
Abstract
Bacterial antibiotic resistance is an emerging global crisis, and treatment of multidrug-resistant gram-negative infections, particularly those caused by the opportunistic human pathogen Pseudomonas aeruginosa, remains a major challenge. This problem is compounded by a lack of new antibiotics in the development pipeline: only two new classes have been developed since the 1960s, and both are indicated for multidrug-resistant gram-positive infections. A promising new approach to combat antibiotic resistance is by targeting bacterial virulence, rather than bacterial viability. The bacterial periplasmic protein DsbA represents a central point for antivirulence intervention because its oxidoreductase activity is essential for the folding and function of almost all exported virulence factors. Here we describe the three-dimensional structure of this DsbA target from P. aeruginosa, and we establish for the first time that a member of this enzyme family is capable of binding small molecules. We also describe biochemical assays that validate the redox activity of PaDsbA. Together, the structural and functional characterization of PaDsbA provides the basis for future studies aimed at designing a new class of antivirulence compounds to combat antibiotic-resistant P. aeruginosa infection. Antioxid. Redox Signal. 11, 921–931.
Introduction
Pseudomonas aeruginosa possesses a diverse array of virulence factors that enable it to evade host defences. Many of the processes that confer virulence or antibiotic resistance are mediated by secreted proteins, including flagellar proteins, type III secretion factors, pili, and enzymes such as proteases and β-lactamases. To function correctly, these secreted proteins must be folded and remain intact in the harsh extracellular environment; for this reason, most incorporate disulfide bonds. Incorporation of disulfide bonds takes place in the periplasm and is catalyzed by enzymes of the Dsb family. The DsbA enzyme from P. aeruginosa (PaDsbA) plays a pivotal role; it is required for the expression of elastase, exotoxin A, protease IV, lipase, and alkaline phosphatase (7, 11, 15, 31, 32, 49). PaDsbA is also required for the formation of a functional type III secretion system, and P. aeruginosa organisms lacking PaDsbA are defective in twitching motility and in the expression of the pilin subunit pilA. Taken together, these findings highlight the intimate relationship between PaDsbA activity and P. aeruginosa pathogenicity and identify PaDsbA as a potential target for the development of a new class of antibacterial agents that target virulence rather than viability (18). This approach has been advocated as a means of generating “more effective drugs with a lower propensity for inducing bacterial resistance” (39).
The DsbA of Escherichia coli (EcDsbA) is the best-characterized member of the DsbA family. However, recent structural and biochemical evidence illustrates that significant differences exist among DsbA homologues (17, 22, 50). For example, the peptide-binding groove identified in the EcDsbA structure (34) and subsequently shown to be used by its redox partner protein DsbB (24) is virtually absent in the structure of Staphylococcus aureus DsbA (SaDsbA) (17). In addition, a hydrophobic patch believed to be important for substrate binding in EcDsbA is not present in SaDsbA.
To investigate the role of PaDsbA further, we determined the 1.5-Å resolution crystal structure of this protein and characterized its redox properties. Our data show that PaDsbA is one of the most oxidizing proteins yet characterized, and the structure of PaDsbA reveals important differences compared with other structurally characterized DsbA proteins, including the unexpected finding that PaDsbA binds small molecules. Collectively, these studies reveal the biochemical function of PaDsbA and suggest a basis for future structure-based drug-design studies targeting this important virulence determinant.
Materials and Methods
Cloning, expression and purification
The codon-optimized Pseudomonas aeruginosa dsbA gene lacking the signal sequence and inserted into the PCR-Blunt vector was obtained from GeneArt (Regensberg, Germany) and subsequently amplified by using forward and reverse primers compatible with ligation-independent cloning (LIC). The amplified gene was then inserted into a modified pET21a vector. This modified vector encodes a leader sequence consisting of an N-terminal maltose-binding protein (MBP) followed by the tobacco-vein mottling virus (TVMV) protease recognition sequence. This plasmid also encodes for TVMV so that the MBP fusion is cleaved during protein folding. Immediately after the TVMV site is an His6-tag, followed by a spacer containing the tobacco etch virus (TEV) protease-recognition sequence and a LIC sequence based on a central SspI restriction site. The cloning protocol introduced three additional amino acid residues (SNA) at the N-terminus of the mature protein when the His6-tag is cleaved with TEV protease. The sequence of the final construct was confirmed to be that of Pseudomonas aeruginosa dsbA (GeneID:877731). The plasmid was transformed into Escherichia coli BL21(DE3)/pLysS for expression by autoinduction (47). In brief, cells were grown with agitation at 200 rev/min for 24 h at 303 K in autoinduction media supplemented with 50 μg/ml ampicillin and 34 μg/ml chloramphenicol. After expression, the cells were harvested with centrifugation, flash frozen in liquid nitrogen, and stored at 193 K. Cells were lysed in 25 mM HEPES, pH 7.5, 150 mM NaCl, 0.5% TritonX-100, protease inhibitor cocktail III (A.G. Scientific, Inc., San Diego, CA) and DNase. The sample was sonicated, and cell debris was removed by centrifugation (41,399 g for 30 min). His6-tagged PaDsbA was purified by using PrepEase High Yield Resin (USB-Millennium Science, Surrey Hills, Victoria, Australia) and eluted with 25 mM HEPES, pH 7.5, 150 mM NaCl, and 500 mM imidazole. The peak fractions were pooled, and the protein concentration was measured at 280 nm by using a NanoDrop ND-1000 (NanoDrop Technologies, Wilmington, DE). Purified recombinant His-tagged TEV (generated from pRK793) (5) was added to a molar ratio of 1:100 to cleave the His tag from PaDsbA. This mixture was dialyzed extensively against 25 mM HEPES, pH 7.5, 150 mM NaCl, at 277 K for 48 h. The mixture was then passed over PrepEase High Yield Resin once more so that the flowthrough contained the cleaved PaDsbA, and the resin retained His-tagged TEV. The cleaved PaDsbA was further purified by gel-filtration chromatography by using a Superdex S-75 column (AKTA; GE Healthcare, Piscataway, NJ).
