on substrate specificity: (a) the classic protein phosphatases, which are tyrosine specific (protein tyrosine phosphatases, PTPs); (b) the dual-specificity phosphatases (DSPs) which can dephosphorylate phosphotyrosine-, phosphoserine-, and phosphothreonine-containing substrates; and (c) serine/threonine (Ser/Thr)-specific phosphatases, which are further divided into two major classes (Fig. 6). Type I phosphatases include protein phosphatase (PP) 1; type II phosphatases include the spontaneously active phosphatases (e.g., PP2A), calcium-dependent phosphatases (e.g., PP2B), and magnesium-dependent (e.g., PP2C) classes of phosphatases.

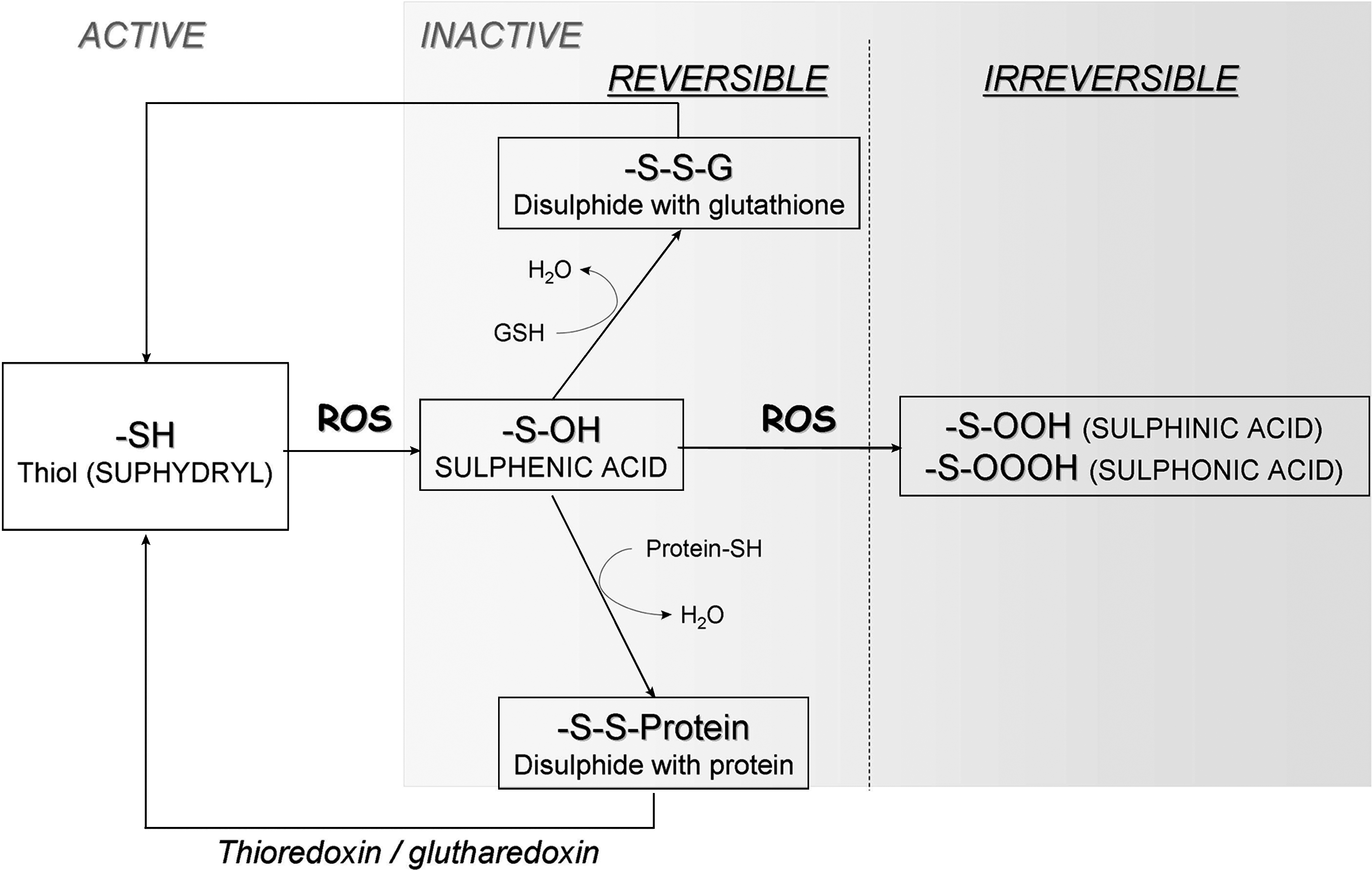
The regulation of cell function through redox-sensitive cysteine residues has been most convincingly demonstrated in PTPs. They are encoded by 38 genes in humans and belong to a larger family of cysteine-dependent phosphatases that totals approximately 100 human members and numerous pseudogenes (for review, see ref. 3). All PTPs contain an essential cysteine residue in the signature motif C(X)5R that exists as a thiolate anion at neutral pH (30). This thiolate anion contributes to the formation of a thiol-phosphate intermediate in the catalytic mechanism of PTPs. Oxidation of the active-site cysteine of PTPs to a sulfenic derivative leads to enzymatic inactivation; however, this modification can be reversed by incubation with thiol compounds (Fig. 7). In some cases, the sulfhydryl group is open to further irreversible oxidation if no cysteine derivatives or thiols are close enough to facilitate the formation of a disulfide bridge. The addition of another oxygen molecule or two additional oxygen molecules results in the formation of sulfinic and sulfonic acid, respectively. These oxidative modifications are irreversible, and the phosphatase will be unable to become active again, even in a reducing environment (Fig. 7). It is highly likely that all phosphatases are sensitive to oxidative inhibition to some degree, as they all require a reduced cysteine for catalysis. Reversible oxidation was first demonstrated for PTP-1B during epidermal growth factor (87) and insulin (97) signaling. The same regulation