
Abstract
Epithelial to mesenchymal transition (EMT) is a fundamental process, paradigmatic of the concept of cell plasticity, that leads epithelial cells to lose their polarization and specialized junctional structures, to undergo cytoskeleton reorganization, and to acquire morphological and functional features of mesenchymal-like cells. Although EMT has been originally described in embryonic development, where cell migration and tissue remodeling have a primary role in regulating morphogenesis in multicellular organisms, recent literature has provided evidence suggesting that the EMT process is a more general biological process that is also involved in several pathophysiological conditions, including cancer progression and organ fibrosis. This review offers first a comprehensive introduction to describe major relevant features of EMT, followed by sections dedicated on those signaling mechanisms that are known to regulate or affect the process, including the recently proposed role for oxidative stress and reactive oxygen species (ROS). Current literature data involving EMT in both physiological conditions (i.e., embryogenesis) and major human diseases are then critically analyzed, with a special final focus on the emerging role of hypoxia as a relevant independent condition able to trigger EMT. Antioxid. Redox Signal. 12, 1383–1430.
Reactive Oxygen Species, Redox Signaling, and Redox Regulation in EMT
EMT in Human Health and Disease
EMT in embryogenesis or Type 1 EMT: A process for dispersing cells in embryos
EMT as a mechanism contributing to re-epithelialization in wound healing
EMT in fibrogenesis and organ fibrosis or Type 2 EMT: The examples of kidney, lung, and liver
Hypoxia as an emerging and independent master condition able to trigger EMT in human diseases
Endothelial to mesenchymal transition: A peculiar form of EMT involved in pathophysiology
I. Introduction
A. Epithelial to mesenchymal transition as a paradigm of cell plasticity: Definition of EMT and introductory remarks
The following distinctive main features of epithelial cells are summarized in Figure 1 (114, 233, 250): a) In normal conditions, they usually form regular layer of cells, often one cell thick, in which neighboring elements are adjoined by means of specialized junctional structures that are referred to as tight junctions, adherens junctions, desmosomes, and gap junctions. Epithelial cells in culture typically form clusters of cells that maintain these specialized junctional structures. b) The intrinsic adhesiveness of epithelial cells allows the three-dimensional organization of a well structured epithelium. c) Epithelial cells are usually polarized in a characteristic apical–basolateral pattern; major determinants of this polarization include: the specific and localized distribution of cell adhesion molecules, mostly cadherins, and some integrins (often defining the apical pole); the already mentioned organization of specialized junctional structures, that make the lateral edge of the epithelial cell to be easily identified; the organized polarization of actin cytoskeleton, and the presence of a basal membrane or lamina that identifies the basal surface. d) Although epithelial cells may show some degree of motility to migrate within the epithelium, moving away from the neighboring cell(s), in normal conditions they do not leave the epithelial environment or layer.

The main features of mesenchymal cells are, by definition, quite different and distinctive: a) They do not form regular layer of cells as well as the stable specialized junctional structures described for epithelial cells. b) Mesenchymal cells can just form adhesions focally with neighboring cells. c) Although mesenchymal cells may be polarized when migrating or interacting with neighboring cells, they lack typical apico-basal polarity as seen in epithelial cells and in culture have a typical spindle-shaped morphology. d) Mesenchymal cells that exhibit high motility when in cell culture can easily migrate within tissues as single cells or by one of the so-defined collective mode of migration (for example, forming a chain of migrating cells).
As elegantly described by Friedl (74), mesenchymal cells can usually migrate within 3D extracellular matrix (ECM) by a continuous cycle of five independent steps: a) localized actin polymerization to filaments that drives cell polarization and results in the formation of a leading pseudopod; b) interaction of the pseudopod with ECM ligands, adhesion to the substrate, and induction of signaling pathways and cytoskeletal modification leading to formation of focal adhesions or focal contacts; c) recruitment of metalloproteinases like matrix metallopeptidase 14 or MMP-14 (also known as MT1-MMP or membrane-type 1 matrix metalloproteinase) to the leading edge membrane to provide pericellular ECM protelysis; d) engagement of actin filaments with cross-linking proteins or contractile proteins such as myosin II, resulting in stabilization and contraction of membrane-anchored actin strands that results e) in local cell contraction and in a slow forward gliding of the posterior part of the cell, with the cell finally moving along the substrate in the direction of the leading edge.
Occasionally, mesenchymal cells can also migrate by adopting a more primitive, ameboid-like type of migration in which cells can be envisaged to “crawl” within the tissue by continuosly adapting their shape to the actual microenvironment and preformed ECM, a type of migration that in the adult organism is usually adopted by leukocytes, hemopoietic stem cells, and some cancer cells (74).
The static postulate of the existence of an absolute dichotomy between epithelial and mesenchymal cells, that has dominated large part of the last century, has been gradually abandoned when several laboratories pointed out that epithelial cells can convert into mesenchymal cells by a process that is now widely accepted and defined as epithelial–mesenchymal transition or EMT.
EMT indeed defines a fundamental process, paradigmatic of the concept of cell plasticity that has been originally described in embryonic development where cell migration and tissue remodelling have a primary role in regulating morphogenesis in multicellular organisms. According to current literature (2, 15, 27, 35, 114, 139, 168, 227, 250, 251, 298), the original definition of EMT was focused on the formation of mesenchymal cells from epithelial cells in different areas of embryos: this process follows the loss of epithelial cell polarization as a result of disappearance of specialized junctional structures, cytoskeleton reorganization, and organelle redistribution and gradual acquisition of typical EMT-related mesenchymal features and behavior (Figs. 2 and 3). The EMT process in embryo development has a natural counterpart since embryonic mesenchymal cells can eventually undergo a reverse transition process, known as mesenchymal to epithelial transition or MET (35), leading them to regain a fully differentiated epithelial phenotype.
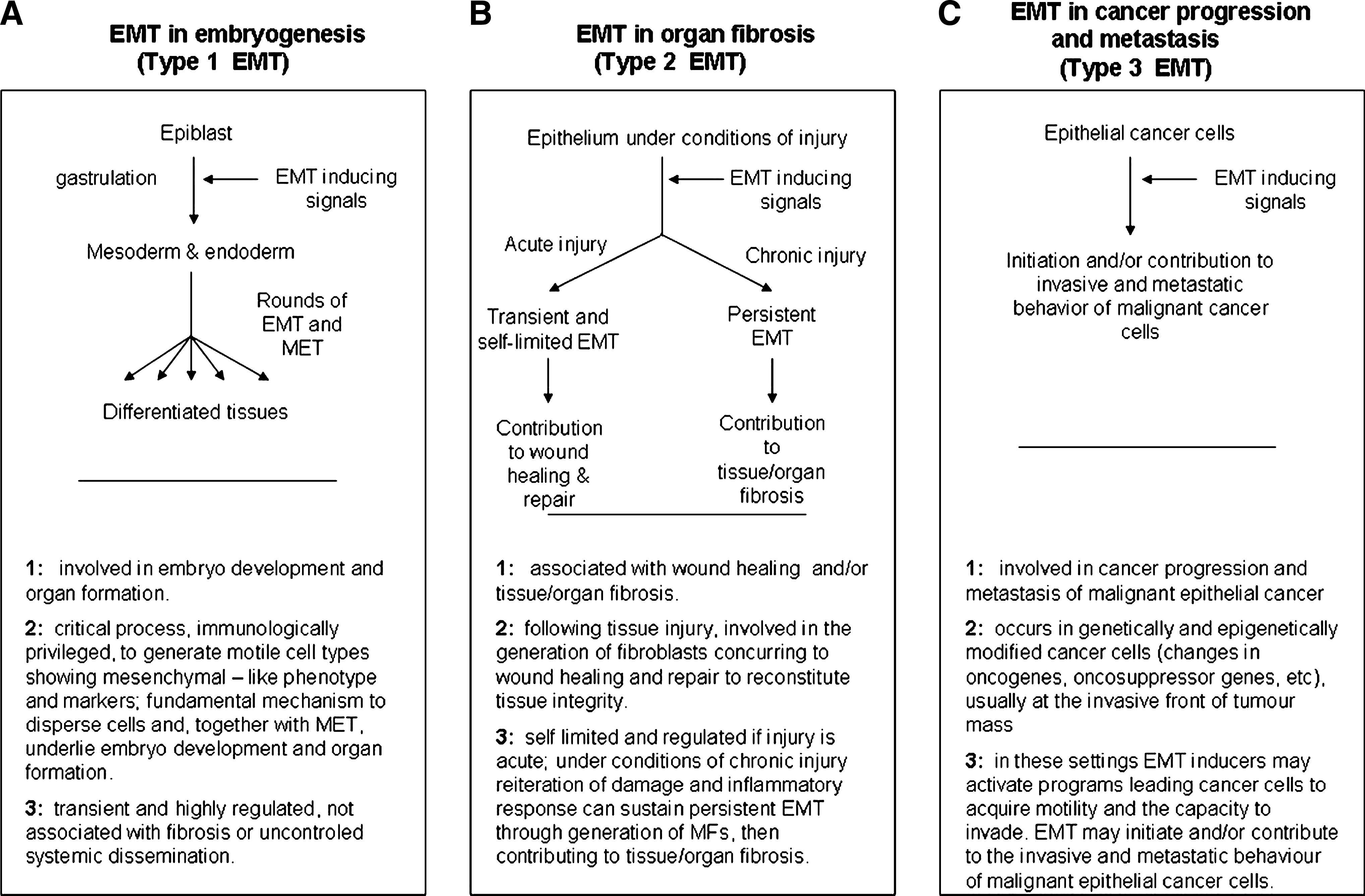
In the previous decade, the EMT process (and then possibly MET) was identified in at least two other well-defined pathophysiological conditions (2, 15, 27, 35, 114, 139, 168, 227, 250, 251, 298), including organ fibrosis and cancer progression and metastasis, leading to the recent suggestion that EMTs may be even classified into corresponding three different subtypes (2, 114, 298): a) Type 1 EMT, involved in embryonic development; b) Type 2 EMT, associated with tissue damage, regeneration, and organ fibrosis; c) Type 3 EMT, involved in cancer progression and metastasis (Fig. 4). However, the reader should note that it is believed that the EMT programs and related major morphological and functional events, detected in either physiological or pathophysiological conditions, are stimulated and regulated by a common set of inducing stimuli, signal transduction pathways, transcription factors, and post-transcriptional mechanisms (2, 15, 27, 35, 114, 126, 139, 168, 227, 250, 251, 298). In this review, in order to facilitate the reader coming across the EMT literature, the traditional way to present the EMT process will be flanked in the title of subsections or, as in Figure 4, by a direct reference to the recently proposed classification.
On these premises, this review offers a comprehensive introduction to current knowledge in the field of EMT. A first section will describe in detail major relevant features of EMT. Following sections will focus on signaling mechanisms that are known to regulate or affect the process, including the recently proposed role for oxidative stress and reactive oxygen species (ROS), as well as to critically analyze current literature data involving EMT in both physiological conditions and major human diseases. The section dedicated to the involvement of EMT in human diseases, with a major focus on organ fibrosis (i.e., the field which is most familiar to authors), will also introduce some note of caution in the interpretation of results. Indeed, not always data provided by literature can unequivocally identify a role for EMT or do not take into account other relevant features in the overall scenario. We believe that caution is necessary to avoid the potentially misleading message, particularly when talking about of chronic activation of wound healing in clinically relevant human chronic diseases, that EMT is the major, if not the only relevant, mechanistic feature involved.
B. Epithelial–mesenchymal transitions under the lens
It has been authoritatively pointed out that the sudden explosion of literature studies in the field is unavoidably leading to some confusion concerning the correct use of EMT (114, 250, 298); accordingly, the suggestion is to restrict this definition just to those studies reporting unequivocal signs of in vitro and, possibly, in vivo transdifferentiation.
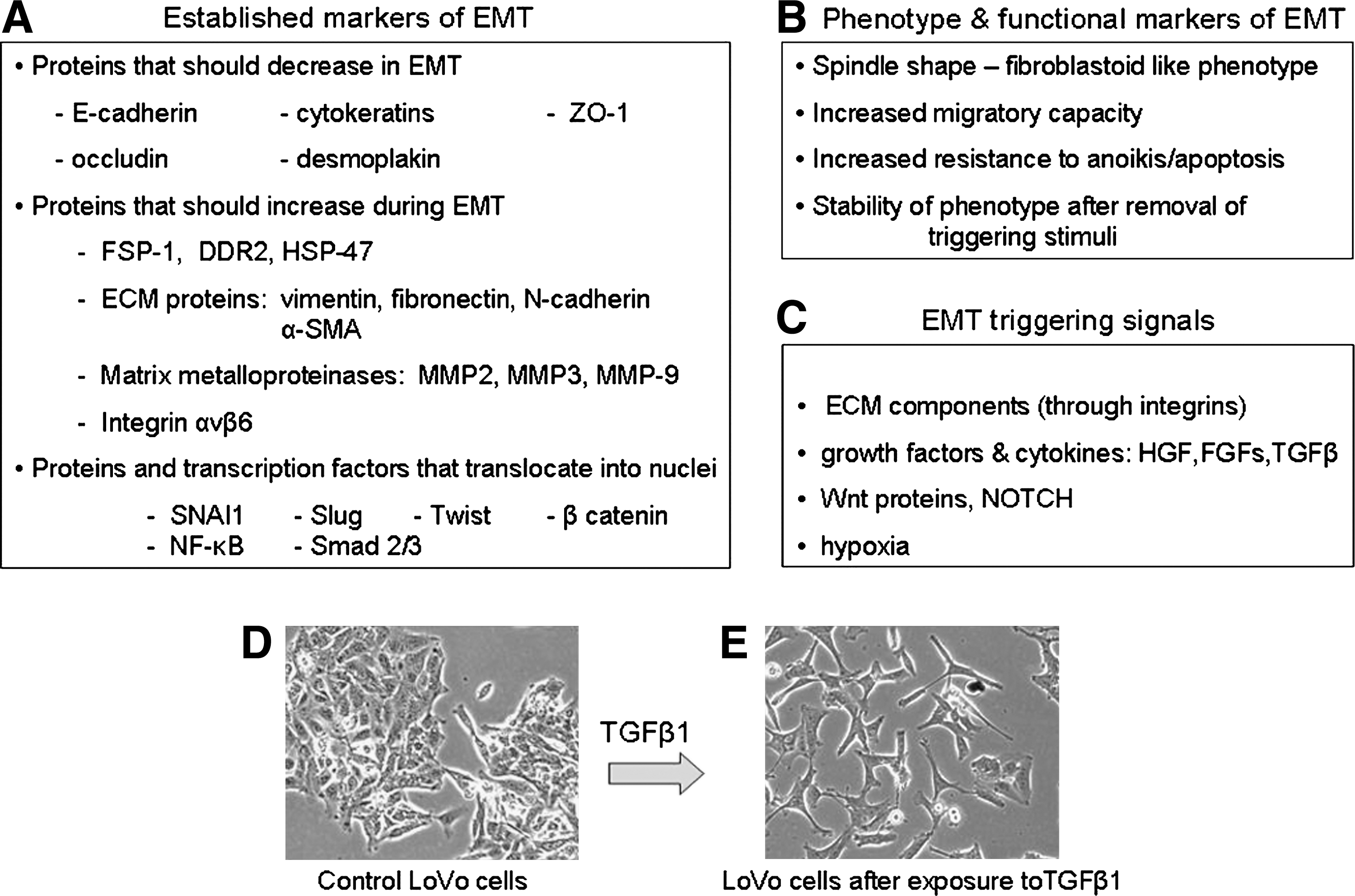
The problem for anyone interested in detecting the process is then apparently simple: to search for defined markers and parameters able to unequivocally recognize EMT. This goal is relatively simple to obtain if cell culture models are used and literature (114, 229, 250, 298) indeed suggests to focus on a number of selected features that are summarized in Figure 2.