Peak fractions containing PaDsbA were pooled and concentrated to 100 mg/ml in 10 mM HEPES, pH 7.5, by using Amicon Ultra centrifugal filter devices with a 10-kDa cutoff (Millipore, Billerica, MA). The protein concentration was again measured at 280 nm, and the purity was assessed with SDS-PAGE analysis.
Disulfide isomerase assay
An in vitro assay monitoring the refolding of scrambled RNaseA was used to measure the isomerase activity of PaDsbA, EcDsbC, and EcDsbA (19). Disulfide-scrambled RNaseA was produced as previously described (17). The isomerization of scrambled RNaseA (40 μM) disulfide bonds to their native conformation was measured by incubation in 100 mM sodium phosphate buffer, 1 mM ethylenediaminetetraacetic acid (EDTA), pH 7.0, 10 μM dithiothreitol (DTT), with 10 μM protein sample. Positive and negative controls consisted of two additional reactions with folded native RNaseA or buffer, respectively. All assays were performed at 298 K, and RNaseA activity was determined by monitoring cCMP hydrolysis at 296 nm at several time points.
Chemical unfolding and refolding
Oxidized PaDsbA was diluted to 1.5 μM in 20 mM Hepes-NaOH, 170 mM NaCl, 0.1 mM EDTA, pH 7.0 buffer containing guanidinium chloride at various concentrations and incubated at room temperature for 24 h. Reduced PaDsbA was unfolded under identical conditions but in the presence of 0.75 mM DTT. Refolding experiments were performed by first unfolding oxidized and reduced PaDsbA stock solutions (37.5 μM) in 20 mM Hepes-NaOH, 170 mM NaCl, 0.1 mM EDTA (pH 7.0), 6 M guanidinium chloride (containing 20 mM DTT for the reduced protein) for 20 h at room temperature, and then taking 20 μl of the unfolded protein solution and mixing it with 480 μl of buffer containing guanidinium chloride at various concentrations. These solutions were then incubated for 24 h at room temperature. Three independent experiments were performed for each transition, which were measured fluorimetrically at 353 nm, by using an excitation wavelength of 280 nm. Data were fitted according to a two-state model (38, 44).
Thermal stability
The thermal-denaturation equilibrium was measured with methods we described previously (17). We confirmed that PaDsbA disulfide oxidation was complete by using the Ellman assay before measuring the temperature-induced unfolding curves. The reduced sample was prepared by adding 0.75 mM DTT (final concentration) to the oxidized sample. Temperature-induced unfolding was measured at 226.5 nm for the oxidized sample and 225 nm for the reduced sample on a Jasco J-810 spectropolarimeter. The experiment was conducted by heating the samples from 25°C to 95°C at 1°C/min (unfolding). Once 95°C was reached, samples were then cooled to 25°C (refolding).
PaDsbA redox potential
The fractions of reduced and oxidized PaDsbA (2 μM) in 100 mM sodium phosphate, 1 mM EDTA, pH 7.0, at 298 K, and different concentrations of excess reduced glutathione (GSH) and oxidized glutathione (GSSG) (51) were determined from the redox-dependent PaDsbA fluorescence at 340 nm (excitation, 280 nm) after equilibrium for 24 h. Three independent experiments were performed to give the estimate of the equilibrium constant. The redox potential was then calculated as described previously (51).