was then demonstrated for low-molecular-weight PTP (LMW-PTP) during platelet-derived growth factor stimulation (23). Both PTP-1B and LMW-PTP rescued their phosphatase activity because of a re-reduction 30 min after receptor activation (12, 18). Very few articles in the literature are able to make a direct link between oxidation of PTP and Nox; nevertheless, it has been reported that the Src-homology 2 domain (SHP-2) PTP is reversibly oxidized in response to PDGF, and that requires association with the PDGFR (100). Because PDGF is involved in Nox1 regulation (69), it is likely that Nox is at least indirectly responsible for SHP-2 redox inactivation. More recently Sharma et al. (136) showed that IL-4 receptors can activate Nox1 and Nox5, thereby increasing ROS production, and consequently inactivate PTP-1B, which physically associates with and deactivates IL-4 receptors (136), thus revealing a role for ROS in cytokine crosstalk.
The dual specific phosphatases (DSP) also can be oxidized. In contrast with the classic PTP member like PTP-1B that forms a sulfenyl amide linkage between the active-site cysteine and an adjacent main-chain nitrogen (131), oxidation of phosphatase and tensin homologue (PTEN) in vitro with H2O2 leads to the formation of a disulfide bond between the active-site cysteine (cys-124) and another cysteine residue (cys-71) that is close by in its three-dimensional structure. This disulfide bond prevents further irreversible oxidation of the cysteine residues and PTEN can retain its phosphatase ability. PTEN has been demonstrated in many studies to be the main phosphatase negatively regulating the PI3K/Akt pathway. PTEN can dephosphorylate the lipid PIP3 to PIP2, preventing the recruitment of PH-containing proteins to the plasma membrane. This results in a decrease in the survival signal transduced by Akt. The importance of PTEN in regulating this pathway is highlighted by its classification as a tumor suppressor. Moreover, Savintsky et al. (134) provided evidence for the degradation of cdc25 phosphatase (another DSP), which is the result of H2O2 -induced disulfide bond formation between the active-site cysteine and another invariant cysteine residue.