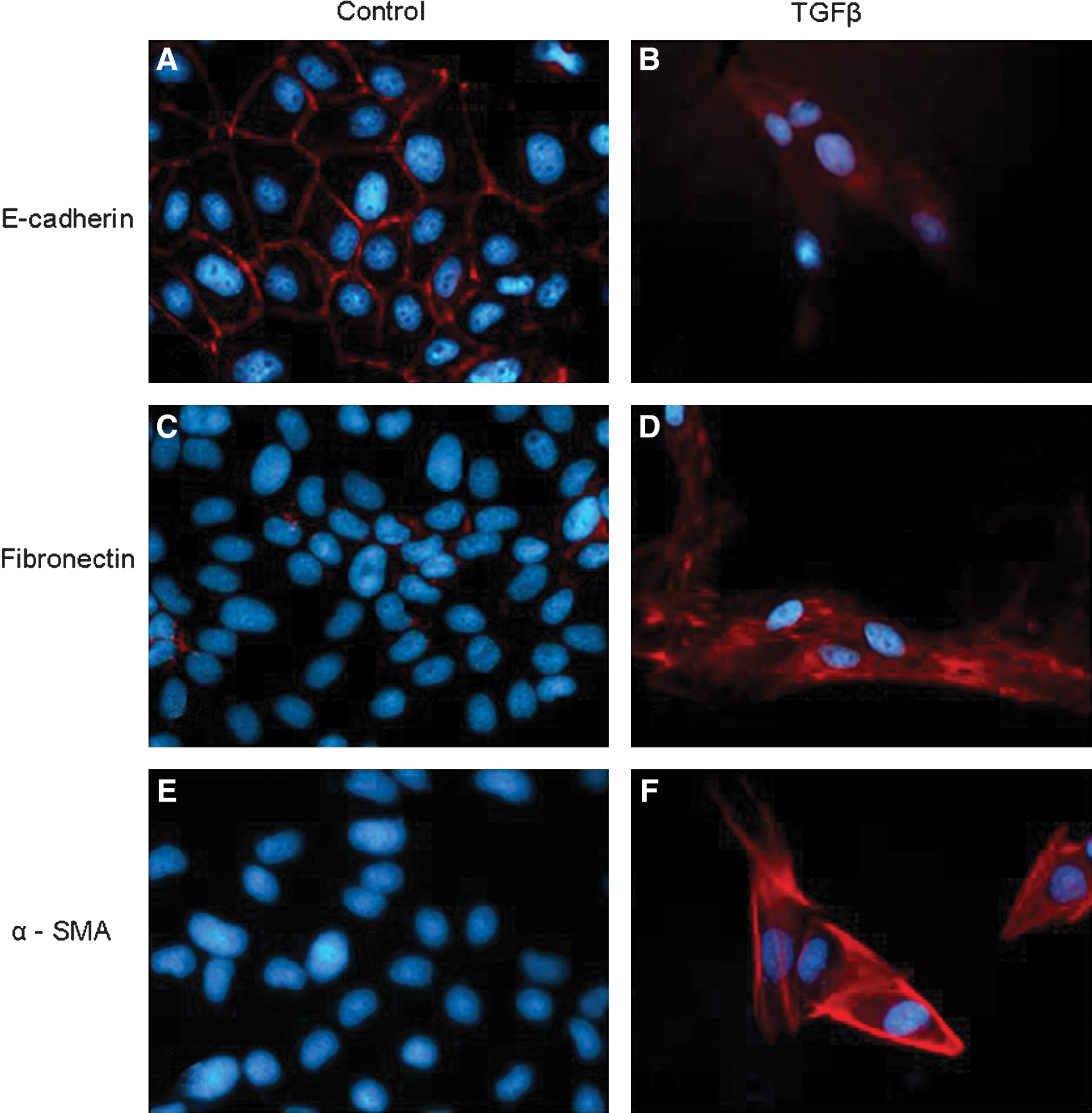
The process of EMT classically involves a series of events that lead epithelial cells to lose their characteristics and acquire those features and properties that are usually attributed to mesenchymal cells. This will encompass a spectrum of changes in intracellular and intercellular architecture as well as cell behavior that can be identified by integrating the morphological approach to identify selected markers of EMT with some specific cell biology assay. As it will be detailed in the section dedicated to signaling pathways regulating EMT, this may also include the search for the involvement of some specific transcription factor or the activation of a defined signaling component.
The initial step of any EMT involves a progressive disruption of specialized junctional structures of epithelial cells that follows exposure to a number of biological effector signals (growth factors, cytokines, changes in ECM, hypoxia, ROS, etc) with a time-dependent “kinetics” of changes that usually requires first the dissociation of tight junctions and then the dissociation of adherens junctions and desmosomes. Along these lines, the most direct approach to appreciate EMT is to follow time-dependent changes in cell morphology: cells of epithelial origin (including cancer cells) in culture should lose cell contacts and acquire an evident fibroblast-like, spindle-shaped morphology, often scattering (i.e., moving away) from original cell clusters (Fig. 3). A commonly associated approach is then to follow changes in those cellular markers indicated in Figure 2. Where markers are concerned (Figs. 2 and 3 and Ref. 298), a minimal requirement for the use of the acronym EMT is to show (by immunofluorescence, immunohistochemistry, Western blot analysis, etc.) that morphological changes are associated with downregulation of E-cadherin protein levels and upregulation of most common mesenchymal markers such as vimentin, fibronectin, α-smooth muscle actin (α-SMA), fibrillar collagen (types I and III), S100 calcium binding protein A4 (S100A4, also known as fibroblast-specific protein 1 or FSP-1) as well as increased activity of defined matrix metalloproteinases (MMPs) like MMP-2, MMP-3, and MMP-9. This approach can be enlarged by searching for more specific molecular markers such as SNAI1 (snail homolog 1, Drosophila) and β-catenin (CTNNB1 or catenin (cadherin-associated protein) beta 1) that may be related to the activation/involvement of specific signal transduction pathways in the EMT process, depending on the specific effector under analysis or the overall prevailing biological signal or mediator in the microenvironment. A final preliminary proof confirming that EMT has occurred is usually offered by convincing evidence of an increased migratory ability of the cell type under analysis, that can be obtained by using the so-called wound-healing assay (WHA), the classic assay of chemotaxis based on one of the several available modifications of the classic Boyden's chamber or, particularly relevant if cancer cells or epithelial cells supposed to give origin to myofibroblasts are analyzed, the classic invasion assay (33, 180, 181).
As a cautionary note, one should keep in mind that the overall scenario of EMT just presented for cell culture and summarized in Figure 2 is a general one and that the resulting spectrum of changes may vary significantly depending on the cell type involved, the effector cytokine or mediator under investigation or the prevailing overall biological signals in a defined and controlled microenvironment. In other words, one should not expect, for example, to detect all the mentioned changes for mesenchymal markers at the same time in the same cell type; moreover, sometimes the mesenchymal-like changes may be limited to increased protein levels that, as may happen for α-SMA or vimentin, do not further assemble into the standard fibrillar form.
In principle, the same panel of markers that are used to detect EMT in a controlled condition of cell culture should be investigated also in vivo under pathophysiological conditions, with disruption of basement membrane being an additional criterion (298). However, the fact that EMT can be induced by a wide variety of effectors or can occur in so many conditions of clinical interest makes it somewhat difficult to unequivocally identify the process in a pathological specimen. Nevertheless, there are markers that are considered as suggestive of in vivo occurring EMT in organ fibrosis or cancer progression, as is the case for S100A4 (or FSP-1). The S100A4 protein, in particular, is part of a large family of Ca2+- binding proteins called S100 (58, 224, 244) that share the common feature of two Ca2+-binding EF-hand motives and usually exist as dimers (245). Detection of FSP-1 has been reported in connection with EMT involvement in a large variety of pathophysiological conditions in either experimental models of disease or human specimens or biopsies, as recently extensively reviewed (229).
II. Molecular Mechanisms Involved in EMT
A. General concepts
Initiation of the EMT process obligatory requires proper “signals” originating from outside the epithelial cells that, in turn, involve an extensive intracellular machinery of signal transduction pathways, transcription factors, target genes, and other regulatory mediators. One should acknowledge that experiments performed on tissue cultures have been instrumental for outlining the molecular and signaling mechanisms involved in EMT induction and regulation. Where initiation signals and signaling pathways are concerned, a number of crucial general concepts should be outlined: a) Several extracellular signals are able to trigger EMT that are indeed not specific but, rather, multifunctional, since they can also induce proliferation or several other adaptative responses, depending on the local microenvironment; the list of signals able to induce EMT includes ECM components (such as collagen and hyaluronic acid), several soluble polypeptide growth factors, including at least hepatocyte growth factor (HGF ), members of the families of platelet-derived growth factor (PDGF ), fibroblast growth factor (FGF ), and transforming growth factor β (TGFB1, TGFB2, TGFB3, usually indicated as TGFβ1, TGFβ2, TGFβ3) as well as different isoforms of WNT (wingless-type MMTV integration site family) proteins (2, 114, 227, 250, 298) or MMP (210, 250) or several members of the family of bone morphogenetic proteins or BMP (83). Recent experimental evidence, with studies mainly performed on cancer cells of epithelial origin, has added to the list the presence of hypoxia as a condition potentially able to induce EMT (33, 107, 141). b) EMT signals from the extracellular environment, apart from hypoxia, are integrated at the membrane level by specific receptors involving related intracellular signal transduction pathways; indeed, in response to outside signals, EMT is carefully modulated (either activated or repressed) by several signaling pathways that show an impressive and very significant degree of cross-talk. c) Most signals and signaling pathways triggering EMT have several end-points in common, including downregulation of E-cadherin expression, as well as of other EMT-associated genes. Some signals also share, as common targets, cellular cytoskeleton and junctional structures. d) The specificity of action of a single extracellular signal able to affect EMT is not absolute or unequivocal but strictly dependent on the tissue context. An excellent example is offered by the action of HGF that during somitogenesis can induce EMT (250) and is widely accepted as a major inducer of EMT in different normal and neoplastic cells (2, 114, 227, 250, 298). However, in other conditions, HGF may operate by counteracting EMT, as shown recently by HGF-mediated inhibition of EMT of tubular epithelial kidney cells towards the myofibroblast phenotype (reviewed in Ref. 149). Moreover, HGF is actually even proposed as a potential therapeutic tool to counteract the pro-fibrogenic action of TGFβ, leading to myofibroblast activation in kidney fibrosis (104, 149, 284, 292).
B. E-cadherin downregulation: The major role of SNAI1 and GSK-3β
The list of signals potentially able to trigger EMT is impressive and continuously growing (for an updated list, see Fig. 5). A detailed analysis of all the molecular mechanisms described in the literature is beyond the scope of this review and the interested reader can refer to several excellent and authoritative reviews in the specific field (2, 114, 227, 250, 298). This section will be then dedicated to a brief overview of all major and well-established mechanisms and signaling pathways involved in EMT. According to this approach, we will first focus on a number of selected growth factors able to interact with their cognate receptors, either tyrosine-kinase receptors or serine-threonine kinase receptors, as well as on ECM stimulation of integrins that are all able to activate downstream signal transduction pathways resulting in two major features (Fig. 6):downregulation of cell–cell adhesion structure components, with E-cadherin being the most relevant gene target; dynamic rearrangement of the actin cytoskeleton that is necessary to accomplish the acquisition of migratory properties.


The crucial event in EMT is represented, without any doubt, by E-cadherin downregulation that is the most relevant step in reducing cell–cell adhesion, eventually leading to destabilization of the epithelial architecture (2, 27, 88, 114, 227, 250, 298). This statement is reinforced at least by the following considerations: repression of E-cadherin gene has been shown to be sufficient for induction as well as completion of EMT; reactivation of E-cadherin gene is a crucial event for the reverse process of MET; most signal transduction pathways and molecular mechanisms involved in EMT ultimately converge on E-cadherin expression.
A central role in E-cadherin gene repression is attributed to the zinc finger transcription factor SNAI1 that is activated by most of the signaling pathways able to trigger EMT (Figs. 6 –8). SNAI1 has been shown to operate as a repressor of E-cadherin gene by binding to the two E-boxes of the human and murine E-cadherin promoter, both sharing an identical core consisting of the sequence 5’-CACCTG (14, 34). A confirmation of the relationships between SNAI1 and E-cadherin expression has been obtained by experiments in which SNAI1 overexpression in different epithelial cells leads to an unequivocal conversion towards a fibroblastic phenotype at the same time that E-cadherin expression is lost (34). Moreover, SNAI1 is also able to act by upmodulating other mesenchymal genes.
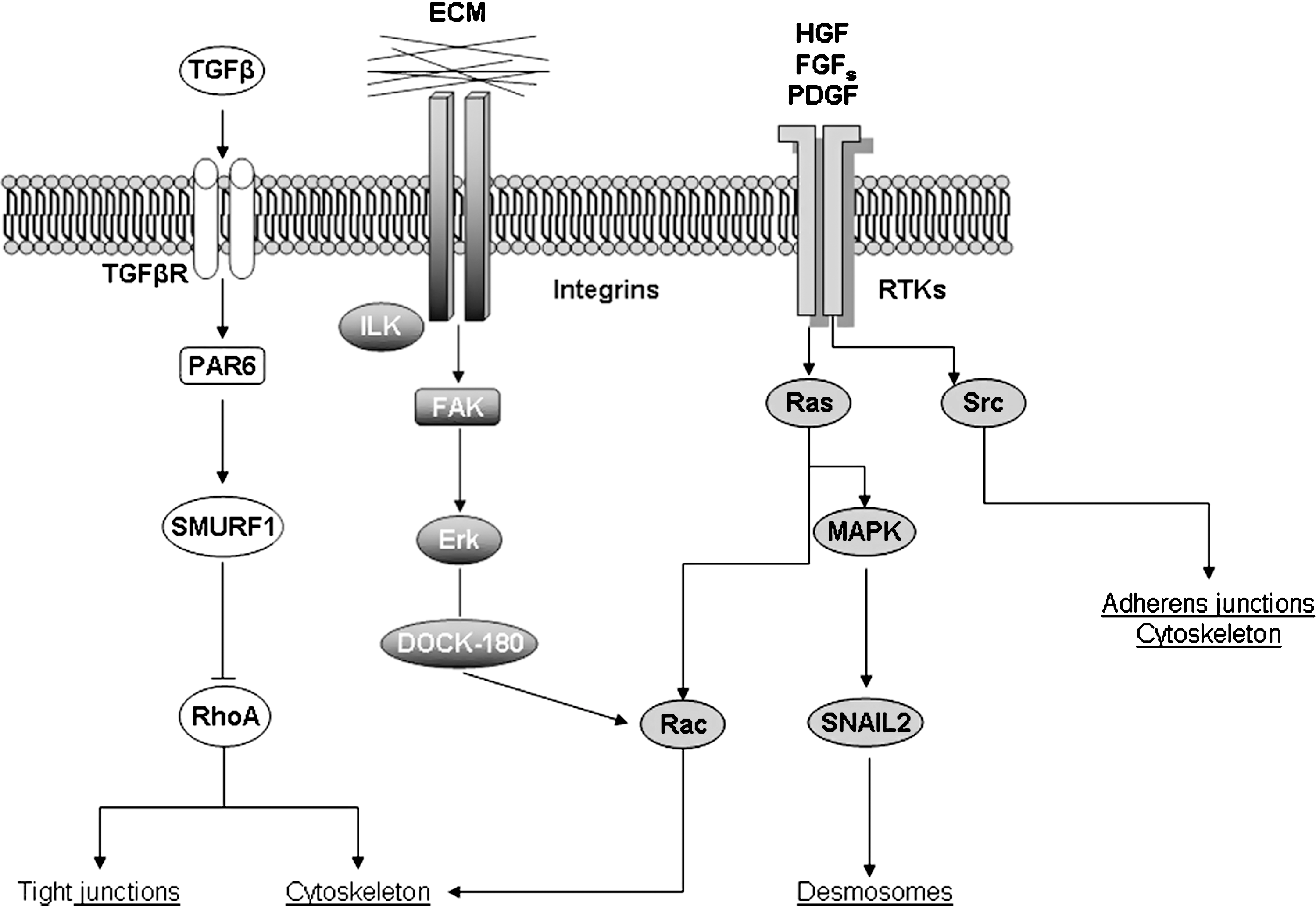
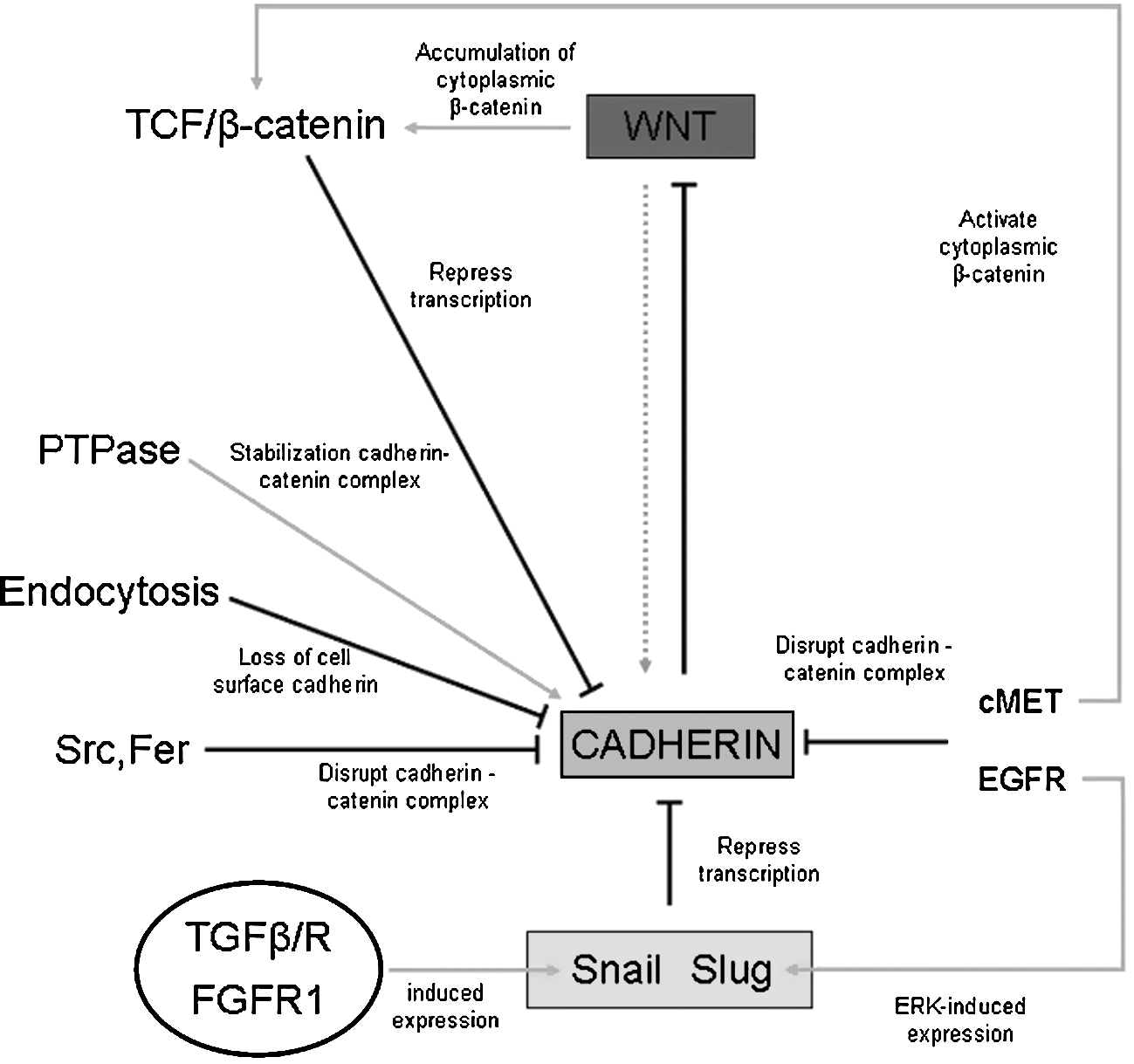
Both SNAI1 transcription and transcriptional activity of SNAI1 are negatively modulated by the activity of glycogen synthase kinase-3β (GSK3B, usually referred as GSK-3β) (8), a kinase which is known to be active in resting epithelial cells (170, 192) and is then a critical determinant of EMT. In particular, experimental data indicate that active GSK-3β can bind and phosphorylate serine residues of SNAI1 at two consensus motifs to dually regulate its function. Phosphorylation of the first motif regulates its BTRC (beta-transducin repeat containing) mediated ubiquitination and subsequent proteasomal degradation, whereas phosphorylation of the second motif controls its subcellular localization (200, 305). Indeed, inhibition of GSK-3β results in the upregulation of SNAI1 and downregulation of E-cadherin both in vitro and in vivo (8, 305).