Insulin-reduction assay
We measured the ability of PaDsbA, EcDsbC, and EcDsbA to catalyze insulin reduction in the presence of DTT by using methods described previously (21). In brief, reaction mixtures were prepared in cuvettes containing 130 μM insulin, and protein catalyst (5 to 10 μM) in 100 mM sodium phosphate buffer, 2 mM EDTA, pH 7.0. Reactions were started with the addition of DTT to a final concentration of 0.33 mM. After thorough mixing, the rate of precipitation was monitored by recording the increased turbidity of the reaction mixture at 650 nm every 30 s. Absorbance measurements were made by using a 1-cm cuvette and a Cary50 spectrophotometer. The noncatalyzed reduction of insulin by DTT was monitored in a control reaction without catalyst.
EcDsbA complementation
For these experiments, we cloned the padsbA gene into the arabinose-inducible pBAD33 vector (14). The constructs were then chemically transformed into the nonmotile E. coli dsbA−
mutant and dsbA−/dsbB−
double-mutant strains [JCB817 and JCB818, respectively (4)] and plated on LB chloramphenicol plates. As positive controls, we used JCB817 and JCB818 cells containing pBAD33::EcDsbA. Triplicates of fresh colonies were stabbed into minimal agar (M63) containing 40 μg/ml of each amino acid, and 1 mg/ml arabinose to induce expression. Additionally, the experiment was performed by using agar without arabinose to monitor background complementation. The experiment also was performed in the absence and presence of
Crystallization and diffraction data measurement
The hanging-drop vapor-diffusion method was used for crystallization of PaDsbA with the commercially available screens Crystal Screens 1 and 2, polyethylene glycol (PEG)/Ion Screen, Index (Hampton Research, San Diego, CA), Wizard Crystal screens 1 and 2 (Emerald BioSystems, Bainbridge Island, WA), the JCSG+ Suite (Qiagen Pty Ltd, Doncaster, Victoria, Australia) and the PACT premier screen (Molecular Dimensions Limited, Newmarket, Suffolk, England). Crystallization trials were set up in 96-well plates by using a Mosquito crystallization robot (TTP Labtech, Melbourne, Cambridge, England). Each drop consisted of 100 nl protein solution and 100 nl well solution. The crystallization plates were incubated at 293 K and imaged by using a temperature-controlled RockImager (Formulatrix, Waltham, MA). Crystals of PaDsbA appeared within 2 days in a condition from the JCSG+ Suite screen comprising 24% (wt/vol) PEG of average molecular mass of 1,500 Da (PEG 1500), and 20% (vol/vol) glycerol. A fine screen around this condition was then set up by using VDXm 24-well hanging-drop plates and 18-mm siliconized coverslips (both from Hampton Research, San Diego, CA). To increase the crystal size, each coverslip held one 2-μl drop containing 1 μl protein solution and 1 μl well solution. The optimized condition consisted of 24% (wt/vol) PEG 1500, and 22% (vol/vol) glycerol. We attempted to solve the structure of PaDsbA by molecular replacement with Phaser (36) and the CCP4 program CHAINSAW, by using as models the coordinates of DsbA structures available at the time: DsbA from Escherichia coli (PDB code 1DSB, Seq ID 27%), Staphylococcus aureus (3BCI Seq ID 21%), Wolbachia pipientis (3F4R Seq ID 8%), Vibrio cholerae (1BED Seq ID 28%), and Nitrosomonas europaea (2IN3 Seq ID 12%). However, these trials were unsuccessful, and we therefore pursued single-wavelength anomalous dispersion (SAD) phasing methods. Selenomethionine (SeMet) PaDsbA was produced by using a protocol similar to that previously described for the production of SeMet CcmG (9). In brief, E. coli BL21(DE3)pLysS cells containing the PaDsbA expression plasmid described earlier were grown at 310 K in M63 minimal media containing 50 μg/ml SeMet (D/L mixture). Cultures were induced at the exponential phase of growth (OD600 = 0.6) with a final concentration of 0.5 mM isopropyl β-
For SeMet PaDsbA data collection, crystals were harvested from hanging drops by using a 0.2- to 0.3-mm CrystalCap Copper Magnetic HT Cryoloop (Hampton Research) and placed in a 1-μl drop consisting of 35% (wt/vol) PEG 1500 and 25% (vol/vol) glycerol for ∼30 s before flash cooling in liquid nitrogen (our crystal-cooling trials indicated that both PEG and glycerol were necessary to achieve adequate cryoprotection). The PaDsbA crystal was maintained at 100 K throughout data collection. X-ray data were collected at the Australian Synchrotron on PX1 beamline 3BM1 (wavelength, 0.953645 Å). The crystal-to-detector distance was 150 mm, and 1-degree oscillation images were collected for a total of 720 degrees, with an exposure time of 5 s each. This collection strategy resulted in a highly redundant data set ideally suited for SAD phasing. Diffraction data were processed and scaled by using the HKL2000 program package (37).