The Ser/Thr phosphatases also are sensitive to redox modifications. It was demonstrated in vitro that H2O2 can reversibly block some of the main Ser/Thr phosphatases, such as PP2A, and PP1α (110, 122). PP1 and PP2A contain redox-sensitive cysteine residues (39, 55). Structure-based analysis has identified a potential disulfide oxidoreductase active site, Cys-X-X-Cys, in members of the PP1 subfamily (39). It is still not clear whether the oxidation of this pair of cysteine residues can result in PP1 inactivation in vivo. The Ser/Thr phosphatases (the main members being PP1, PP2A, PP2B, and PP2C) dephosphorylate serine and threonine, which are the main phosphorylation events in the transduction of the PI3K/Akt survival pathway. Various studies demonstrated links between these phosphatases and the PI3K/Akt pathway. For example, the calcium-activated PP2B has been shown to be a direct phosphatase of Akt, Gsk-3β, and Bad (76, 88, 165) (Fig. 8). PP1α has been shown to dephosphorylate Akt and Bad, while PP2A has been demonstrated to be a key Akt phosphatase. PP2A can dephosphorylate Akt on both threonine 308 and serine 473, blocking the PI3K/Akt pathway (Fig. 8). Immunoprecipitation studies have shown that PP2A can colocalize with Akt. Recently, a novel phosphatase belonging to the PP2C family of phosphatases was shown to dephosphorylate Akt on serine 473 only. This novel phosphatase, PHLPP, contains a PH domain that localizes it in the vicinity of activated Akt. Expression of PHLPP in cells derived from a small cell lung cancer that have constitutively active Akt (phosphorylated on serine 473 and threonine 409) resulted in a 50% decrease in phosphor-serine 473 Akt levels. This resulted in a correlating decrease in the phosphorylation levels of an Akt substrate and an increase in the basal levels of apoptosis in this cell line (44). These experiments highlight the importance of the phosphorylated serine residue in Akt signaling and demonstrate a clear link between the increased activity of PHLPP with an induction of apoptosis via the PI3K/Akt pathway. Inactivation of PP2A was shown in cells treated with tumor necrosis factor-alpha (TNF-α) or interleukin-1 (55), which can both induce H2O2 production. TNF-α has been shown to regulate Nox1, Nox4 (168), and recently NOXO1 (83) and IL-1 were shown to enhance Nox2 expression, which can provide a direct link between Nox-generated ROS and inactivation of PP2A.
Activation of transcription factors
Abundant evidence exists for the regulation of gene expression by ROS. Nox-dependent ROS have been shown to induce, for example, the expression of TNF-α (118), TGF-β1, and angiotensin II (56). Most studies investigating the mechanism of ROS-upregulated mRNA have shown that the upregulation can occur through redox-sensitive second-messenger systems [MAPK activation (50)] or through transcription factors including NF-κB, hypoxia-inducible factor (HIF)-1α, p53, and AP-1, which contain redox-sensitive cysteine residues in their DNA-binding domains (147). In most cases, thiol oxidation of these proteins inhibits their DNA-binding activities.
p53
p53 is a transcription factor as well as a tumor-suppressor gene. Recent work demonstrated how the redox potential of a tumor cell can be elevated during carcinogenesis, as a consequence of the loss of wild-type p53. However, under normal physiologic conditions, p53 was recently shown to be able to reduce the redox potential within a cell by regulating the expression of antioxidative genes such as glutathione peroxidase, SOD2, and the mammalian sestrins sestrin1 and sestrin2 (sestrins are involved in the regeneration of oxidized peroxiredoxin). This differential control of redox levels within a cell is attributed to the levels of p53 itself. Thus, low levels of p53 suppress ROS within the cell whereas higher levels of p53 promote ROS accumulation. Studies in p53−/− mice have demonstrated that the antioxidant function of p53 may directly contribute to the prevention of tumor development (109, 130). In cancer cells deficient in wild-type p53, lack of the antioxidative properties of p53 has the ability to increase the redox stress within the cell, which in turn can increase the rate of mutagenicity and also modulate redox-sensitive pathways. This may have important implications in the activation of redox-sensitive survival pathways. For instance, Li et al. (91) showed that, in endothelial cells that express Nox2 and Nox4, Nox2-derived ROS participate in cell-cycle regulation and apoptosis through modulation of p21cip1 and p53 expression (91).