Such a direct relationship between GSK-3β, SNAI1, and E-cadherin expression is relevant for at least two reasons: it implies that sustained activation of GSK-3β is a mechanism by which resting epithelial cells can avoid EMT. As we will see, several signal transduction pathways that are able to trigger EMT (within classic inducers those involving polypeptide growth factors and related receptors, integrins, and integrin-linked kinase or ILK, WNT/β-catenin) indeed converge on GSK-3β inhibition as a critical step to control SNAI1 nuclear translocation and then E-cadherin downregulation (see Fig. 6 and later in this section for more details).
SNAI1 is able to repress E-cadherin promoter at least by two mechanisms. The N-terminal SNAG domain of SNAI1 can interact directly with histone deacetylase 1 and 2 (HDAC1 and HDAC2), as well as with the corepressor mSin3A, then mediating the repression by recruitment of chromatin-modifying activities, forming a multimolecular complex to repress E-cadherin expression (198). Alternatively, SNAI1 can form a ternary complex by recruiting the cytosolic AJUBA protein that, in turn, can function as a scaffold protein to bind the protein arginine methyltransferase 5 (PRMT5); this ternary complex recognizes the proximal promoter region of E-cadherin gene exerting a silencing activity (102).
Although SNAI1 is likely to have a predominant role, other repressors of E-cadherin promoter have been identified, such as SNAI2 (a member of the SNAI family, also designated as Slug), the basic helix–loop–helix factors E47 and Twist, as well as the two-handed zinc factors ZEB1 and SIP1 (also known as ZEB2) (201). Very recently, the E2-2 basic helix–loop–helix transcription factor has been added to the list of factors able to induce full EMT, as shown in Madin–Darby canine kidney cells (MDCK). Both isoforms of E2-2 (E2-2A and -2B) were able to induce EMT by E-cadherin repression; however, the latter event mediated by E2-2 is indirect and independent on proximal E-boxes of the promoter (238).
At this point, it seems appropriate to refer the reader to some cautionary notes concerning E-cadherin downregulation, SNAI1 and other repressors. As recently discussed by Klymkowsky and Savagner (see Ref. 126 and references therein for a more detailed analysis), a number of published studies suggest that the overall scenario may be more complex, particularly when cancer cells are concerned. As early as 1992, a report on NBT-II rat bladder carcinoma cells showed that EMT-related modifications induced by FGF, including overexpression of vimentin in motile cells, were not accompanied by a loss of E-cadherin expression or a reduction of the intercellular adhesiveness (24). Similarly, other studies have reported that in certain cancers (mainly breast and colon carcinomas) SNAI1 and E-cadherin were found to be co-expressed (16, 43).
C. Major signaling mechanisms related to growth factors acting on tyrosine kinase receptors
Where growth factors (such as EGF, FGF, PDGF, HGF, IGF ) and related tyrosine kinase receptors (RTKs) are concerned, it is well known that RTKs dimerize after ligand binding and autophosphorylate on tyrosine residues which, in turn, act as docking sites for SH2 domains containing proteins such as growth factor receptor-bound protein 2 (GRB2), phosphoinositide-3- kinase (PI3K), and Src (SRC) that, once recruited, can stimulate their respective downstream signaling pathways (153, 243). This includes also the activation of Ras that does not possess SH2-domains but is activated following Grb-2-mediated recruitment of the guanine nucleotide exchange factor Sos (SOS, son of sevenless homolog, Drosophila). Sos, in turn, allows activation of Ras by converting the GDP to the GTP-bound form and then the activation of the Ras–Raf (RAF1)– MEK1 (MAP2K1–ERK1/2 (MAPK1, MAPK3) cascade. This ultimately leads to MAPK nuclear translocation and regulation of gene expression by means of phosphorylation of several transcription factors such as SNAI2 and those belonging to Ets family, including Jun and Fos (25, 46, 153, 243). The connection with EMT is immediate with SNAI2, which is a known repressor of E-cadherin (50, 201) but also relies on the fact that AP-1 and Ets factors are believed to represent putative mediators of EMT (25, 49, 103). Moreover, activated MAPK can directly suppress the activity of GSK-3β by phosphorylation (55, 73, 163), thus potentially upregulating SNAI1 functions.
Following interactions between polypeptide ligands and related RTKs, PI3K is also activated and can generate the second messenger phosphatidylinositol-3,4,5-triphosphate (PIP3) which, in turn, leads to activation of the serine/threonine kinase Akt (AKT1, also defined as protein kinase B or PKB) that is known to have a role in cell cycle progression, cell proliferation and survival. However, PI3K activation is also considered a major molecular effector of EMT (138) since, through Akt, can phosphorylate and inactivate GSK-3β. This is relevant because inactivation of GSK-3β will prevent GSK-3β-dependent phosphorylation of both SNAI1 and β-catenin, then preventing their proteasomal degradation. This allows nuclear translocation and transcriptional activity of SNAI1 and β-catenin that can both promote EMT (see later concepts related to the involvement of WNT/β-catenin signaling pathway in EMT) (11, 20, 305). The same mechanism can be elicited also by HGF that can lead to a transient decrease in GSK3β activity and a parallel selective increase in the uncomplexed pool of β-catenin (192). Moreover, it has been also reported that the Met receptor tyrosine kinase can operate in a more direct way to regulate intracellular localization of β-catenin by phosphorylating a specific β-catenin tyrosine residue (Y142) and then preventing α-catenin interaction (20). In addition, the PI3K/Akt pathway can also activate Rho GTPases and cooperates with TGFβ signaling to affect EMT (11, 19).
Growth factors that interact with RTKs, through the involvement of Ras and PI3K mediators as well as other EMT-inducing signaling pathways, also affect the activity of the Rho family of small GTPases, including cell division cycle 42 GTP binding protein or Cdc42 (CDC42), ras homolog gene family Rho (RHO) proteins, and Rac. These proteins are crucially involved in the regulation of E-cadherin-based cell adhesiveness as well as in the rearrangements of the actin cytoskeleton (i.e., by regulating assembly/disassembly equilibrium) that are essential for changes in cell shape and the acquisition of migratory properties, the last step of EMT (65, 97, 101, 118, 307). A related example is offered by the reported PDGF-BB-dependent induction of EMT in proepicardial cells, a condition leading to smooth muscle cell differentiation and requiring ras homolog gene family member A or Rho-A (RHOA) mediated actin reorganization and p160 Rho-kinase activity (151). Moreover, activation of RhoA seems to be critical in cancer cells to mediate EMT (17, 65, 118) and it should be underlined that increased Rho activity is also playing a role in mediating TGF-β-dependent EMT (11, 19).
Another relevant mediator of EMT is represented by Src, a cytoplasmic tyrosine kinase downstream of both growth factor receptors (including RTKs) and integrins with a well-known role in the control of cell growth, cell adhesion, and cell motility (6). In normal epithelial cells, low levels of activity of Src family kinases are required to maintain the integrity of epithelium. However, activated Src has been shown to be able to phosphorylate focal adhesion kinase (FAK) that, in turn, leads to activation of MAPK and in the phosphorylation of myosin light chain kinase (MYLK). MYLK will of course phosphorylate myosin light chains, an event which is believed to correlate with disorganization of E-cadherin-mediated cell–cell contacts as well as with the acquisition of a mesenchymal-like phenotype and the ability to migrate. MYLK can be also phosphorylated by a RhoA-dependent pathway involving ROCK signaling (see Fig. 7 that shows different pathways converging on adhesion switch and dinamyc changes leading to migratory capacity). Activated Src can also induce endocytosis of E-cadherin through activation of the E3 ubiquitin ligase Hakai (77) or through the ARF family GTPase ARF6 (ADP-ribosylation factor 6) (190).
D. TGFβ, Wnt/β–catenin, and ECM-related signaling in EMT
TGFβ (see also Fig. 8) can be considered as one of the most potent inducers of EMT and its action is mediated by interactions with Type I and Type II TGFβ-related serine-threonine kinase receptors (TGFßRI and TGFßRII). After binding of the ligand, TGFßRI and TGFßRII can form an heteromeric complex with TGFßRII being then able to trans-phosphorylate TGFßRI; phosphorylated TGFßRI, in turn, can phosphorylate the related effectors represented by cytoplasmic Smad proteins (Smad2 and Smad3). Phosphorylated Smad2/3 will form an heterodimeric transcription complex with Smad4 that, upon nuclear translocation, binds to chromatin and regulates the expression of several target genes involved in the control of cell proliferation, apoptosis, cell migration, and cell differentiation, including those related to EMT (35, 157, 169). However, TGFβ can also signal by Smad-independent mechanisms that include activation of MAPK (through activation of Ras), PI3K, and integrin-linked kinase (ILK) pathways.
Where relationships between TGFβ and EMT are concerned, heterodimeric Smad transcriptional complexes can act by affecting the expression of several other transcription factors. The following pertinent relevant findings have been reported (35): a) TGFβ, through Smad signaling, is able to transcriptionally repress Id genes that are known inhibitors of differentiation and EMT (128); this Id repression is required for subsequent downregulation of E-cadherin and ZO1 (TJP1, tight junction protein 1, zonula occludens 1), then resulting in EMT. b) TGFβ can transcriptionally induce SNAI1 expression through either Smad complexes or via activation of ERK and PI3K pathways (202). c) TGFβ, through Smad complexes, can transcriptionally induce the expression of high mobility group A2 (HMGA2) chromatin associated protein that, in turn, induces expression of transcriptional regulators of E-cadherin promoter such as SNAI1, SNAI2, and TWIST (253). d) Smad complexes can interact with ZEB1 or ZEB2, then forming repressor complexes on the E-box region of the E-cadherin gene as well as of other genes (232). e) TGFβ, through Smad signaling, has been reported to be able to activate directly LEF1 (172, 173) or indirectly via Wnt signaling pathway (134,135), a mechanism that again results in E-cadherin downregulation. f) TGFβ, through Smad complexes, can transcriptionally induce the expression of a number of mesenchymal genes, including vimentin, N-cadherin, fibronectin, α-SMA, and plasminogen activator inhibitor 1 or PAI-1 (reviewed in Refs. 172,173, 271,292). g) Activated TGFßRI can interact with several proteins leading to signaling pathways that regulate dissolution of tight junctions; examples are represented by interaction of TGFßRI with occludin, PAK-1, and PAR6 (PARD6A). The case of PAR6 is of particular interest. PAR6 operates as a scaffold protein for the assembly of polarity-regulating proteins like Rho, aPKC, and PAR3 (PARD3), then affecting the assembly of tight junctions. PAR6 is able to form a complex with TGFßRI and occludin, and exposure of cells to TGFβ can recruit also TGFßRII to this complex; TGFßRII can phosphorylate PAR6 that in turn binds to SMURF1, a E3-ubiquitin ligase, leading to ubiquitination-mediated degradation of RhoA and then contributing to dissolution of tight junctions through subsequent depolymerization of filamentous (F )-actin (35, 186). h) TGFβ induction of EMT has been reported to require and/or signal also through integrin-linked kinase (ILK) (144), a kinase that interacts with the cytoplasmic domains of β1 and β3 integrins, is activated through cellular interactions with ECM and growth factors and is able to downregulate E-cadherin expression (183).
The Wnt/β–catenin pathway is another major signaling mechanism involved in EMT that is able to transduce within the nucleus signals that link cell–cell contacts and cell adhesion to the cellular response. As elegantly reviewed some years ago (174), an intense convergence of Wnt, β-catenin, and cadherin pathways exists that has a major role in regulating gene expression and interactions with neighboring epithelial cells during differentiation, including EMT. The 92 kDa protein β-catenin has a central role in this scenario since it may exist in three different functional forms: as a complex with E-cadherin that is involved in the regulation of cell adhesion (β-catenin binds the cytoplasmic tail of E-cadherin in order to link it to α-catenin and then to F-actin cytoskeleton); as a part of a multisubunit complex also formed by axin, adenomatous polyposis coli (APC) and GSK-3β, in which β-catenin undergoes a GSK-3β-dependent serine-threonine phosphorylation, being then targeted by BTRC for ubiquitination and proteasomal degradation; finally, as a transcriptional complex formed with TCF/LEF transcription factors. Where the Wnt/β–catenin pathway is concerned, the signaling is activated when one of the several Wnt isoforms can bind plasma membrane Frizzled (FZ) receptors; this is followed by recruitment to the plasma membrane and activation of Dishevelled (DVL) that, in turn, phosphorylates and inactivates GSK-3β. Inactivation of GSK-3β is crucial because it allows the formation of the transcriptional complex β-catenin/TCF/LEF than can translocate into the nucleus in order to activate an extensive list of target genes that transcribe for proteins involved in EMT like fibronectin, vimentin, matrilysin, SNAI2, Ets, and Jun (46, 174). Alternatively, the β-catenin-related signaling can be activated by all those mechanisms, resulting in downregulation of E-cadherin, leading to an accumulation of β-catenin in the cytoplasm, or by those signaling pathways that are able to converge to phosphorylate GSK-3β, including activation of ILK-, PI3K/Akt-, or MAPK/Ras pathways. Since GSK-3β inactivation is a major mechanism leading to SNAI1 upregulation, β-catenin-related signaling will then result in destabilization of the epithelial phenotype and EMT triggering (8, 46, 174, 305). The resulting overall scenario delineates an intense cross-talk between different signaling pathways, with GSK-3β representing then a crucial cellular crossroad. In addition, the scenario is even more complex for at least three more reasons (174, 285): a) The activation of the Wnt/β–catenin pathway is negatively regulated by a number of proteins that can bind Wnt isoforms on the external plasma membrane surface, then preventing their binding to Frizzled receptors, including Wnt inhibiting factor 1 (WIF1), Frizzled-related protein (FRZP), or to the co-receptor LRPAP1 (low density lipoprotein receptor-related protein associated protein 1) as Dickkopf (DKK). b) A further element of complexity is introduced by the regulation of the cadherin–catenin complex that results from the balance of the activity of tyrosine kinases (i.e., both RTKs and cytoplasmic TKs) and of protein tyrosine phosphatases (PTPs). Phosphorylation of specific tyrosine residues of β-catenin (Y654, Y142) by RTKs and cytoplasmic TKs will lead to dissociation of the cadherin–catenin complex, then releasing β-catenin into the cytoplasm, whereas aspecific phosphorylation of β-catenin by casein kinase II or tyrosine dephosphorylation by PTPs will result in the opposite effect. c) β-Catenin may be involved in EMT through a Wnt-independent mechanism, as shown recently by a study performed on human HT-29 cells undergoing PDGF-BB-dependent EMT (285). This study has revealed that PDGF-BB can induce EMT through nuclear phosphorylation of a specific tyrosine residue (Y593) of p68 RNA helicase that, in turn, resulted in β-catenin nuclear translocation by blocking β-catenin phosphorylation by GSK-3β and displacing Axin from β-catenin.