Structure determination and refinement
The structure of PaDsbA was solved by SAD phasing. PaDsbA contains six Met residues, and SeMet PaDsbA crystals were predicted to have one molecule in the asymmetric unit, according to the Matthews analysis (35). The locations of all six selenium atoms were determined by using Phenix AutoSol (1), and the model was improved with iterative model-building, density modification, and refinement by using the Phenix AutoBuild wizard. This was followed by cycles of interactive manual refitting of the model by using the program COOT (10) and refinement with phenix.refine (1). During the later stages of refinement, glycerol molecules and water molecules were built into (Fo − Fc) difference maps. Some surface residues with weak electron density were modeled with reduced occupancies for the side-chain atoms (Asn2, Asp4, Asp5, Glu14, Ser16, Lys26, Tyr41, Arg62, Glu87, Glu96, Lys100, Glu101, His102, Lys103, Lys104, Glu109, Asp113, Lys122, Glu123, Lys140, Lys143, Glu173, Lys185, Lys191, Lys192), and some residues were modeled with alternate conformations (Ser17, Ser22, Cys37, Cys40, Ile47, Glu52, Ser83, Glu110, Ser127, Mse138, Glu139, Mse156, Lys161, Lys177). Table 1 provides the statistics for the x-ray data collection and final refined model. The coordinates and structure factors of SeMet PaDsbA have been deposited (PDB code 3H93).
Values in parentheses refer to the highest-resolution shell.
Results
PaDsbA is active in the insulin-reduction assay
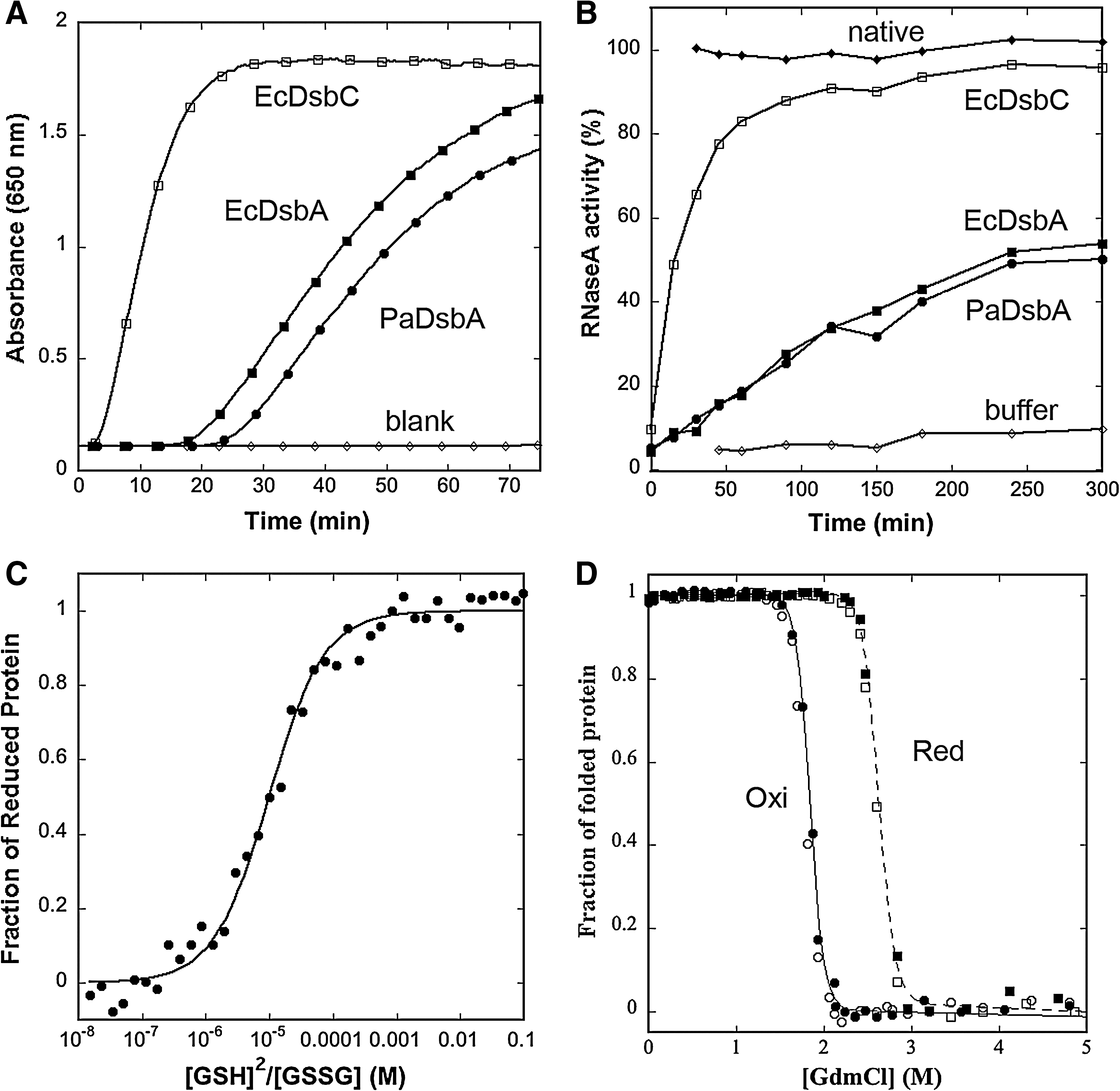
Before obtaining the crystal structure of PaDsbA, we examined the redox activity of the purified protein to confirm that the protein was active and to characterize it more fully than had previous studies (49). First, we used the insulin-reduction assay (21). In this assay, oxidoreductase enzymes reduce the interchain disulfides of insulin, causing precipitation of the insoluble insulin B-chain. Precipitation is monitored as an increase in absorbance at 650 nm. We found that the purified PaDsbA we produced was as active as Escherichia coli DsbA (EcDsbA) in this assay, but less active than the disulfide isomerase E. coli DsbC (EcDsbC) (Fig. 1A), in agreement with an earlier report (49). We therefore set about a more-comprehensive analysis of the redox activity of PaDsbA.