NF-κB
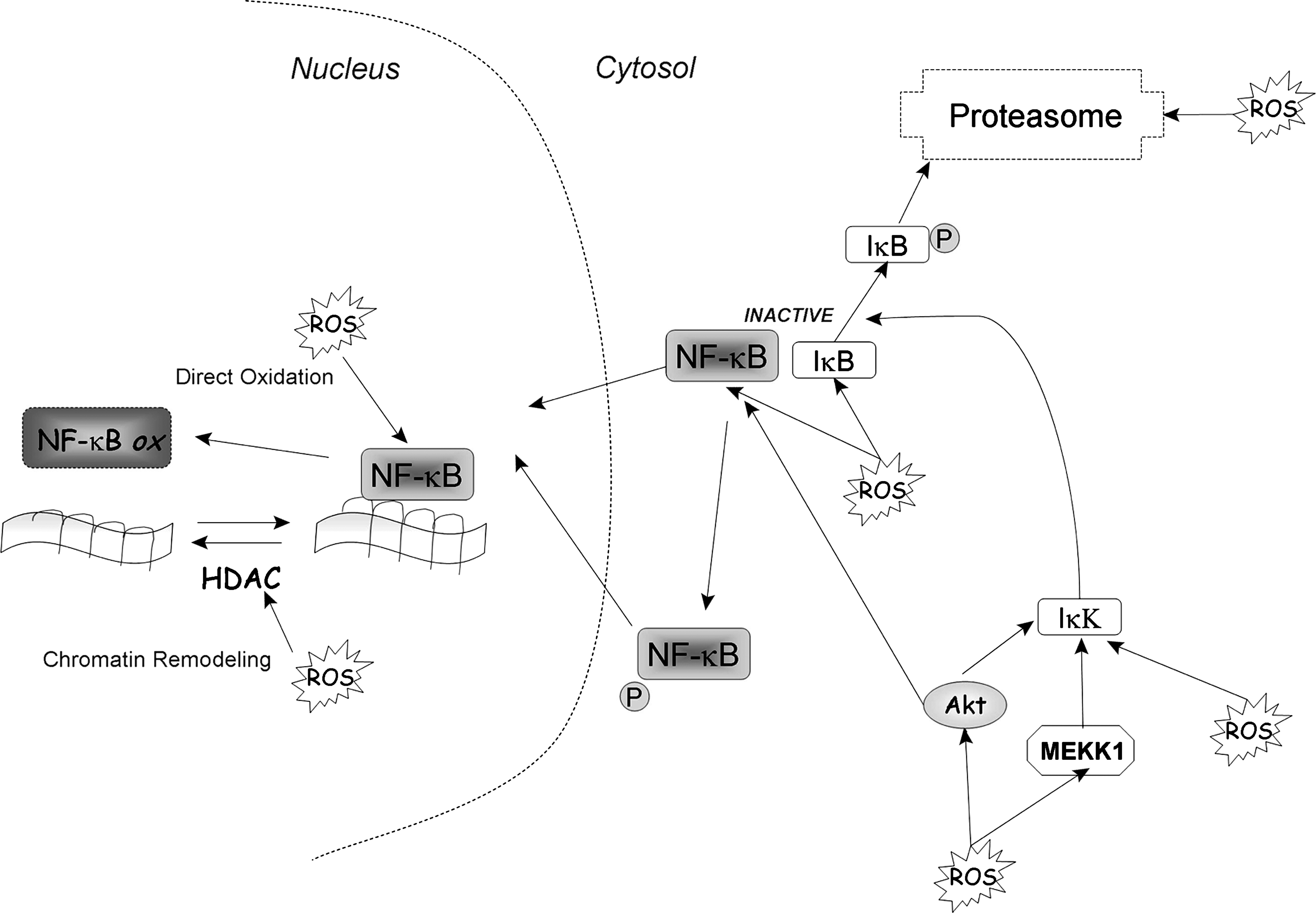
The heterodimeric protein NF-κB is a ubiquitous redox-regulated transcription factor that remains sequestered in the cytoplasm as an inactive complex with its inhibitory counterpart IκB. Exposure to oxidative stimuli leads to phosphorylation and subsequent proteasomal degradation of IκBα, thereby releasing free NF-κB dimers for translocation to the nucleus. Experimental evidence suggests that ROS have paradoxic effects on NF-κB regulation. ROS can either activate or inhibit NF-κB activity, depending on the ROS levels, types of stimuli, and cell types. A moderate level of ROS generally leads to NF-κB activation (Fig. 9). Conversely, a high level of ROS could inactivate NF-κB, leading to cell death. In the nucleus, direct oxidation of the redox-sensitive cysteine 62 of the p50 subunit inhibits its availability to bind DNA (153). This oxidation is reversible, and DNA binding can be restored. Besides direct structural modification, DNA-binding activity of NF-κB can be modulated by chromatin remodelling (121). Thus, the enzyme histone deacetylase (HDAC), which catalyzes the removal of an acetyl group from histone, can be inactivated by oxidative stress, allowing histone acetylation, chromatin uncoiling, and increased accessibility for NF-κB (120). In the cytosol, NF-κB is sequestered as a complex formed with its inhibitor IκB; its activation is then regulated by phosphorylation of NF-κB itself or phosphorylation of its inhibitor. Under certain conditions, H2O2 is able to activate NF-κB activity directly through phosphorylation of IκB-kinase (IκK) (64) or indirectly through activation of Akt or MEKK1 (or both), which then phosphorylates and activates IκK. The transactivation of NF-κB induced by Akt or MEKK1 has been shown to be really important in NF-κB antiapoptotic effects (107, 158). Active IκK phosphorylates IκB and liberates active NF-κB from the complex to translocate to the nucleus. Phosphorylated IκK is then degraded by the proteasome. Because the proteasome system is also redox sensitive, ROS can regulate NF-κB activity by affecting IκK stability. One example of the direct effect of Nox on NF-κB was shown using siRNA against Nox2, which decreased insulin-induced O2 •− production and inhibited the turn-on of NF-κB as well as p38MAPK and ERK 1/2 (132).