Other relevant signals able to trigger and modulate a spectrum of cell responses like proliferation, survival, differentiation, and migration, then including also EMT, can be conveyed by matrix degrading proteases, extracellular matrix (ECM) components, and by integrins, the latter operating as ECM receptors.
Matrix degrading proteases can belong to the family of metalloproteinases (in particular MMP2 and MMP9) cysteine proteinases or urokinase-type plasminogen activator (u-PA) system and may participate to EMT and migration by a number of mechanisms that can be briefly summarized as follows (88): a) By degrading ECM, an event that, apart from favoring migration, may alter the extracellular milieu and, in turn, affect and/or modulate cells responses. Moreover, matrix degradation will also favor the release of growth factors and survival factors stored in the ECM that may be able to trigger EMT. b) By proteolysis of extracellular E-cadherin domains that may result in loss of cell–cell adhesion, dissociation of β-catenin from cadherin–catenin transmembrane complexes as well as formation of E-cadherin fragments able to favor migration or, in cancer cells, invasion (150, 155). c) Some selected MMPs can directly elicit EMT in target cells; this has been shown for MMP3, also known as stromelysin-1, which is a stromal metalloproteinase found to be upregulated in many human cancers. MMP3 has been reported to induce classic EMT changes in the nontumorigenic mouse mammary epithelial cell line SCp2 by eliciting first a cleavage of E-cadherin, resulting then in dissociation of cell adhesions and relocalization of β-catenin. However, when further investigating the phenomenon, Radisky and coworkers found that the underlying relevant event was a MMP3-dependent upregulation of the expression of a splice-variant of Rac1 that, by increasing intracellular levels of reactive oxygen species (ROS), was resulting in upregulation of SNAI1 transcription factor as a major determinant of EMT (209, 211). This mechanism will be discussed later in the section about the role of redox signaling in EMT.
Where extracellular matrix components are concerned, an induction of EMT by collagens (types I, III, IV, and V) was early suggested by using NBT-II rat bladder carcinoma cells (260). Guarino then proposed (89) that peritumoral ECM may favor EMT and then cancer cell invasion, suggesting the contact of epithelial cells with an interstitial type of collagen as the putative relevant switch. This has been indeed confirmed when cells cultured on fibrillar type collagens (collagen type I and III) showed evident EMT changes associated with increased motility and invasiveness (160). These findings also disclosed the role of integrins and, later, of the serine/threonine integrin-linked kinase (ILK), a signaling protein stimulated by both integrins and growth factor receptors. ECM-induced stimulation of integrins results in their clustering at adhesion sites and in the subsequent recruitment and activation of a number of signaling protein mediators, including focal adhesion kinase (FAK), Src, Ras, PI3K, Rho GTPases, and ILK (81), then eliciting different signaling pathways potentially able to trigger EMT. In particular, after being recruited at the adhesion site, FAK undergoes autophosphorylation, leading to binding and activation of Src that, in turn, phosphorylates FAK at tyrosine residues: this event results in both binding and activation of PI3K, thus leading to activation of the PI3K–PIP3–Akt pathway, and formation of SH2-binding sites for the Grb2–Sos complex, thereby resulting also in Ras–MAPK activation (81). FAK-dependent activation of Src can also lead to phosphorylation of the docking protein paxillin, which, in turn, can associate with the adaptor Crk in a paxillin–Crk–DOCK1 (also known as DOCK180) signaling complex that in the end leads to the activation of Rac (263).
Another way leading to EMT has been shown in mammary epithelial cells, where β3-integrin was identified as a critical mediator for TGFβ-induced EMT by a mechanism involving first formation of β3-integrin/TGFβRII complexes that blocked TGFβ-mediated growth arrest and increased -mediated invasion and EMT. Dual β3-integrin/TGFβRII activation induced tyrosine phosphorylation of TGFβRII, a phosphotransferase reaction mediated by Src in vitro that ultimately led to MAPK activation and EMT (79).
In this scenario of connection between ECM, integrins, and TGFβ, the last actor to be mentioned is represented by ILK, a kinase which interacts with the cytoplasmic domain of β-integrins and, when activated, can directly phosphorylate several downstream signaling targets, including Akt and GSK-3β. ILK-mediated inactivation of GSK-3β activity (an event reinforced also by Akt activation) is once again an event able to lead to both SNAI1 upregulation and nuclear translocation as well as stimulation of β-catenin nuclear translocation as a β-catenin/TCF/LEF transcriptional complex (8, 192). The effectiveness of ILK has been unequivocally confirmed by studies in which ILK overexpression resulted in increased β-catenin nuclear translocation, formation of β-catenin/TCF/LEF complex, and EMT induction (178, 239).
E. Emerging molecular mechanisms involved in EMT triggering
Recently, different laboratories have postulated a role for Notch signaling in both development and neoplastic EMT (104). The activation of the transmembrane receptor Notch by the Jagged ligand has been shown to result in Notch nuclear translocation and activation of target genes, including hairy/enhancer-of-split-related transcriptional repressor HEY1 which would promote E-cadherin downregulation and EMT. Interestingly, the same repressor HEY1 is involved also during TGFβ–Smad3 signaling, suggesting a joint functional role for Smad3, HEY1, and Jagged-1 during TGFβ-induced EMT (290). These results have been integrated by another study showing upregulation of SNAI1 by TGFβ through Notch signaling in heart development and endothelial cell transformation (254) and by a more recent observation indicating that, in vascular endothelial cells, Notch can signal by upregulating SNAI2, but not SNAI1, leading to VE-cadherin downregulation (177).
A role in EMT has been proposed also for the Hedgehog–Patched–Gli (Hh) pathway that may have a role particularly in cancer cells. The Hedgehog signaling pathway is essential for numerous processes during embryonic development and members of this family of secreted proteins (designated as Sonic or Shh, Desert or Dhh and Indian or Ihh in mammals) have been described to control cell proliferation, differentiation, and tissue patterning in a dose-dependent manner (197). In the absence of ligand, the Hh signaling pathway is inactive because the transmembrane protein receptor Patched-1 (PTCH1) inhibits the activity of Smoothened (SMO), a seven transmembrane protein.
The effector transcription factor Gli1 (GLI1, GLI family zinc finger 1), a downstream component of Hedgehog signaling, is prevented from entering the nucleus through interactions with cytoplasmic proteins, including Fused (FU) and suppressor of Fused homolog (SUFU). Activation of the pathway requires binding of any of the three major mammalian ligands to PTCH1 that results in de-repression of SMO, thereby activating a cascade that leads to the translocation of the active form of GLI1 to the nucleus to activate (197): a) transcription of genes codifying for the proteins involved in the pathway; b) Hedgehog interacting protein or HHIP, a Hedgehog binding protein that attenuates ligand, and genes that are involved in controlling cell proliferation (cyclin D, cyclin E, c-Myc, and components of the epidermal growth factor pathway), and in angiogenesis (components of the PDGF and VEGF pathways); c) induction of SNAI1 that can result in E-cadherin repression, then allowing EMT to occur (104).
Another signaling mechanism able to induce EMT requires interaction between endothelin 1 (EDN1, also indicated as ET1) and its receptor type A (EDNRA). Activation of EDNRA by EDN1 can trigger a PI3K-dependent and ILK-mediated signaling pathway leading to GSK-3β inhibition, SNAI1 and β-catenin stabilization, and transcriptional programs that control EMT (9). Alternatively, ET-1 has been shown to act as a relevant mediator of EMT in lung alveolar epithelial cells, acting through ETaR-mediated TGF-β1 production (110), a mechanism that may be involved in pulmonary fibrosis and suggests potential roles for AEC-derived ET-1 in the pathogenesis of other alveolar epithelium-mediated lung diseases.
Transcriptional regulation of genes involved in EMT may be under the control of additional putative factors that recognize as a major target the gene encoding for FSP-1 (265). This gene is upregulated early during the course of EMT and is controlled by a proximal cis-acting promoter element called fibroblast transcription site-1 (FTS-1) (182). FSP-1 transcription has been reported to follow formation of a complex involving FTS-1 and the proteins CArG box–binding factor-A (CBF-A) and KRAB-associated protein 1 (KAP-1). Indeed, kidney epithelial cells engineered to conditionally express recombinant CBF-A (rCBF-A) activate the transcription of FSP1 and undergo EMT. Moreover, the FTS-1 response element also exists in the promoter of other genes involved in EMT, including Twist, SNAI1, E-cadherin, β-catenin, ZO-1, vimentin, α1(I) collagen, and α–SMA.
BMPs, as members of the TGF-ß family of signaling proteins, are secreted ligands that, by binding specific receptors defined as bone morphogenic receptors type I and type II (BMPR1 and BMPR2), signal through autocrine and paracrine mechanisms to regulate cell proliferation and differentiation. The receptors are differentially expressed on organs and cell types and the presence of both types I and II receptors is essential for pathway activation. BMP ligand binding facilitates the heteromeric association of the type I and II receptors and receptor activation occurs through the phosphorylation of the type I receptor by the type II receptor. BMPR1 propagates a signaling cascade by phosphorylating Smads 1, 5, and 8, which results in the association of these Smad proteins with Smad 4. Association with Smad 4, in turn, enables the nuclear translocation of these complexes and the transcriptional activation of target genes. BMPs and their receptors, similarly to TGFβ, have been reported to induce EMT in embryo and fetal development as well as in cancer progression (reviewed in Ref. 10). It should be noted, however, that BMP-7 has been shown also to antagonize TGFβ-induced EMT in renal cells following renal cell injury (299).
F. The emerging role of microRNAs (miRNAs) in EMT
MicroRNAs are a large family of small and noncoding RNAs, evolutionary conserved in metazoan species, that modulate gene expression post-transcriptionally. miRNAs are synthesized by RNA polymerase II as longer transcripts, then processed by Drosha RNAse III endonuclease into ∼70–nt stem loop pre-microRNAs and transported into the cytoplasm by exportin 5 (105, 287). Pre-microRNAs are then processed by Dicer to provide the final ∼ 22–nt mature miRNAs that, by binding to target mRNAs, can induce either their selective degradation, when perfect or near perfect complementarity exists, or translational repression if complementarity is imperfect (105, 140, 287). miRNAS have been shown to be involved in regulation of embryogenesis and organ development (270, 286) as well as oncogenesis (66). Where miRNAs and cancer were concerned, several laboratories have provided data implicating miRNAs either as promoters or suppressors of metastasis (152, 247).
Very recently, different laboratories have reported that several miRNAs can selectively affect EMT in either normal cells as well as in tumor cells. A first relevant study has been performed on MDCK cells treated with TGFβ or transfected with the protein tyrosine phosphatase Petz (PTP-Pez), both reliable non-neoplastic models of EMT (87). The authors reported that all five members of the microRNA-200 family (miR-200a, miR-200b, miR-200c, miR-141, and miR-429) and miR-205 were significantly downregulated in cells that underwent EMT following TGFβ exposure or PTP–Pez transfection. Further experimental manipulations indicated that these miRNAs could cooperatively regulate E-cadherin expression by targeting the classic repressors ZEB1 and SIP1, as well as that inhibition of these miRNAs was sufficient to induce EMT. Moreover, expression of miR-200 family was lost in invasive cancer breast cell lines with mesenchymal phenotype. The same concepts were reinforced by very close results provided by two other studies that employed different normal and neoplastic cell lines (132, 193). Repression of miR-200 family was also recently proposed as a relevant mechanism contributing to PDGF-D-mediated EMT and increased invasiveness of prostate cancer cells (129,130).
Similar studies identified at least other three miRNAs related to EMT: a) miR-155 was found to be mechanistically involved in TGFβ-induced EMT in the normal murine mammary gland (NMuMG) epithelial cells. Indeed, the knockdown of miR-155 suppressed TGFβ-induced EMT and tight junction dissolution, as well as cell migration and invasion by targeting RhoA (131); b) miR-29a, that was found upregulated in mesenchymal cells; when overexpressed miR-29a suppressed the expression of tristetraprolin (TTP), a protein involved in the degradation of messenger RNAs with AU-rich 3′-untranslated regions, and led to EMT and metastasis in cooperation with oncogenic Ras signaling (80); c) miR-21, again upregulated in TGFβ-induced EMT, in human carcinomas as well as in a model of kidney injury and fibrosis, operates by specifically targeting the tissue inhibitor of metalloproteinase-3 (TIMP-3), likely inhibiting then degradation of ECM components (2, 291).
The emerging overall scenario is then extremely interesting because indicates that upregulation or downregulation of different miRNAs may be critical for the regulation of the epithelial phenotype as well as EMT and tumor progression; in the latter case miRNAs may then be able to act as either oncogenes or tumour suppressors, depending on the context.
III. Reactive Oxygen Species, Redox Signaling, and Redox Regulation in EMT
A. Introductory concepts: From oxidative stress to redox homeostasis and redox signaling
Current biomedical literature often refers to the relevance of oxidative stress, reactive oxygen species (ROS), redox homeostasis, and redox signaling in physiological as well as pathophysiological conditions. However, for a reader not directly involved in redox research, it is not always immediately clear which is the real meaning of these definitions and which are the major implications for complex biological systems and cellular or tissue responses. This is pertinent to the present review since ROS, although their role is likely to be at present undervalued, are already mentioned in several authoritative reviews on the EMT process (114, 139, 168, 207, 250) and proposed as putative mediators or modulators of the EMT process. In order to introduce available literature data on the argument, it seems appropriate to offer first a number of potentially useful selected informations and critical concepts. In this section the following major messages may serve as introductory remarks: a) Molecular oxygen (O2) is essential for the survival of all aerobic organisms; indeed, aerobic energy metabolism relies on oxidative phosphorylation, a vital process by which oxido-reduction energy of mitochondrial electron transport is eventually converted to the high-energy phosphate bond of ATP. All aerobic organisms use O2 as the final electron acceptor for mitochondrial cytochrome c oxidase that, in turn, represents the terminal functional element of mitochondrial multicomponent NADH dehydrogenase enzymatic complex able to catalyze the four-electron reduction of O2, leading then also to H2O formation. b) During mitochondrial oxidative phosphorylation and other electron transfer reactions, a number of partially reduced and highly reactive O2 metabolites are generated that are collectively referred to as reactive oxygen species (ROS), including superoxide anion (O2
•−), hydrogen peroxide (H2O2), and hydroxyl radical (•OH). c) Starting from the first identification of ROS in biological material (44), early research in the field was mainly focused on adverse cytotoxic and genotoxic effects exerted by ROS and related reactive intermediates, resulting in the concept of oxidative stress, originally envisaged as a condition representing the outcome of oxidative injury to biologically relevant macromolecules (nucleic acids, proteins, lipids, and carbohydrates) and potentially able to lead to irreversible cell injury (31, 32, 93, 94, 156, 159, 235, 236). By definition, an oxidative stress is likely to occur any time ROS, as well as other free radicals or nonradical intermediates, are generated in the extracellular or intracellular environment at levels exceeding antioxidant defences (235). d) The early view of ROS and oxidative stress as adverse events was gradually implemented by a number of seminal studies (106, 161, 212, 249, 269) and several others in the following years (39, 48, 61, 179, 237 and references therein) showing that ROS and nitric oxide were able to act as fine tuners or regulators of signaling pathways and cellular responses in both physiological and pathological conditions, with parallel studies also indicating a homologous role for the oxidative stress-related aldehydic intermediate 4-hydroxy-2,3-nonenal (HNE) (67, 195, 206, 261). e) Within the last two decades, the following overall scenario has emerged: changes in redox status, generation of ROS and other oxidative stress-related reactive intermediates were not simply related to toxicity and genotoxicity but also actively involved in the modulation of signal transduction, gene expression, proliferation and, more generally, functional response of target cells. In other words, aerobic organisms adapted themselves to coexist with these potentially dangerous chemical entities and developed strategies and mechanisms, evolutionary conserved, to employ them under physiological conditions. f) Redox research is, at present, placed at the forefront of biomedical research and an impressive amount of evidence suggest that increased and/or sustained levels of oxidative stress and related mediators play a major role in the pathogenesis of clinically relevant human diseases, including atherosclerosis, cardiovascular diseases, and diabetes (68, 95, 117, 191, 203, 275), cancer (156, 276), aging (31, 61, 94, 237), neurodegenerative disorders (125, 158, 306), chronic liver (3, 69, 179, 194, 289) and lung diseases (40, 213, 214), to name just a few.