PaDsbA has weak disulfide isomerase activity
The Dsb family of proteins vary in their ability to catalyze disulfide isomerization. We measured the ability of PaDsbA to isomerize, or shuffle, incorrect disulfides of scrambled RNaseA. We found that, under the conditions of our assay, PaDsbA yielded ∼50% active RNaseA after 300 min (Fig. 1B). By comparison, the isomerase EcDsbC yielded ∼90% activity. In this assay, the activity of PaDsbA is more similar to that of EcDsbA, a strong thiol oxidant with a very oxidizing disulfide. The next step was to determine the redox potential of PaDsbA, to identify whether it too is highly oxidizing.
PaDsbA is highly oxidizing
The redox potentials of DsbA proteins vary tremendously; the most reducing DsbA characterized to date is Wolbachia pipientis α-DsbA1 (Wpα-DsbA1), with a value of −163 mV (27), and the most oxidizing are the neisserial NmDsbA proteins [redox potentials, −80 mV (28)] and E. coli DsbL (EcDsbL), the specialized DsbA from pathogenic E. coli [redox potential, −95 mV (13)]. We used the standard assay for measuring redox potential at pH 7.0 and 298 K, from the equilibrium constant with glutathione. The data yielded an equilibrium constant (Keq) for PaDsbA of 9.96 ± 0.88 × 10−6 M, corresponding to an intrinsic redox potential (E o′) of −94 mV (Fig. 1C). PaDsbA is thus 10 times more oxidizing than EcDsbA [Keq = 12 × 10−5 M; E o′ = −122 mV (23)] and as oxidizing as EcDsbL (13).
The active-site disulfide bond of PaDsbA is destabilizing
Disulfides typically stabilize protein structures. However, for DsbAs, the active-site disulfide has been shown to destabilize [e.g., EcDsbA (52); Wpα-DsbA1 (27)] or to have no effect [e.g., Staphylococcus aureus DsbA (17)] on the stability of the protein. We performed guanidinium chloride–induced unfolding/refolding equilibria experiments on PaDsbA to determine the ΔG stab . The transitions were fully reversible and cooperative, and all unfolding curves fitted a two-state model (Fig. 1D) (38, 44). The oxidized form of PaDsbA destabilizes the protein compared with the reduced form (ΔΔG ox/red 13.1 kJ/mol; reduced PaDsbA ΔG stab = −69.7 ± 5.5 kJ/mol; cooperativity, 26.6 ± 2.2 kJmol/M; midpoint of transition, 2.61 M; oxidised PaDsbA ΔG stab = −56.6 ± 3.9 kJ/mol; cooperativity, 30.9 ± 2.1 kJmol/M, midpoint of transition, 1.83 M). On the basis of ΔΔG values, the disulfide form of the PaDsbA active site is as destabilizing as that of EcDsbA [ΔΔG ox/red 14.8 kJ/mol; oxidized EcDsbA ΔG stab , −33.5 ± 1.2 kJ/mol vs. reduced EcDsbA ΔG stab = −48.3 ± 2.8 kJ/mol (23)].
We also performed temperature-induced unfolding experiments on PaDsbA to determine Tm, the transition midpoint of folded to unfolded protein, as monitored by the far-UV CD signal. These results also show that oxidized PaDsbA destabilizes the protein compared with the reduced form (Tm values, 345.8 ± 0.08 K and 355.8 ± 0.19 K, respectively), but we found that the unfolding was not reversible for PaDsbA under the conditions we used. Conversely, EcDsbA unfolding by using this approach was reversible (Tm values: unfolding oxidized 341.7 ± 0.18 K; unfolding reduced 350.9 ± 0.21 K; refolding oxidized 338.4 ± 0.65 K; refolding reduced 349.1 ± 0.43 K).