AP-1
AP-1 is another redox-sensitive transcription factor that plays a critical role in both cell growth and apoptosis. Under specific conditions, AP-1 activation can lead to cell death, whereas under other circumstances, AP-1 can promote cell proliferation. The AP-1 family consists of several proteins, including Jun, Fos, Maf, and ATF subfamilies (6). Activation of AP-1 is regulated at both the transcript and protein levels. The MAPKs play a major role in controlling activation of AP-1 proteins through phosphorylation. All three classes of MAPKs are involved in regulation of AP-1. c-Fos is a substrate of ERK, and ATF2 is regulated by JNK and p38 kinases (68). Oxidative stress can activate c-Jun and ATF2 through phosphorylation by JNK and p38, respectively. Redox regulation of the JNK and p38 pathway is well described (for review, see refs. 154, 155). Like that of NF-κB, transcriptional activation of AP-1 can be regulated by chromatin remodelling (102). Oxidative stress is known to promote AP-1 activity through histone acetylation by inhibition of HDAC (120). Furthermore, the intracellular level of AP-1 can be regulated by redox-mediated mechanisms at the levels of transcription and protein turnover. Recent reports show that expression of c-Jun can be transcriptionally repressed by HDAC or degraded by the proteasome through MEKK1-induced ubiquitination. This downregulation of c-Jun plays an important role in apoptosis induction by oxidative stress (166, 167). As described for NF-κB, several binding sites for AP-1 have been shown on Nox family members [e.g., the 5' region of human Nox1 (82) gene and p67 phox ]. Binding sites for AP-1 transcription factors were found in the first intron of p67 phox , and they are essential for p67 phox expression (46, 92).
HIF
Hypoxia-inducible factor (HIF) was first described as a transcription factor that regulates cellular metabolism under hypoxic stress. However, HIF also can be activated by nonhypoxic stimuli. Active HIF requires heterodimeric formation of two subunits, HIF-α and HIF-β, which then translocate to the nucleus. Under normoxia, a variety of stimuli stabilize HIF, in part through increased ROS/RNS production. However, attenuation of ROS by antioxidant molecule or by genetic downregulation of Nox4 was found to decrease HIF expression. (52, 77, 137). Under hypoxia, both ROS and RNS were found to inhibit HIF-1 DNA-binding activity and HIF-1 accumulation; however, the mechanisms are still unclear (17).
Caspases
The redox regulation of transcription factors and signal-transduction pathways may affect cell survival through a series of molecular processes resulting in the activation or inhibition of cell death–execution molecules. Under certain conditions, ROS may affect directly the activity of those cell-death effectors. Several apoptotic effectors are redox sensitive, and their functions can be directly modulated by intracellular ROS; these include caspases, the Bcl-2 family of proteins, and cytochrome c.
A hallmark of apoptosis is the activation of caspases, which requires sequential proteolysis of the initiator caspases and effector caspases. Apoptotic stimuli can trigger caspase activation through either the extrinsic (death receptor–mediated activation) or the intrinsic (mitochondria-mediated activation) pathways. Activation of the caspase cascade ultimately leads to the cleavage of different substrata, such as PARP, DNA fragmentation, and cell death. ROS can directly affect caspase function. The reduced state of the cysteine in the active site is necessary for the catalytic activity of caspases; thus, depending on the degree of intracellular oxidative stress, caspases can be activated or inhibited. By using different concentrations of exogenous H2O2, Hampton et al. (58) demonstrated that a low dose of H2O2 can activate caspases and induce apoptosis, but a high dose of H2O2 inhibits caspases, and cells undergo necrosis. A recent study by Peshavariya et al. (116) showed that in endothelial cells, Nox2 and Nox4 have two different roles. Although inhibition of Nox4 has no effect on caspase activation, Nox2 inhibition with siRNA caused an increase in caspase 3/7 activity. Therefore, in endothelial cells, Nox4-derived H2O2 promotes proliferation, whereas Nox2 maintains the cytoskeleton and prevents apoptosis to support cell survival.