B. ROS, free radical and nonradical reactive intermediates in biological systems: How they are generated and major properties
ROS is a collective term that includes a number of reactive and partially reduced O2 metabolites, with some of them being free radicals, such as O2
•− and •OH (31, 156). Free radicals are reactive molecular species with an unpaired electron in their outer orbital that can undergo redox reactions by interacting with surrounding biological macromolecules in order to regain a more stable, nonradical, condition. H2O2, the third most relevant molecule included in ROS definition, is more properly a pro-oxidant and nonradical molecule. ROS can be generated within living cells by the following major sources (39, 48, 61, 179, 237, 249): a) Mitochondria (31, 156, 179, 237). It has been calculated that approx. 1%–5% of the electrons flowing through the electron transport chain can be diverted to form O2
•− at the level of Complex I (NADH/ubiquinone oxidoreductase) and Complex III (ubiquinol/cytochrome c oxidoreductase). O2
•− is then converted by a mitochondrial isoform of superoxide dismutase (mtSOD) into H2O2 that can cross mitochondrial membranes and then reach the cytoplasm (Fig. 9). b) Plasma membrane NADPH oxidase (NOX) (7, 137). This multi-subunit complex is known to be expressed by professional phagocytic cells (macrophages, neutrophils, and eosinophils) as well as by a number of nonphagocytic cells playing a critical role in human diseases, including myofibroblast-like cells and cancer cells (68, 179, 194, 276). NOX of professional phagocytes and of nonphagocytic cells are similar in their structure, being formed by two membrane bound components (p22phox and gp91phox/Nox2 or another member of the NOX family of protein) forming the flavocytochrome b558, and four cytosolic components (p40 phox, p47phox, p67phox, and the GTPase Rac1/2), that following stimulation, are recruited to the plasma membrane where they interact with Cyt b558 leading to increased activity and then generation of O2
•− that is then converted into H2O2. Where redox signaling is concerned (39, 48, 61, 179, 237, 249), the major difference is that nonphagocytic NOX, that is constitutively active and produces a very low level of ROS, can significantly increase both activity and ROS generation, as detailed later, in response to a number of growth factors, cyto- and chemokines, and other conditions. c) Several enzymes involved in redox reactions (31, 39, 48, 61, 156, 179, 237, 249). The list of enzymes able to generate ROS (mostly O2
•− that is rapidly converted by a SOD isoform into H2O2) during their catalytic activity is quite impressive and include several oxidases, peroxidases, cytochromes, mono- and di-oxygenases, with the following being the most relevant examples: isoforms of the cytochrome P450 superfamily, involved in the metabolism of endo- and xenobiotics, including ethanol, steroid hormones, drugs, and chemoterapics; xanthine oxidase; the isoforms of nitric oxide synthase (NOS); the isoforms of cyclooxygenase (COX); 5-lipoxygenase (ALOX5), a mixed function oxidase involved in the synthesis of leukotrienes from arachidonic acid in response to stimuli that are also able to stimulate NOX, particularly growth factors and cytokines; peroxisomal oxidases, that can generate directly H2O2 when metabolizing various substrates (glycolate -, D-amino -, ureate -, fatty acid-CoA - and L-α-hydroxyacid oxidases); lysyl oxidase (LOX), that again generate H2O2 when catalyzing the formation of aldehyde precursors of cross-links in collagen and elastin.
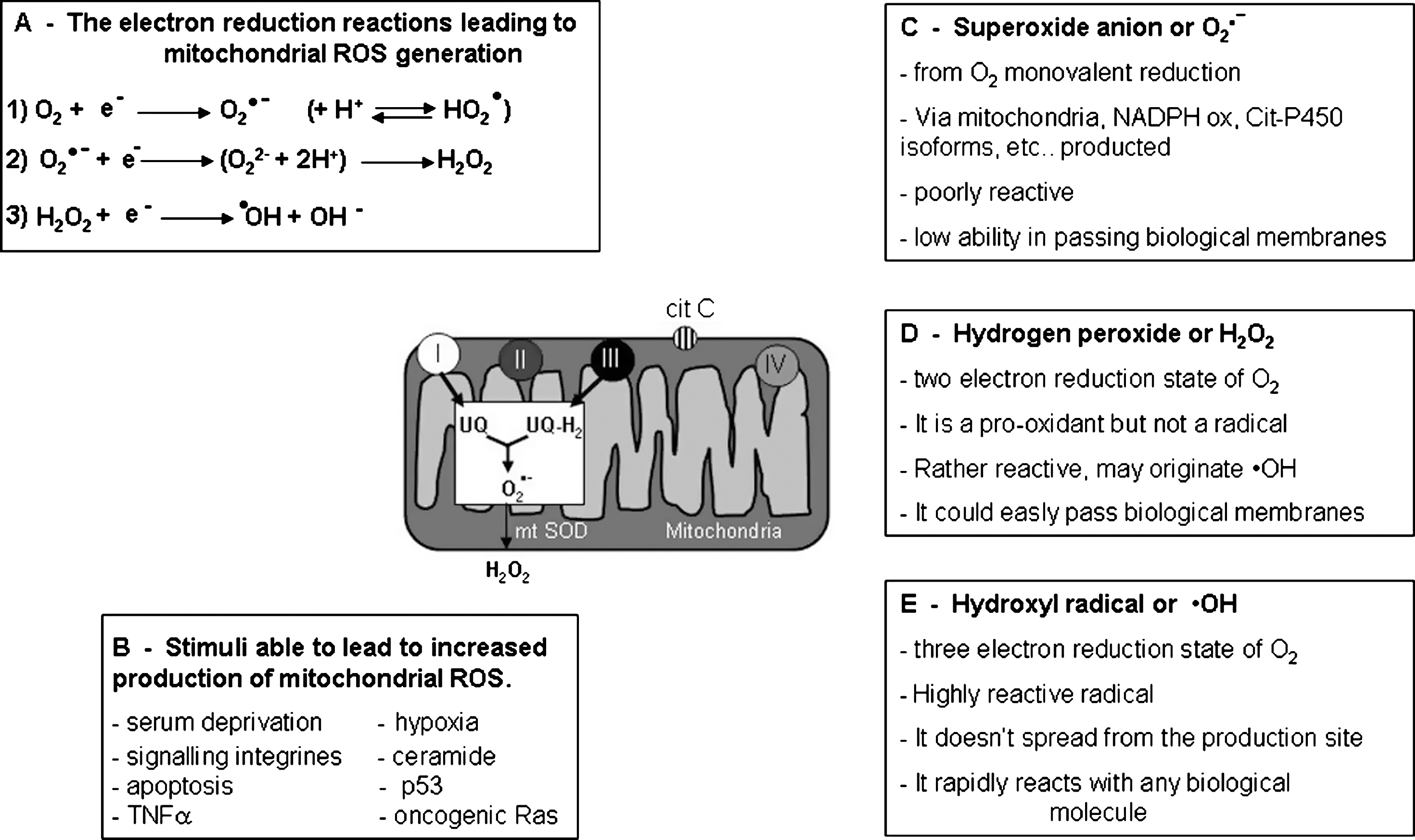
Where the major ROS in biological systems are concerned (Fig. 9), there are a number of properties that a reader should keep in mind (31, 39, 48, 61, 156, 179, 237, 249). O2 •−, the result of univalent reduction of triplet state molecular oxygen, is usually generated intracellularly by mitochondria, enzymes, or in auto-oxidation reactions. The relevant point is that O2 •− is a relatively unreactive intermediate, being able to act at best as a mild reactant in physiological conditions, and has a rather poor ability to cross biological membranes. Only the interaction of O2 •− with NO to give peroxynitrite (ONOO−) is able to transform superoxide into a very reactive intermediate. Moreover, in a living cell O2 •− is usually rapidly converted into H2O2 either enzymatically by SOD isoforms or nonenzymically.
H2O2 is a rather different ROS that represents a two-electron reduction state of molecular oxygen and originates mainly from enzymatic dismutation operated by SOD isoforms or, more rarely, from nonenzymic dismutation of O2 •− or from direct reduction of O2. The most relevant features of this potent nonradical oxidizing agent are represented by the fact that H2O2 easily diffuses across biological membranes and, in aqueous solutions, can affect redox state of inorganic ions, including transition metal ions. If not efficiently removed by either catalase or glutathione peroxidase, H2O2 can give rise to the very reactive and damaging •OH when interacting with O2 •− (Haber–Weiss reaction), or in the presence of divalent metal ions like iron and copper; when Fe2+ is present, the latter reaction is also defined as Fenton's reaction (or Fe2+-catalyzed Haber–Weiss reaction). Indeed, •OH, which may be considered as a three-electron reduction state of O2, can be formed during Haber–Weiss or Fenton reactions or by decomposition of peroxynitrite. •OH has a very short half-life (10−9 sec) and high reactivity, a property that prevents its diffusion from the cellular site of generation and leads it to interact and damage any surrounding macromolecules, including amino acids (potentially leading to protein inactivation/denaturation), carbohydrates (leading to degradation), lipids (leading to lipid peroxidation), and nucleic acids (potentially leading to formation of adducts, such us with deoxy guanidine, and/or mutations).
A few other reactive free radical or nonradical intermediates, as well as their most relevant properties, should be briefly recalled. Nitric oxide (NO) is the most obvious one on the basis of its relevance in both physiological and pathological conditions (61, 179, 187). NO is a small hydrophobic molecule that crosses cell membranes without needing channels or receptors, that can be generated through the conversion of L-arginine in citrulline by three types of NOS: a) eNOS or endothelial NO synthase, bound to plasma membranes and known to be strongly activated by the entry of calcium through membrane-bound receptors (61, 179, 187); b) iNOS or inducible NO synthase (first identified in macrophages and then in other cells), an isoform that generates low levels of NO and is upregulated by pro-inflammatory cytokines and/or LPS; c) nNOS or neuronal NO synthase.
NO can regulate vascular tone, cell adhesion, vascular permeability, and platelet adhesion (61, 187) as well as induce several potentially toxic effects, although many of them are more likely mediated by oxidation products, included in the definition of reactive nitrogen species (RNS). In particular, although efficient systems exist being able to minimize generation of O2 •− and NO, under pro-inflammatory conditions, simultaneous production of O2 •− and NO can be strongly activated leading to the formation of significant amounts of the powerful oxidant peroxynitrite (ONOO−) that can cause significant injury to different cellular structures. ONOO− can act directly as a strong oxidant (by interacting with thiol groups, iron–sulfur centers, and the active site -SH groups in tyrosine phosphatases) or indirectly, by decomposing into highly reactive radicals. Moreover, ONOO− can also react with proteins (leading to tyrosine nitration or direct reactions with specific amino acids), lipids (lipid peroxidation), and nucleic acid (oxidative modifications in nucleobases). ONOO− can also result in mitochondrial damage and even in the induction of irreversible cells death, either apoptosis or necrosis.
A final mention is for 4-hydroxy-2,3-alkenals (HAKs) and F2-isoprostanes, that are end-products of the process of lipid peroxidation, a very common free radical-initiated event that involves oxidative decomposition of ω-3 (22:6) and ω-6 (18:2, 20:4) polyunsaturated fatty acids of membrane phospholipids and leads to significant changes in both structure and functions of biological membranes (31, 67, 156, 167, 179). This process, initiated by the interaction of a ROS or other free radicals with polyunsaturated fatty acids and exacerbated by the presence of divalent metal ions, leads initially to the formation of lipid radicals (L•) that, in turn, can react with available O2 to generate lipid peroxyl radicals (LOO•). From this point the propagation phase of this chain reaction take place leading LOO• to interact with other lipid molecules, resulting in the generation of lipid hydroperoxides (LOOH). LOOH, in turn, undergo a degradative breakdown leading to generation of other radical species (LO• and LOO•), to further propagate lipid peroxidation, and of a number of aldehydic end-products like malonyldialdehyde (MDA), 4-hydroxy-2,3-alkenals (HAKs) of different chain length (67) as well as to F2-isoprostanes (167). 4-Hydroxy-2,3-nonenal (HNE), the most active HAK in biology and pathophysiology (195, 206, 261), as well as F2-isoprostanes (so defined because of their PGF2-like structure) are relatively stable and lipid soluble compounds that can easily diffuse from the site of generation and cross biological membranes. Moreover, both HNE (195, 206, 261) and F2-isoprostanes (45) have been proposed to act as mediators able to affect redox state, signal transduction, and cell responses, and their detection in biological fluids or in tissues is today considered as one of the best way to evaluate in vivo occurring oxidative stress (92).
C. Antioxidant defenses
As already anticipated, ROS, RNS, HAKs as well as other free radical or nonradical reactive intermediates may interact with any relevant biological macromolecule, giving rise to events that can either lead to cytotoxic consequences or contribute to redox regulation and signaling, as summarized in Figure 10. Because of the relevance of the impact of redox reactions on living cells, nature has developed and refined a number of mechanisms that are designed to regulate intracellular levels of ROS, oxidants as well as any other related reactive intermediate and then also to protect biological macromolecules from oxidative stress. These mechanisms are collectively indicated with the general definition of antioxidant defenses and relies on antioxidant enzymes, small antioxidant molecules, proteins able to bind transition metal ions or able to undergo redox cycles as well as natural and synthetic chain breaking antioxidants. Figure 11 and the following text offer a brief synthesis of most relevant antioxidant defences in a living cell. The interested reader can refer to authoritative and comprehensive reviews for more details (18, 91, 288).


Where protection from ROS and oxidants is concerned, the following categories of naturally occurring components of the antioxidant defence system may be outlined: a) Antioxidant enzymes. This category includes the following major enzymes: 1) superoxide dismutase (SOD) isoforms, that operate by transforming O2
•− into H2O2; three major isoforms have been characterized that are the cytoplasmic Cu/Zn - SOD, the mitochondrial Mn-SOD, and the so called ec-SOD, that is secreted in extracellular environment; 2) catalase (CAT) that is within peroxisomes and is responsible for the removal of H2O2; 3) glutathione (GSH) peroxidase (GPX) isoforms that are able to remove H2O2 as well as other organic hydroperoxides; 4) glutathione reductase, which is an enzyme designed to recover oxidized glutathione. b) GSH and other small molecules. This category includes several molecules (see Ref. 91), mainly endogenous, and the following should be mentioned: 1) GSH, the most abundant and relevant water soluble antioxidant, that is acting as a substrate for H2O2-removing enzyme like GPX and dehydroascorbate reductase as well as a scavenger of •OH (leading to the thiyl radical GS• that is not harmless) or as a thiol in regenerating oxidized -SH groups of proteins; 2) ascorbic acid (vitamin C), a cofactor for several enzymes that can operate as an electron donor and then as a reducing agent; ascorbate can also scavenge (i.e., interact directly with) •OH. But one has to briefly mention, that, depending on the overall concentration, ascorbate may become deleterious by reducing Fe3+ to Fe2+ and, in the presence of H2O2, can lead to generation of significant amount of the dangerous •OH; 3) uric acid, which is present in blood plasma and has been reported to scavenge singlet oxygen, •OH, and peroxyl radicals. c) Molecules able to sequestrate transition metal ions. Transition metal ions such as iron and copper can exacerbate ROS generation, then the naturally occurring protein able to dispose these ions, including ferritin, transferrin, ceruloplasmin, metallothionein, and lactoferrin, should be recalled not only for their most obvious role in iron and copper homeostasis but also as molecules that may prevent ROS production via the Fenton reaction by “sequestering” redox active metal ions. d) Thioredoxins and glutaredoxins systems (18, 288). These are two analogous systems based on thiol/disulfide exchange proteins that are involved both in antioxidant defense and in redox signaling. Thioredoxins (TXNs) are a family of 12 kDa proteins, including TXN-1 and TXN-2, possessing a catalytic site in which two cysteine residues can be reversibly oxidized to form disulfide bridges. TXNs undergo NADPH-dependent reduction by the enzyme TXN-reductase and, in turn, they can reduce oxidized cysteine groups on proteins. By this intramolecular disulfide–thiol exchange, TXNs can act as hydrogen donors contributing to the control of redox state. TXNs (mainly TXN-1) may supply reducing equivalents to a number of TXN–peroxidases (peroxiredoxins) and also play a role in redox signaling by modulating kinases or transcription factors forming with heterodimers. Glutaredoxins (GLRXs, the cytosolic GLRX-1 isoform and GLRX-2, the latter existing both as a mitochondrial and nuclear isoform) also belong to the Trx superfamily of thiol/disulfide exchange proteins and act as reductants of protein–SG mixed disulfides. Similarly to what described for TXN system, GLRXs have a role in redox regulation and the GLRX system is composed by GLRX isoforms, GSH reductase, GSH, and NADPH.