PaDsbA complements EcDsbA in vivo
By using an in vivo complementation assay with an arabinose-inducible PaDsbA expression plasmid, we showed that PaDsbA complements EcDsbA in an E. coli DsbA-null strain. This agrees with results of others (49) who used a PaDsbA expression plasmid in an E. coli DsbA-null strain that did not require arabinose for expression. Furthermore, we used an E. coli strain lacking both DsbA and DsbB and found that PaDsbA does not complement this strain unless the media is supplemented with cystine. These results suggest that PaDsbA interacts with EcDsbB, and that this interaction is required in vivo for reoxidation of PaDsbA. A summary of the results is presented in Table 2.
Structure of PaDsbA
Hanging-drop crystallization trials were conducted on purified native PaDsbA by using a Mosquito nanoliter crystallization robot. Initial crystallization conditions were optimized, and the resulting crystals were found to diffract to high resolution. SeMet-labeled protein crystals were then used to determine the crystal structure by SAD methods, making use of high-resolution, high-redundancy data collected at the Australian Synchrotron. The resulting 1.5-Å resolution structure reveals one molecule of PaDsbA in the asymmetric unit. The refined model matches the data very well, with Rfactor and Rfree values of 11.0 and 15.2% respectively (Table 1). The final crystallographic refinement parameters, electron-density maps, Ramachandran plot (41), and MolProbity analysis (30) indicate that the PaDsbA crystal structure is of very high quality.
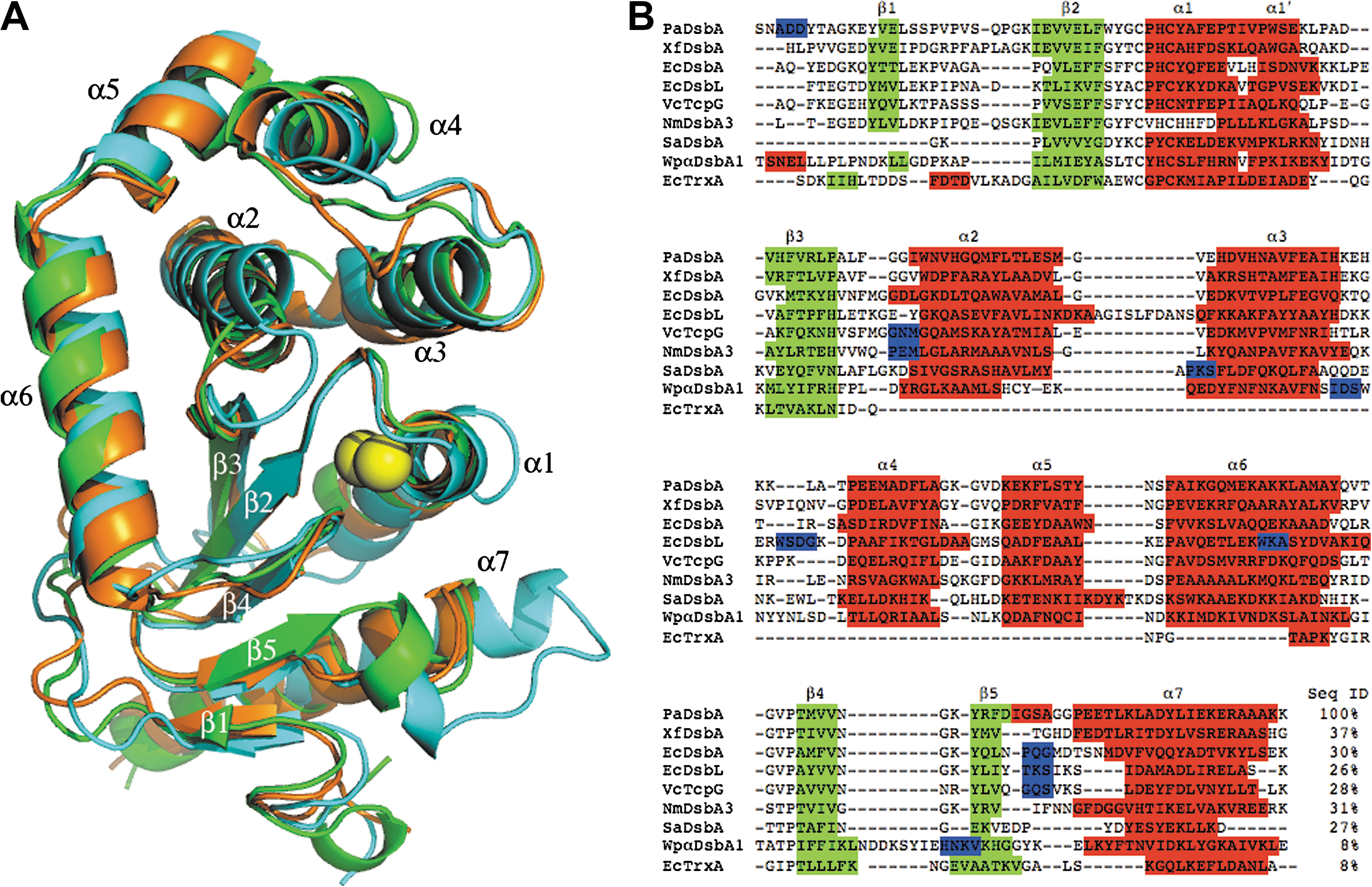
In agreement with all other structurally characterized DsbA molecules, PaDsbA contains a thioredoxin (TRX) fold (33) with an inserted α-helical domain. A Dali search (20) indicates that the closest structural homologue to PaDsbA is a DsbA from the plant pathogen Xylella fastidiosa (XfDsbA, 37% sequence identity, PDB code 2REM). A structural superposition of PaDsbA and its homologues XfDsbA and EcDsbA resulted in overall root-mean-squared deviations of 1.6 Å (186 Cα aligned) and 2.2 Å (188 Cα aligned), respectively (Fig. 2A). Structural differences between DsbA homologues are highlighted in a sequence alignment (Fig. 2B), showing, for example, that long loops present in EcDsbL ((13) between α2 and α3) and WpαDsbA1 ((27) between β4 and β5) are unique to these molecules. Also evident from Fig. 2 is that the “bulge” in EcDsbL that connects helices α3 and α4 is absent in PaDsbA, but present in WpαDsbA1 and XfDsbA. The most obvious region of variability among the structurally characterized DsbA proteins is in the loop connecting β5 and α7 (Fig. 2B), which is involved in binding substrates and the redox-partner protein DsbB. We note that the cis-Pro loop residues of PaDsbA are GVcisPT, whereas those of EcDsbA and EcDsbL are GVcisPA (Fig. 2B).