Relevance to Disease
Most of the studies quoted to date have come either from cell-culture experiments, in which various elements of the survival pathways were artificially expressed, or from a totally in vitro situation, in which cell lysates only were used in the experiments. The question therefore arises whether H2O2 produced from Nox can act as a survival molecule in disease models. Nox proteins are already known to play a large role in a prodeath manner in certain diseases (for review, see ref. 13), particularly chronic granulomatous disease, in which a mutation in Nox2 or p22 phox leads to a loss of immune function in those with the disease. But in the last decade or so, more evidence has accumulated for their contribution to prosurvival cell signaling in certain situations. Vaquero et al. (159) showed that H2O2 generated by Nox4, in particular, is prosurvival in pancreatic cancer cells, which has since been confirmed in several studies (35, 101, 169). This also was found to be true of other cancer models, for example, Nox1 is increased in prostate cancer (93) and in certain colon cancers (43), and Nox5 is involved in hairy cell leukemia (66) and prostate cancer (16). This has also been found in hepatocytes expressing Nox1, 2, and 4 (103); astrocytes, in which Nox4 is important (94, 139); and in various cell types of the cardiovascular system (31). Even in monocytes and macrophages, ROS generated by Nox proteins are found to have a prosurvival effect (162).
By understanding precisely how the production of H2O2 is generated by Nox, we can begin to use this knowledge to control the pathway, and so help in the development of therapeutics. These proteins are of course regulated at a transcriptional level [reviewed in (13)], but this is outside the scope of this review. Overall, a subtle change in the level of Nox activity is likely to be the key, and not some overall inhibition/activation. Therapeutics, based on the information in this review, might be able (a) to stop Nox activity relatively quickly, so that only a short burst would occur; or (b) to generate a small burst of H2O2 some time before a known large oxidative stress were to happen. We are slowly getting glimpses as to how this might be possible. It is feasible that manipulating the two main controlling factors of Nox activation, which are the phosphorylation of p47 phox and activation of Rac, or their regulators, could control Nox activity with more finesse than by targeting Nox alone. This will be possible only for Nox1 and 2. In the case of Nox5, Duox1, and Duox2, this control would be through controlling calcium levels and also phosphorylation of the proteins involved. For Nox4, the most work is to be done to determine its regulators, and therefore how to control it in disease situations. The downstream targets of H2O2 could also be targeted, once they have been identified in individual diseases.
Concluding Remarks
The dual nature of ROS has recently emerged, showing that they are not necessarily destructive but can also be closely regulated molecules acting as second messengers in many different signaling pathways. H2O2 is one of the main ROS capable of acting as a second messenger because of it is relative stability. The Nox family of proteins plays a major role in the production of H2O2, which mediates the regulation of survival pathways. Nox proteins are multimeric proteins, with different subunits, such as p47 phox and p67 phox , which allows a very tight and quick regulation of H2O2 production. Although much is known about the archetypical Nox, Nox2, and its closest homologue, Nox1, much remains to be learned about the regulation of the newer members of the Nox family; for example, very little is known about any regulatory proteins associated with Nox4.
When considering the role of H2O2 as a survival molecule, it is important not only to determine its source, but also to understand how H2O2 interacts with its target molecules. The main way that H2O2 affects cell-signaling pathways is through oxidation of specific molecules. If the concentration of H2O2 is high, then these oxidation processes may lead to irreversible damage, followed by cell death. However, this is not always the case, and H2O2 is capable of reversible inhibition of many enzymes, including phosphatases. As a consequence of survival pathways transducing their survival signal through phosphorylation of key proteins, phosphatases are potent negative regulators. The PI3K/Akt survival pathway is a good example of such, as multiple steps are negatively regulated by phosphatases.
It is important to realize that ROS-mediated regulation of cell survival is a very complex cascade of events. Activation of Nox proteins will modulate ROS production, which will activate various intracellular signaling pathways, inducing survival factors such as insulin, PDGF, TNF-α, and Bcr-Abl. In turn, those survival factors have been shown to be Nox regulators producing complex feedback loops. A comprehensive understanding of the regulation of the redox regulation of survival pathways, as well as of the regulation of ROS production by Nox, is essential when considering ROS as survival molecules and eventually developing redox-based therapeutic strategies in the many ROS-related diseases.
Footnotes
Acknowledgments
The first two authors contributed equally to this work. This work was supported by Science Foundation Ireland, Children Leukemia Research Project, and the Irish Cancer Society.