This brief section on antioxidant defenses can be completed by mentioning those natural and synthetic antioxidants that are able to protect from lipid peroxidation. According to Halliwell and Gutteridge (91), “an antioxidant is any substance that, when present at low concentrations compared to those of an oxidizable substrate, is able to significantly delay or inhibit oxidation of that substrate.” Of course, this generic definition may also include primary antioxidants (that is, free radical scavengers, then molecules able to interact directly with, and/or to block the initiating free-radical, like mannitol) and synthetic molecules able to bind transition metal ions (for example, desferrioxamine, a synthetic compound able to bind iron). However, most authors who use the term antioxidant have in mind the so-called chain breaking or secondary antioxidants, with α-tocopherol (vitamin E) being the naturally occurring prototype. These natural or synthetic molecules have a chemical structure designed to intercept radical intermediates produced during sustained lipid peroxidation such as peroxyl- or alkoxyl-radicals, then preventing (i.e., breaking) the perpetuation of hydrogen abstraction in the chain reaction.
D. Redox homeostasis and redox signaling
According to current literature, redox signaling is a definition that can be used to indicate any condition, in physiology or pathophysiology, in which a process can be regulated or modulated by a signal that is delivered through redox chemistry (39, 48, 61, 179, 237, 249). Any significant increase in intracellular levels of ROS in a biological system can result, by definition, in an alteration of the so-called redox homeostasis and, as always happens in any complex system reacting to the presence of defined reactants, single cells and multicellular organisms have developed highly specific redox sensors and mechanisms that are the basis of oxidant scavenging and ROS signaling systems. In simple words, redox signaling represents the response or part of the cellular response designed to reset the original state of redox equilibrium. As proposed by different authors (61, 179, 249), one can easily envisage the following intuitive scenario: a) Generation of very low or steady-state levels of ROS and other reactive intermediates. This is a physiological condition of unstimulated cells in which redox homeostasis is controlled specifically by catalase, Trxs, SODs, and GPXs as well as by naturally occurring antioxidants such as GSH, vitamin E, β-carotene, ascorbate, and urate, as well as by less specific but much more abundant, antioxidant components that are represented by amino acids, peptides, and proteins. This means that within the cells there is no significant unbalance of pro-oxidants vs. antioxidant defenses and then the cell apparatus does not respond by means of a redox signaling. b) Generation of a relatively low and transient increase in intracellular levels of ROS. Here a shift in redox balance can occur but it is time- and concentration- limited and redox signaling will then primarily operate through redox-sensitive signaling pathways and transcription factors (39, 48, 61, 179, 237, 249, see later) in order to upregulate transcription of genes encoding for products that will reset in the due time redox homeostasis (for example, antioxidant enzymes, TXNs, and GLRXs, cystine transport system to sustain synthesis of GSH, etc.). c) Generation of higher levels of intracellular ROS and other reactive intermediates. Levels of intracellular ROS and other reactive intermediates may be very high and/or persistently increased within a cell during acute tissue injury or in tissues undergoing chronic injury. The target cell, depending on the specific agent or condition involved, the overall severity and/or duration of the injurious process, may face two different scenarios. The first one can be dramatic for the cell: if levels of ROS or of reactive intermediates are very high (that is, severe oxidative stress) they can significantly damage macromolecules, alter cellular structures and functions, eventually leading to irreversible injury and cell death. A more interesting condition is the one that is likely to occur in conditions of chronic injury, in which levels of oxidative stress are significantly higher than normal but unable to induce irreversible cell injury; in such a condition cells and/or tissues may still reach an equilibrium or, as elegantly defined by Dröge (61) a quasi-stable state, a definition that implies a shift of the intracellular redox state to higher levels of ROS and a chronically deregulated condition in which redox signaling can upregulate patterns of gene expression and cell responses that are believed to significantly contribute and/or sustain the development of chronic diseases and even cancer progression (39, 48, 61, 179, 237, 249, 276).
The scenario offered is of course a didactic and oversimplified one, and it is conceivable that in a tissue undergoing acute or chronic injury, inflammation, and wound healing, the different conditions may coexist, with an overall scenario in which the development of a disease is resulting from the sum of both ROS-dependent damaging effects and changes in gene expression.
But how are cells able to respond to an altered redox status? The point is that mammalian cells possess redox sensors which are redox-sensitive specialized proteins, able to sense or evaluate intracellular levels of ROS by means of a redox-based mechanism affecting one or more residues/domains within its three-dimensional structure. These redox sensors that have been evolutionary developed and refined from those occurring in prokaryotic cells and yeast, are able then to transform the redox change/s into a specific setting for antioxidant activity-related transcription and much more. In higher eukaryotes, redox regulation of transcription as well as of signaling elements like protein phosphatases, relies on properties and strategies similar to those described in bacteria or yeast (cysteine-based oxidation/reduction cycles) and then evolutionary conserved. Mammalian cells, for example, still express thiol-peroxidases affecting H2O2-dependent signaling but, as already mentioned, they also express TRxs and GPxs that are also involved in the modulation of signal pathways.
In an oversimplified scheme, three different mechanisms exist that may explain how an increase of intracellular ROS can trigger transcription of redox sensitive genes (146): a) Redox reactions may involve directly either signaling components or transcription factors; this is the case, for example, of redox reactions involving transcription factors such as Ref1 (Redox-factor-1), a ubiquitous reductase having cysteine residues (Cys65 and Cys94) that are critical for redox-dependent modification of several transcription factors, including AP-1 (activator protein-1), NF-κB (nuclear factor κB), p53, ATF/CREB (activating transcription factor/cAMP-response element-binding protein), and HIF-1α (hypoxia-inducible factor 1α). Ref1 acts by reducing -SOH groups and/or oxidized cysteine residues or disulfide bonds present on transcription factors that, under these oxidized conditions, have reduced or absent DNA-binding activity; the reduced transcription factors then become able to bind their related sequences on DNA. b) Redox reaction may affect nuclear translocation of transcriptional regulators that are maintained into an inactive form in another cellular compartment; a typical example is Nrf-2 (nuclear factor (erythroid-derived-2)-like-2), which is a transcription factor able to bind to the so-called ARE (antioxidant responsive elements) regulatory sequences on the promoter of several genes encoding for enzymes involved in detoxification (glutathione S-transferases, NAD(P)H quinone oxidoreductase, the multidrug resistance-associated protein and cysteine-glutamate exchange transporter), resulting in upregulation of their transcription. Nrf-2 is usually inactive when bound to KEAP-1 (Kelch-like ECH associated protein-1), a sensor protein rich in cysteine residues that forms a complex with cullin-3 and Nrf-2 to target the latter for proteasomal degradation. Exposure to oxidative stress, resulting in oxidation of selected cysteine residues (Cys151, Cys273, and Cys288) together with other reactions, modifies KEAP-1, leading to arrest of Nrf-2 ubiquitylation and degradation and allowing Nrf-2 to detach from KEAP-1 and translocate into the nucleus. c) Redox-sensitive transcription can be modulated by alterations of the so-called “redox buffers.” Indeed, several transcription factors as well as DNA modifying enzymes are sensitive to the most relevant reduced/oxidized molecular redox pairs, such as GSH/GSSG, NADPH/NADP, and NADH/NAD.
Before analyzing principles of redox signaling, one has to realize that in mammalian cells ROS-specific responses like those regulated by p53, AP-1, NF-κB, c-Myc, FOXO, and other factors, can be seen as part of long-term differentiation programs that operate an integration between ROS protection and multiple metabolic/adaptative responses. More properly, higher eukaryotic cells have developed strategies that, by diverting the original defensive design of redox signaling, use intracellular ROS produced within cells (but also those entering cells from the extracellular environment as in inflammatory conditions) to modulate several signaling pathways such as, in particular, those downstream to growth factor, cytokine, and chemokine receptors. The critical point is that redox changes and ROS may affect simultaneously different signaling pathways, then resulting in the modulation of major metabolic or adaptative responses of cells. This is believed to play a critical modulatory role in several physiological or pathophysiological conditions, including many chronic diseases of clinical relevance (39, 40, 48, 61, 125, 158, 179, 213, 214, 237, 249, 276, 306).
A well-established concept in redox signaling is that signal transduction elicited by the interaction of peptide factors (growth factors, cytokines, chemokines, or other ligands) with their respective receptors can be enhanced or modulated by intracellular ROS generation. This is achieved because peptide ligands can trigger not only their specific signal transduction pathway but also activation of NOX, the latter resulting, by a series of mechanisms, in a positive feedback on signal transduction itself; moreover, one should consider that the same positive feedback can be also elicited whatever the source of ROS, then including ROS released by mitochondria or generated by other intracellular sources as well as those entering the cell from the extracellular environment. Along these lines, H2O2 is the best candidate because it has a rather long half-life, a relatively low reactivity, and an intrinsic ability to cross biological membranes. Since available literature concerning redox signaling is now impressive (more details can be found in Refs. 39, 48, 61, 179, 237, 249), here only a brief introduction to the most established concepts will be presented that may be useful to introduce the putative role of ROS and redox changes in EMT.
As mentioned, the starting point relies in the fact that ROS can be generated as a consequence of ligand–receptor interaction to affect receptor-mediated signaling pathways. ROS have been shown to mediate a positive feedback on signal transduction elicited by growth factors like PDGF, EGF, or NGF that results in a reinforcement of the signal transduction pathway elicited by activation of the receptor tyrosine kinases (RTKs). As schematically depicted in Figure 12, activation of RTKs by their specific polypeptide ligands can also involve activation of p21Ras and of Rac, then leading to a parallel activation of a subunit of nonphagocytic NOX, likely a gp91phox analogue. Pertinent to this review, TGFβ1, which operates by binding receptor serine/threonine kinase (RS/TK) and involves Smads and Src kinases, has also been described as leading to activation of a NOX in nonphagocytic cells to generate O2
•− that will then spontaneously or enzymatically dismutate into H2O2. Although the type of receptors involved differs, a similar mechanism has been described also for other ligands such as IL-1, TNF, angiotensin II, thrombin, and insulin. The resulting overall scenario that is likely to be critical for cancer cells or cells involved in diseases characterized by chronic inflammation and wound healing, is the following: these nonphagocytic cells, due to the pathological condition, will be affected by signals received from the extracellular environment, like peptide factors engaging their receptors or extracellularly generated ROS, that will ultimately lead to an increase in intracellular levels of ROS that, in turn, will modulate intracellular signaling pathways by one or, likely, two or more of the following mechanisms: a) ROS can inhibit protein tyrosine phosphatases in order to enhance signaling pathways (38, 39, 51, 61, 179). Protein tyrosine phosphatases (PTPs) operate as negative regulators of RTKs–mediated signaling, responsible for switching off the activated receptors by means of dephosphorylation (255, 256). However, it is well known that ligand-induced activation of RTKs, as in the case of PDGF and other growth factors, can lead to PI3K-mediated activation of Rac that, in turn, is recruited to the NOX complex inducing increased generation of ROS. The underlying mechanism relating ROS and PTPs inhibition is relatively simple: ROS such as H2O2 can oxidize a critical and redox-sensitive cysteine residue within the active site of PTPs, leading to inactivation of the PTPs itself. This inactivation or inhibition can be either reversible or irreversible, depending on the actual intracellular levels of ROS and then the degree of oxidation of –SH group of the active PTP, but in any case will result in a reinforcement of RTKs downstream signaling of variable duration. It is relevant to underline that this mechanism has been described also in cells exposed to radiation, metals, alkylating agents, and environmental oxidants, then to conditions that may even activate RTKs in a ligand-independent manner or RTK trans-activation (268). Moreover, significant changes in intracellular thiol/disulfide redox state are likely to further affect the system since the relative- or time-limited depletion of reducing agents may prevent reversion of oxidized/inactive PTPs to the reduced/active form. b) ROS are able to activate MAPK cascades or specific protein kinases (39, 48, 61, 179, 237, 249, 276). Literature offers several examples of protein kinases that can be activated by ROS, but some of these findings have been obtained by using quite high levels (that is, in the millimolar range) of ROS like H2O2 that may be not reached in a biological environment. Studies performed by employing more realistic conditions have identified a restricted number of molecular targets and pathways that are activated by mild oxidizing conditions or by mild shift in the thiol/disulfide redox state. This include signaling components of the Src family of protein tyrosine kinases (p59fyn and p56lck), JAK2, c-Jun NH2-terminal kinases (JNKs), p38MAPK, and, in some cells, ERK1/2. Within the several available, the serine/threonine kinase protein kinase C α or PKC-α is an interesting example of redox-sensitivity. PKC-α, as well as other PKC isoforms, are usually activated by diacylglycerol or phorbol esters for whom PKC has a specific binding site in an evolutionary conserved cysteine-rich region. According to this characteristic, all these kinds of PKC isoforms can be activated by H2O2 in a way that involves tyrosine phosphorylation in the catalytic domain. Two other peculiar redox-dependent mechanisms have been disclosed by studies designed to investigate ROS-dependent activation of JNKs. In a first series of studies, the redox sensitivity of the activation of apoptosis signaling-regulating kinase 1 (ASK-1), a kinase that, in turn, can lead to activation of MKK3/6, MKK4/MKK7, and then of JNKs and p38MAPK, finally results in the phosphorylation of activating transcription factor 2 or ATF-2, c-Jun, and p53. ASK-1 is usually inactive as the consequence of being associated to a TXN protein that binds to the NH2-terminal domain of ASK-1, then inhibiting its kinase activity. An increase in intracellular ROS can induce TXN dimerization and dissociation from ASK-1 that is followed by multimerization of ASK-1, activation of its kinase activity, and then of the downstream signaling, leading to activation of JNKs and p38MAPK (85, 147). A second mechanism leading to ROS-dependent activation of JNK (a potentially relevant mechanism able, if sustained, to induce apoptosis) has been described by the group of Karin for TNF-mediated cell death, with ROS being able to inhibit JNK phosphatases, which are the specific JNK-inactivating enzymes (116). c) Increased levels of ROS can activate specific transcription factors. Literature suggests that several transcription factors (TFs) can be considered as redox sensitive, with NF-κB and AP-1 being the two best characterized examples. NF-κB has been the first TF identified as redox sensitive (230), and it is known to be involved in inflammatory reactions, in the control of cell growth and survival to apoptosis (248), and, possibly, also to necrotic cell death (23). NF-κB is a rather generic definition including, in mammalian cells, c-Rel, RelA (p65), RelB, NF-κB1/p50, and NF-κB2/p52 proteins, all being able to recognize DNA sequences called kB sites (23, 82, 248). NF-κB is also involved in maintaining mitochondrial integrity and in regulating antioxidant activity (82, 230). Redox-dependence of NF-κB relies on different described mechanisms of activation that may vary in different cells (82); activation of NF-κB may depend either on H2O2-dependent activation through the classical IKK-dependent pathway, or it may involve an atypical phosphorylation of the tyrosine 42 residue of IκBα by so-called spleen tyrosine kinase Syk (a mechanism independent on IκB kinase or IKK). Interestingly, literature data indicate that all cytokines leading to NF-kB activation are likely to cause intracellular generation of ROS that are then responsible for IKK activation and IκBα degradation, with IL-1, TNF, and LPS being the best characterized examples. This in principle suggests that ROS, produced intracellularly as a part of the response induced by inflammatory cytokines, are likely to contribute to reinforce the signal. Where the redox-sensitivity of AP-1 is concerned, a similar scenario has been outlined. As is well known, AP-1 is a dimeric (homo- or heterodimer) TF typically formed by c-Jun and c-Fos and involved in several physiological and pathophysiological processes. Activation of AP-1 has been detected in the presence of low levels of ROS, and at least two mechanisms may lead to its redox-dependent activation: the first involves oxidative activation of JNKs that, in turn, phosphorylate specific serine residues (ser63 and ser73) of the NH2-terminal transactivation domain of c-Jun, a domain that is essential for functional activation (146, 147, 248); the second is likely to involve a mild shift in the redox state by different oxidants or ROS (61, 249).