Glycerol-binding sites
Unexpectedly, the electron-density map revealed the presence of four bound glycerol molecules, located in discrete locations on PaDsbA (Fig. 3). Two glycerol molecules are bound to the active-site face of the enzyme, and two more are bound to the opposite face. Three of the four glycerol molecules bind to the TRX fold, and one of these three (Glyc-1) is located close to the catalytic cysteines. The Glyc-1 binding site lies on the face of β-strands 1 and 5. Glyc-1 is modeled in two subtly different conformations, both held in place by hydrogen bonds to the main-chain carbonyl oxygen of Lys10 (after β1) and the side chain of residue Arg163 (β5). In addition, five ordered water molecules form interactions with Glyc-1. This site is important not only because of its proximity to the active-site cysteines, but also because this region is involved in binding substrates (43) and the essential partner protein DsbB (24) (Fig. 4).


Glyc-2, the second glycerol molecule that binds on the active-site surface of PaDsbA, is located between the C-terminal end of α6 and the truncated loop joining β3 and α2. Glyc-2 forms hydrogen bonds to the carbonyl oxygen of Leu66 and to nearby water molecules. It also forms a hydrogen bond to a symmetry-related PaDsbA through the side chain of Lys124. Of the two glycerol molecules bound on the opposite face of PaDsbA (Figs. 3 and 4), Glyc-3 is located in an area rich in acidic and basic residues and is within hydrogen-bonding distance of the side chains of Asp180 and Arg187 on helix α7 and to three ordered water molecules. Finally, Glyc-4 is located in a relatively hydrophobic pocket between the loop joining helices α2 and α3 and the loop connecting α4 and α5 (Figs. 3 and 4). It forms hydrogen bonds with the main-chain carbonyl oxygen of Gly119 and the amide nitrogen of Asp121, located in the α4-α5 loop, and with three ordered water molecules.
Electrostatic surface features of PaDsbA
The surface electrostatics of DsbA proteins vary tremendously. For example, EcDsbA and the DsbA from Vibrio cholerae, VcTcpG, have a hydrophobic patch and a hydrophobic groove surrounding the active-site cysteine residues (22, 34), whereas EcDsbL (13) and Wpα-DsbA1 (27) are basic, and the DsbA from Staphylococcus aureus is polar (18). PaDsbA has a smaller hydrophobic patch than EcDsbA, and its surface is more basic, although not so basic as EcDsbL. The β5-α7 loop that forms the hydrophobic groove of PaDsbA is also truncated compared with EcDsbA (Fig. 2B).
Discussion
Pseudomonas aeruginosa is a particularly nasty opportunistic human pathogen that causes serious nosocomial infections (42) and places significant economic stress on health care systems (6). Increasing reports are appearing of multidrug-resistant strains (40, 46), and P. aeruginosa possesses numerous pathways to evade host defense mechanisms and to damage cells (18), including the ability to inject virulence proteins directly into host cells during infection through a type III secretion system (8, 12). PaDsbA plays a pivotal role in these virulence mechanisms by assembling and stabilizing virulence factors, including the secretion system (7, 15, 49). Pseudomonas aeruginosa also produces type IV fimbriae that allow the bacteria to adhere to and colonize host cells and to allow twitching motility for spreading within a host (16). Malhotra et al. (32) showed that dsbA mutations abolish twitching motility in P. aeruginosa. Taken together, these recent studies provide compelling evidence that PaDsbA plays a central role in P. aeruginosa virulence, and that blocking the activity of PaDsbA will, in turn, block the virulence of P. aeruginosa.
In this study, we performed a comprehensive analysis of PaDsbA showing that it is highly oxidizing, but a weak disulfide reductant and disulfide isomerase (Fig. 1). We found that the active-site disulfide of PaDsbA destabilizes the structure; this characteristic likely drives PaDsbA-catalyzed thiol-disulfide exchange with substrate proteins. In some ways, PaDsbA shares characteristics with the well-characterized DsbA from E. coli. However, PaDsbA is much more oxidizing, in that its redox potential is 28 mV higher than that of EcDsbA. The DsbL protein (13), a DsbA found in pathogenic strains of E. coli, and the neisserial NmDsbA (28) proteins are other DsbA proteins known to have highly oxidizing disulfide bonds. However, DsbL (48) and NmDsbAs (45) appear to have a limited range of substrates, and their highly oxidizing nature may be a consequence of the requirements to oxidize specific substrates. Conversely, PaDsbA is thought to have a broad range of substrates (7, 15, 16, 32, 49), so its highly oxidizing nature is unlikely to be required for the same reason. We speculate that the unusually high redox potential of PaDsbA may be required in P. aeruginosa to allow very rapid oxidative folding/reoxidation processes to enable a broad range of virulence factors to be produced rapidly and efficiently. However, we cannot rule out the possibility that the value we measure simply represents the upper end of a natural spectrum of highly oxidizing redox potentials for DsbA proteins.
DsbA enzymes from Gram-negative bacteria generally lack a well-defined, buried active-site cavity; instead, they have a relatively broad surface and often a hydrophobic groove surrounding the exposed active site (34). These types of surface features are not considered to be especially conducive to the binding of small molecules. We were, therefore, surprised that the electron-density map for PaDsbA clearly identified the binding of four glycerol molecules (Fig. 4). Although these glycerol molecules play no physiologic role in PaDsbA function, they show that PaDsbA can recognize some small molecules. This significant finding opens the way to develop small-molecule inhibitors of PaDsbA as potential antivirulence agents. Glyc-1, in particular, is interesting, as it is located in the same binding site that recognizes the essential partner redox protein, EcDsbB (24) (Fig. 4). Given its small mass and expected low binding affinity, glycerol is unlikely to inhibit PaDsbA. This is supported by our preliminary data, indicating that inclusion of glycerol had no effect on PaDsbA activity in EcDsbA complementation, insulin reduction, and scRNaseA assays (not shown). However, a tightly binding small molecule that binds to the same region as Glyc-1 is likely to interfere with PaDsbA activity, and hence P. aeruginosa virulence. Two of the glycerol molecules (Glyc-1 and Glyc-2) are prime targets for elaboration and are bound to the enzyme sufficiently close to each other to be linked into one larger ligand. All four glycerol molecules are bound in sizeable clefts, with volumes ranging from 260 (Glyc-1) to 720 Å3 (Glyc-3) [calculated with PDBsum (29), Fig. 3]. The clefts are not fully used by the glycerol molecules, so that a potential exists to elaborate the individual molecules into larger (and potentially tight-binding) inhibitors.
In conclusion, we determined the high-resolution crystal structure of DsbA from P. aeruginosa. This is the first reported structure of a DsbA with small molecules bound, providing potential starting points for inhibitor development. All four of the bound glycerol molecules provide important information about drug-binding sites on PaDsbA that will be valuable for future studies aimed at developing therapeutics to combat this opportunistic pathogen.
Footnotes
Acknowledgments
We thank the Australian National Health and Medical Research Council (grant to J.L.M., M.J.S., and B.H.; fellowship to J.L.M.) and The University of Queensland (fellowship to S.R.S.) for financial support. We acknowledge use of the Australian Synchrotron and the support of PX1 3BM1 beamline scientists Julian Adams and Tom Caradoc-Davies; and we thank Karl Byriel and Gordon King for their support and access to the UQ ROCX Diffraction Facility.
Author Disclosure Statement
This research was funded entirely by government and university research grants. No competing financial interests exist.