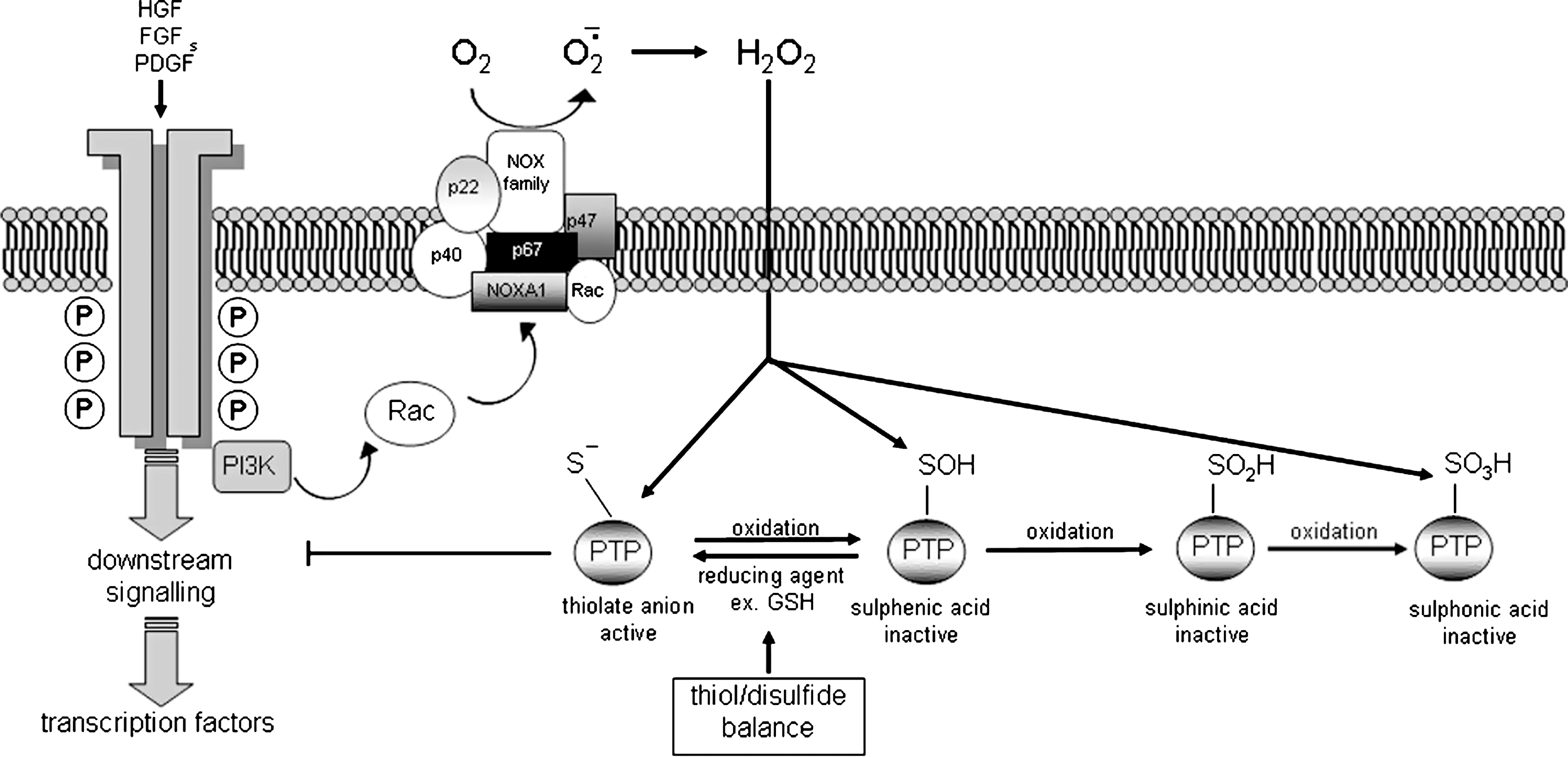
E. ROS and EMT: A link that may be relevant in chronic inflammatory/fibrotic diseases and cancer
According to the basic principles of redox signaling discussed above, and the nature of molecular mechanisms that are believed to be responsible for EMT (described in Section II), one may easily state that, at least theoretically, there are several ways in which ROS and changes in redox homeostasis may contribute to the process of EMT. However, when reviewing the literature about EMT in normal and pathological conditions, one realizes that not so many reliable studies have provided such a direct “link” between ROS generation and EMT triggering. This is quite surprising because, as already mentioned, there is no doubt that ROS are involved in cancer as well as in a number of chronic inflammatory diseases that have a relevant impact on human beings (61, 68, 95, 117, 158, 179), with ROS having a well-defined role in cancer cell biology and cancer progression (see 276 and refs. therein) as well as in fibrogenesis, as extensively described for the liver (see 179 and refs. therein) but with features and mechanisms that are common to other relevant chronic conditions resulting in organ fibrosis. Even more surprisingly, authoritative and recent reviews about EMT (114, 207, 250) seem to restrict the relevance of ROS on two conditions, including MMP-3-(211) and hypoxia-dependent (33) induction of EMT.
As a consequence, in this section a rapid overview of the existing literature data that already establish a direct connection between ROS and EMT will be offered together with a more speculative, redox signaling-centered, attempt to envisage the putative impact of ROS on EMT triggering (summarized in Fig. 13).

The first study to be mentioned (166), in our view, is a conceptually very straightforward one: mouse NMuMG cells (mammary gland epithelial cells) were exposed in culture to repeated treatment with low (i.e., not cytotoxic) doses of H2O2 up to 4 days, a protocol designed to mimic a condition of chronic inflammation that is very common in several human diseases and is also causally associated with carcinogenesis. Although the authors did not even mention EMT in their study, in these experimental conditions NMuMG cells underwent phenotypic conversion to a fibroblastoid-like phenotype that was associated with dissolution of cell–cell contacts, E-cadherin redistribution in the cytoplasm, and upregulation of a set of integrin family members (α2, α6, and β3 integrins) as well as increased expression and/or activity of several MMPs, including MMP-2, MMP-3, MMP-9, MMP-10, and MMP-13. The chronic H2O2 treatment also resulted in persisting activation of ERK1/2 and p38MAPK (but not of JNKs) as well as of the small GTPase Rac1; moreover, the treatment was sufficient also to induce an increased invasive behavior of NMuMG cells that was prevented by MMPs inhibitors, possibly the most relevant finding. This study established a first direct link between extracellular generation of ROS and EMT.
In a previous section, when describing mechanisms regulating E-cadherin downregulation, we outlined the major roles of SNAI1 and GSK-3β in mediating signals leading to EMT. Moreover, we also emphasized the role of extracellular signals represented by a number of growth factors and cytokines, with TGFβ being one of the most potent factors able to trigger a full EMT program. Along these lines, there are different lines of research that have mechanistically linked intracellular generation of ROS to the involvement of these relevant EMT-triggering mechanisms and signals.
A first line of research proposed ROS as intracellular mediators of TGFβ1-induced EMT in the rat proximal tubular epithelial cell line NRK52E (218), used as a cell culture model to investigate the proposed relevance of TGFβ1-dependent EMT as a mechanism contributing to tubulo-interstitial fibrosis in kidney (176). TGFβ1, as shown previously in other cells and also in MDCK cells (301), was found to increase intracellular levels of ROS that, in turn, were found to contribute significantly to Smad 2 phosphorylation, activation of p38MAPK and ERK1/2, increased expression of αSMA, and fibronectin and E-cadherin downregulation. This TGFβ1-dependent scenario was prevented by pretreating cells with either N-acetyl-cysteine or agents affecting NOX activity (e.g., apocynin) and almost fully reproduced by simply exposing cells to H2O2, with the apparent exception of Smad 2 phosphorylation. Conceptually related findings, as we will see in more details later in the section dedicated to organ fibrosis and EMT, are those proposing a close relationship between oxidative stress, TGFβ1- and angiotensin-related upregulation of NOX2, and increased intracellular generation of ROS and EMT, as a critical sequence of events contributing to kidney fibrosis associated with chronic allograft nephropathy (56, 57). Very recently, another study has identified an interesting mechanism relating TGFβ1, ROS generation and EMT induction in AML-12 murine hepatocytes (304). TGFβ1 has been reported to induce a dramatic decrease in ferritin heavy chains by repressing the translation of its mRNA, an event resulting in a very significant rise in intracellular labile iron pool and related increased generation of intracellular ROS. Of relevance, both inhibition of ROS generation as well as forced overexpression of ferritin heavy chains were able to suppress EMT.
The role of ROS as intracellular mediators of EMT induction emerged from another line of research designed to understand the mechanisms underlying induction of EMT by MMP-3 (also known as stromelysin 1) and performed on the model of nontumorigenic mouse mammary epithelial cell line SCp2. Repeated exposure of these cells to MMP-3 (150) resulted in loss of E-cadherin, nuclear translocation of β-catenin, activation of TCF/LEF transcriptional activity, development of anchorage independence, formation of tumors when injected in vivo, as well as several other common features of EMT, including upregulation of SNAI1 (reviewed in 209). In a very interesting study (211), mentioned earlier in this review, the authors found a close relationships between MMP-3, intracellular generation of ROS (detected using the dichloro-dihydrofluorescein diacetate or DCF-DA technique), SNAI1 upregulation, and EMT induction. Both SNAI1 upregulation and EMT were prevented by using redox quenching agents like dimethyl-sulphoxide (DMSO) and N-acetyl-cysteine (NAC). Moreover, EMT as well as increased cell scattering and invasiveness were reproduced simply by exposing SCp2 cells to hydrogen peroxide (209, 211). More precisely, MMP-3-induced increased generation of ROS, eventually leading to SNAI1 upregulation, was reported to be dependent on the increased expression of an alternatively spliced form of Rac1 (211). Although Rac1 isoforms are usually believed to be involved in the regulation of ROS generation by NADPH oxidase complex, the authors reported that another way to prevent MMP–induced EMT was to transfect cells with expression plasmids encoding for SOD2 isoform (but not SOD1 or catalase), then suggesting that a crucial point was represented by ROS release by mitochondria. This hypothesis of a mithochondrial release of ROS was confirmed by other researchers who were able to trigger EMT in the HK-2 line of human immortalized proximal tubular kidney cells by adding aldosterone. Both increased expression of SNAI1 and EMT were prevented by NAC as well as by rotenone, an agent able to block selectively ROS release by mitochondria, but not by the NOX inhibitor apocynin (302).
Data relating intracellular generation of ROS of mitochondrial origin to SNAI1 nuclear translocation and induction of EMT were recently confirmed and further extended by our group in a study designed to investigate the role of hypoxia as an independent stimulus able to trigger EMT in cancer cells of epithelial origin (33). Hypoxia-induced EMT relies on a biphasic scenario involving an early and redox-sensitive switch and a late phase, associated with increased migration and invasiveness, mostly dependent on HIF-1α and VEGF. The early and transient increased generation of ROS released by mitochondria of hypoxic cells has been related to early nuclear translocation of SNAI1 (within 6 h) as well as to very early (i.e., 15 min of hypoxia) phosphorylation and inactivation of GSK-3β. The latter crucial event lasted for several hours, was prevented by rotenone and DPI, and was easily reproduced by simply adding hydrogen peroxide even in normoxic conditions. During hypoxia, EMT was also sustained by long-lasting nuclear translocation of β-catenin (33). A more general message from the study, apart from other relevant implications related to hypoxia, was that a rise in intracellular levels of ROS may mediate EMT triggering by determining phosphorylation/inactivation of GSK-3β, a crucial step known to affect negatively transcriptional activity of both SNAI1 and β-catenin. These findings also imply indirectly that an increase in intracellular ROS is potentially able also to positively amplify Wnt/β–catenin signaling, a major pathway that has also been involved in EMT. Indeed, ROS-dependent inhibition of GSK-3β is likely to represent a second additional redox-dependent mechanism able to modulate Wnt/β–catenin signaling other than the one previously described by Funato and coworkers (78), the latter based on redox-sensitive inhibitory binding of DVL to the thioredoxin-like protein nucleoredoxin.
Coming back to ROS, GSK-3β, SNAI1, and EMT triggering, it is relevant to recall that in one of the two original studies describing the crucial role of GSK-3β as an endogenous inhibitor of SNAI1 transcription (8), the authors also reported that GSK-3β was able to inhibit NF-κB pathway. This finding provided a connection between such a relevant enzyme (that is a real intracellular crossroad for different converging signaling pathways) and the previous report from the same laboratory indicating NF-κB as a critical factor able to drive SNAI1 expression and EMT in a way that was also related to ERK signaling (12). The involvement of NF-κB, that is not only cytokine- and growth factor- dependent but also the paradigmatic redox-sensitive transcription factor, further supports the concept that intracellular levels of ROS may indeed regulate E-cadherin expression through a mechanism involving then GSK-3β, NF-κB, and SNAI1. This concept may be even reinforced by data showing that ROS-dependent GSK-3β phosphorylation/inactivation during early phase of EMT was prevented by blocking ERK and PI3-K signaling pathways (33).
The putative relationships between ROS, NF-κB, SNAI1, and EMT are also strongly suggested by other data that can be briefly summarized as follows: a) By using cells overexpressing the constitutively active p65 unit of NF-κB (MCF10A/p65) a direct relationship between NF-κB and downregulation of E-cadherin and desmoplakin as well as with upregulation of vimentin was found (41), a relationships that under chronic exposure to TNF was able to induce the EMT-like phenotype in normal MCF10A cells through the involvement of other two relevant transcription factors for EMT like ZEB1 and ZEB2. b) The group of Karin, in a study in which relationships between ROS and SNAI1 were confirmed, has shown that transfection of BEAS-2B human bronchial epithelial cells with a Ikkβ-kinase mutated vector (resulting in inhibition of Ikkβ activity) resulted in a rapid triggering of EMT and in increased migration/invasiveness (37). c) A recent study has further confirmed the relationships between ROS, SNAI1 expression and E-cadherin downregulation, by providing evidence in MCF-7 cells that SNAI1 mRNA is stabilized by HuR during hydrogen peroxide-induced SNAI1 expression (59) and that this event was causally related to hydrogen peroxide-induced cell migration. d) In another recent article, ROS have been shown to induce hypermethylation of the E-cadherin promoter by increasing SNAI1 expression; in particular, SNAI1 induced DNA methylation of the E-cadherin promoter by recruiting histone deacetylase 1 and DNA methyltransferase 1 (145). e) Finally, a very recent study has provided convincing evidence that SNAI1 is stabilized by the inflammatory cytokine TNFα through the activation of the NF-κB pathway (277).
In order to complete this section dedicated to the relationships between ROS and EMT, we propose a more general speculative scenario (summarized in Fig. 13) in which all the previously reported established data are combined with those major mechanisms that have emerged in the last decade from redox signaling studies.
IV. EMT in Human Health and Disease
A. EMT in embryogenesis or Type 1 EMT: A process for dispersing cells in embryos
The concept of epithelial to mesenchymal transition (EMT) was originally introduced more than 40 years ago by studies performed on chick embryos (96, 257, 258) and became of general value in 1982 when Greenburg and Hay published their seminal study (86) showing that even epithelial cells from embryonic and adult anterior lens (i.e., from tissues that in vivo do not form usually mesenchyme) if cultured in three-dimensional collagen gels, became elongated, lost their polarity, detached from the explants, acquired a mesenchymal-like phenotype, including formation of pseudopodia and filopodia as well as the ability to migrate. EMT in embryogenesis (2, 114, 298) should be considered as a fundamental process occurring at multiple steps of embryonic development in order to enable the conversion of various types of epithelial cells into mesenchymal cells, allowing the necessary migration of these cells that, in turn, may maintain the mesenchymal phenotype or, as rapidly inferred, may undergo the reverse program, defined as Mesenchymal to Epithelial Transition (MET), allowing the formation of new epithelial structures in the embryo (2, 26, 50).
The relevance of EMT is suggested by the fact that such a program has been found to underlie a variety of tissue remodeling events during embryo development, including fundamental steps such as mesoderm formation, neural crest development, heart valve development, secondary palate formation, and male Mullerian duct regression, to name just a few (2, 250, 283). These findings overall outlined a major concept: mesenchymal cells do not derive only from the mesoderm primary germ layer, but also from epithelial endodermal cells through EMT, that then can regulate the early stages of development of most living organisms (with the apparent exception of the two phyla of Porifera and Cnidaria). As a matter of fact, in the absence of EMT, embryo development cannot proceed past the stage of blastula, with EMT being the main responsible for the evolutionary conserved plasticity of epithelial cells and then for remodeling of epithelial sheets which include events in embryogenesis termed invagination, evagination, intercalation, branching, and multilayering. EMT is critical in allowing the formation of the three-layered embryo through gastrulation as well as in the formation of structures such as, in addition to those already mentioned, vertebrae, the craniofacial structures, and the neural derivatives (2). In this review we will recall only EMT involvement in some of the major steps in embryo development.
Mesoderm formation from the primitive ectoderm, which is initiated during gastrulation, should be considered as the earliest event involving an EMT program during embryogenesis, as originally described in the fruitfly (Drosophila melanogaster) and lower vertebrates (amphibian and avian) (121) and then confirmed also in mammals (266). In particular, in mammals and birds the induction of mesoderm is observed in the so-called primitive streak of the primitive ectoderm. Mesoderm formation begins with invagination of epithelial cells of the primitive ectoderm, an event that is limited to a small population of epithelial cells and involves a number of progressive morphological changes, including the narrowing of apical compartments, the redistribution of organelles to the apical location, and the bulging of the basal compartments. The few epithelial cells involved start then progressively to lose their cell–cell adhesions, undergo full EMT and, following basement membrane breakdown (i.e., a critical aspect in regulating gastrulation EMT), migrate through the acellular space which is located under the primitive ectoderm in order to eventually populate new areas of the embryo that will develop into mesoderm and endoderm (2, 168, 171, 250).
Where the role of EMT-related specific signaling pathways in mammalian mesoderm formation is concerned (reviewed in Refs. 2, 168, 171, 250), studies on mouse gastrula indicate a major role for signaling through FGF receptor-1 (FGFR1) that, in turn, controls SNAI1 expression and then E-cadherin transcription, with inactivation of SNAI1-1 leading to complete inhibition of EMT. The Wnt signaling pathway is also involved in the process, as indicated by the fact that expression of a truncated β-catenin construct in oocytes induced a premature EMT in the epiblast that is concomitant with SNAI1 transcription. Moreover, the Wnt-β–catenin canonical pathway also regulates the formation of the node, which is the most critical transient embryonic structure that serves as an organizing centre for subsequent development, including the formation of the primitive streak, the anterioposterior axis, and the somites (2, 168, 171, 250).
Neural crest formation is another EMT-involving process in vertebrate embryogenesis (2, 62, 250). Neural crest is a transient but very relevant embryo structure that appears in the ectoderm at the interface between the neural plate and lateral ectoderm; precursors cells from this structure are able to migrate over long distances in the embryo and, by following precise migratory routes at each axial level, can give rise to several derivatives, including craniofacial structures (cartilage, bone, muscles), melanocytes, adrenal medulla, and cells of the sensory and autonomic nervous systems.
Presumptive neural crest cells emerge from neural epithelium as cells with rounded and pleiomorphic shapes that move away from those of the neural tube while losing N-cadherin-mediated cell–cell adhesion (259). Migration of mesenchymal-like neural crest cells to the different final appropriate destination sites requires disruption of the basal lamina and is likely to be critically affected by the type and levels of ECM components, as suggested for fibronectin and hyaluronic acid (205); indeed, it has been shown that the onset of migration is preceded by the appearance of high levels of both ECM components in presumptive neural crest area. Multiple signaling pathways have been shown to cooperate in order to regulate EMT during neural crest determination and segregation and the overall scenario is then very complex (2, 250). EMT in neural crest territory, as recently reviewed (2, 250), is likely to be induced by BMPs, Wnt, and FGF, whereas neural crest segregation and delamination (in mammals) involves Foxd3 that, in turn, requires the concomitant expression of Sox9, SNAI2, and SNAI1 in order to allow EMT to proceed. In particular, SOX9 is able to both inhibits apoptotic cell death (possibly through SNAI1) and to specify the neural crest cell lineages. However, none of the mentioned genes has proven to be the master regulatory one for EMT occurring during neural crest formation.
Another step of embryo development in which EMT has been reported to occur is heart valve formation. Cardiac valves originate from the so-called endocardial cushion that is an early structure which is formed when primitive myocardial cells start to secrete abundant ECM that physically separates myocardium from endocardium. This cushion is then infiltrated by mesenchymal cells that originate from an endocardial cell layer through an EMT process that is guided by signals released by the atrioventricular myocardium (162, 217). Endocardial EMT is regulated through interactions between TGFβ and NOTCH signaling pathways, with endocardial cells undergoing decreased expression of N-CAM, losing cell–cell adhesion, and invading the endocardial cushion, a sequence of events that is believed to lead to the formation of cardiac septa and valves (2, 204, 250, 283). In particular, a nice study has revealed a complex scenario of interactions between Notch and TGFβ–BMP pathways: the transcriptional repressor SNAI2 represents a direct target for Notch signaling pathway whereas TGFβ–BMP-related pathways mainly upregulate SNAI1. However, Notch and TGFβ pathways can synergically upregulate SNAI1 expression despite the fact that SNAI1 is not a direct target of Notch (177).
As a final example of involvement of EMT in embryogenesis, formation of the secondary oral palate can be proposed. In order to be formed, secondary palate requires fusion of palatal shelves that are covered at the leading edge by epithelial cells. These epithelials cells, early after fusion that is required to form the medial epithelial seam, undergo an EMT program and contribute to the formation of the mesenchymal compartment of the palate (72). Palatal EMT in mouse embryo has been suggested to be mainly driven by members of the TGFβ superfamily, with a major role described for TGFβ3 (172, 173).
B. EMT as a mechanism contributing to re-epithelialization during wound healing
Wound healing is a complex multistep process that involves different cells, including cells of epidermis and dermis, vasculature, and immune system (4, 250), as well as an initial inflammatory response that results in the abundance of several cytokines and growth factors. In particular, ligands for EGF receptor (EGFR), including EGF, heparin-binding EGF-like growth factor (HB-EGF ), and transforming growth factor α (TGF α) have been suggested to play a major role together with keratinocyte growth factor (FGF7) and TGFβ1. These polypeptide ligands, as well as mechanic stimuli, are responsible for the activation of basal and suprabasal keratinoctes and re-epithelialization, the crucial step for successful wound healing and repair resulting in sealing of the epidermal wound and re-establishment of barrier function. During re-epithelialization, migrating keratinocytes undergo a series of morphological and functional changes that are reminiscent of EMT. Indeed, keratinocytes at the leading edge of the wound undergo an evident reorganization of the actin cytoskeleton and junctional structures, resulting in loss of polarity and disruption of cell–cell contacts and partial or complete degradation of basement membrane. The same cells then acquire the ability to migrate from the edge of epidermal wound to the de-epithelialized area. However, re-epithelialization in wound healing is seen as a partial EMT process since it involves cells that are both cohesive and motile (4). Indeed, migrating keratinocytes remain part of a cohesive cell sheet, as they retain some intercellular junctions; during the final stages of re-epithelialization keratinocytes undergo further changes progressively regaining epithelial characteristics.
Concerning the role of EMT, the group of Savagner has published in recent years relevant studies that have disclosed a major role for SNAI2 in regulating re-epithelialization (5, 226). SNAI2, but not SNAI1, is typically overexpressed in mouse and human keratinocytes at the edge of epidermal wound; its critical role in re-epithelialization has been established either by using skin explants from SNAI2 knockout mice (resulting in a compromised re-epithelialization) or in human keratinocytes that, manipulated in order to overexpress SNAI2, showed increased cell spreading and desmosome disruption (226). However, marking a consistent difference between standard EMT process and partial EMT seen in re-epithelialization, E-cadherin expression did not appear to be significantly decreased in migrating keratinocytes, as shown by RNA quantification after microdissection and immunolocalization.
SNAI2 has been recently reported to be a direct downstream mediator of EGFR in human keratinocytes (133), that is relevant for re-epithelialization since EGFR is known to be upregulated during wound healing. Moreover, increased SNAI2 transcription following EGF treatment of human keratinocytes (i.e., a condition able to strongly stimulate migration of keratinocytes) has been found to mechanistically depend on phosphorylation of Erk5 (5). Finally, it has been also shown that induction of keratinocyte migration by several ligands able to signal through EGFR (TGFβ1, KGF, TGF α, EGF, staurosporine) involves autocrine HB-EGF expression and, surprisingly, critically requires glycogen synthase kinase 3α (127).
C. EMT in fibrogenesis and organ fibrosis or Type 2 EMT: The examples of kidney, lung, and liver
In recent years several laboratories have proposed that EMT may have a role in organ fibrosis (114, 139, 210, 227, 229, 250, 298), where fibrosis may be defined as the result of chronic and uncontrolled activation of wound healing response as it can be appreciated in a number of fibroproliferative diseases in which progressive fibrogenesis (i.e., the process) with the time can result in the progressive accumulation of ECM components, derangement of tissue and vascular architecture, and eventually organ failure (179, 196, 279). The list of fibroproliferative diseases can potentially include diseases affecting all tissues and organ systems, although progressive kidney diseases, pulmonary fibroses, chronic liver diseases leading to cirrhosis, and cardiovascular diseases are those clinical conditions representing a worldwide leading cause of morbidity and mortality. A detailed analysis of cellular and molecular mechanisms sustaining fibrogenesis is out of the scope of the present review but one may assume that progressive fibrogenesis, due to the chronic activation of the wound healing reaction, is likely to be characterized by the following key features (summarized in Fig. 14): a) the persistence of cell injury and tissue damage, associated with a variable degree of necrosis and apoptosis; b) a complex inflammatory infiltrate including activated mononuclear cells and cells of the immune system that is usually followed and/or associated to formation of new blood vessels (i.e., angiogenesis); c) the involvement of ECM-producing cells, including fibroblasts and myofibroblast-like cells (usually referred to as myofibroblasts or MFs) that can potentially originate from different cellular sources (see later in this section); d) marked changes in the quality and quantity of tissue ECM associated with very limited or absent possibilities of remodeling in the presence of a persistent attempt of tissue regeneration.

Moreover, both activation/recruitment of fibroblast or MFs and fibrogenesis as a process, should be considered to be sustained by a number of epigenetic factors/mediators including at least several cytokines, growth factors, and chemokines, as well as hypoxia and ROS, which largely overlap with those stimuli potentially able to trigger EMT.
Coming back to the problem of the origin of fibroblasts and MFs involved in fibrogenesis in different tissues, two rather distinct views or scenarios should be considered before to start to analyse the role of EMT in organ fibrosis. A first scenario is the one delineated by most of investigators involved in the field of fibrogenesis and adult organ fibrosis that suggests the following potential cellular sources for ECM-producing cells and/or MFs (196, 279 and references therein): a) Resident tissue fibroblasts that are interpreted as residual embryonic-like mesenchymal cells left over from organogenesis. b) Blood-borne mesenchymal progenitors that have been defined as “fibrocytes”, exhibiting a fibroblast- or myofibroblast-like phenotype and expressing CD34, CD45, and collagen type I. c) Epithelial cells through the process of EMT. d) Organ specific precursor cells, such as in the liver where extensive literature suggests that resident hepatic stellate cells (HSC) are the main sources of MFs (76, 108, 196 and references therein). e) Bone marrow-derived mesenchymal stem cells (MSC), as recently shown for liver fibrogenesis (221, 262), with MSC differing significantly from fibrocytes being CD34 and CD45 negative and being characterized by expression of CD44, CD90 and CD 105.
A second and somewhat alternative view is the one proposed by Kalluri and Neilson some years ago (115) and that has been substantially recalled more recently by Schneider et al. (229). If we refer to the five potential sources of ECM/producing cells and/or MFs, the view of Kalluri and Neilson can be briefly (more details are in the original study, Ref. 115 and refs. therein) summarized as follows: a) tissue fibroblasts are unlikely to be derived from embryonic mesenchymal cells since primary mesenchymal cells are negative for FSP-1; b) bone marrow-derived, CD34− and FSP-1+ fibroblasts might derive from an endosteal EMT niche (with endosteal lining cells being FSP-1+) transitioning to CD34− and FSP-1+ bone marrow stromal cells (109) that, in turn, may evolve into circulating fibrocytes (1); c) FSP-1+ ECM-producing cells mostly derive locally in tissues following EMT, as essentially inferred by studies on kidney fibrogenesis (109, 115); d) fibroblasts (this is the common definition used by most of researchers supporting a major role for EMT in adult fibrogenesis) are heterogeneous in terms of biochemical differences, phenotypic variability, and respond differently to cytokines and ECM, depending on their tissue origin; Kalluri and Neilson suggest that fibroblasts originated from EMT may differentially express a profile of genes, receptors, or signaling pathways as a memory of their previous life as mature epithelium.
As a reader can easily appreciate, the two views are theoretically quite different but have in common a notion: ECM-producing cells in adult fibrogenesis are heterogeneous. Because of the extensive literature in this field, we will limit our analysis to selected conditions of adult fibrogenesis.
The most convincing evidence for a significant role of EMT in organ fibrosis (recently defined as Type 2 EMT, Refs. 114, 298) has been provided by studies on renal fibrogenesis associated to chronic kidney diseases (CKDs) which are characterized by progressive loss of kidney function and ECM increased deposition, leading to widespread fibrosis. In particular, tubulo-interstitial fibrosis is interesting because deterioration of kidney function is largely dependent on the severity and extent of interstitial lesions. Moreover, whatever the initial cause, interstitial fibrosis is characterized by the appearance of α-SMA-positive MFs as main effector cells. Historically, the first findings supporting a role for EMT in kidney fibrogenesis were provided by Strutz and coworkers (240, 241) using an animal model of anti-tubular basement membrane (TBM) disease. These authors showed that in the ongoing disease tubular epithelial cells could express the antigenic marker FSP-1 that in normal conditions is expressed by fibroblasts but not by epithelial cells. In the following years several lines of evidence confirmed the presence of EMT in renal fibrosis both in animal models of CKDs as well as in human kidney biopsies (148, 242, 297, and references therein). In animal models, which of course allow to follow the kinetics of events, several tubular epithelial cells were found to co-express tubular markers and α-SMA, suggesting that they were cells in transition towards the mesenchymal phenotype; moreover, these cells lost the epithelial marker E-cadherin and started to express α-SMA and to produce collagen type I. In most of the models EMT was apparently limited to proximal tubular epithelium, and it has been suggested that EMT may represent a sort of reverse embryogenesis in which the tubular epithelial cells transform back into a mesenchymal phenotype from which they originate. However, as recently reviewed (240), it is likely that all tubular epithelial cells may possess the capacity to undergo EMT. Moreover, also glomerular parietal epithelial cells have been reported to undergo EMT in two rat models of CKDs like 5/6 nephrectomy and the model of antiglomerular basement membrane (GBM) antibodies (175,176).
Iwano and coworkers (109), by using a model of obstructive injury to tubular epithelium (i.e., the model of unilateral ureteral obstruction), showed a mechanistic link between tubular epithelium, EMT, and kidney MFs: they genetically tagged renal proximal tubule in order to follow their fate and movements and they found that LacZ-tagged epithelial cells with time exhibited abnormal/degenerated morphology, became disorganized, and, as FSP-1 and HSP-47 positive cells, started to move into the interstitial space. In this study, the authors, by performing a careful cell count of interstitial fibroblasts in both normal and injured kidney during experimental fibrosis, reported that local EMT and bone marrow contributed to 36% and 15% of kidney fibroblasts, respectively. However, as recently suggested by Strutz (242), the contribution of EMT to the formation of MFs and then fibrosis may be less relevant in other experimental models.
Coming back to the model of obstructive injury to tubular epithelium, a time-dependent analysis of the origin and dynamics of renal MFs activation revealed a biphasic pattern, in which MFs come mainly from local activation of interstitial fibroblasts at the earlier stage, whereas at the later experimental stage (14 days from obstruction) predominantly derive from tubular cells following EMT (148).
Where human studies are concerned, analysis of 133 biopsies from patients affected by different CKDs was performed by means of immunohistochemistry or in situ hybridization (216). In this study, evidence was reported for a presence of a significant, although numerically limited, number of tubular epithelial cells being positive for vimentin or α-SMA as well as for HSP-47, prolyl-4- hydroxylase, collagen type I and III; moreover, the authors observed loss of epithelial antigens from 8% to 10% of the tubular cross sections. However, the most interesting findings was the detected significant relationships between the number of tubular epithelial cells with EMT features with serum creatinine values and the degree of interstitial injury, suggesting that these features may be of value even in the assessment of disease severity. A positive correlation between EMT markers (mainly FSP-1) and renal function (i.e., serum creatinine levels) was also reported by others in biopsies from patients with tubular atrophy and interstitial fibrosis (267), as well as in IgA nephropathy, lupus nephritis, and chronic allograft failure (reviewed in 242). More controversial is the correlation between EMT and proteinuria, but available evidence (240 and reference therein) suggests that proteinuria alone (i.e., a condition able to result in direct tubular toxicity and inflammatory response) is not sufficient to induce EMT or, at best, is a weak inducer of EMT. Although in a rat experimental model of chronic allograft nephropathy, EMT was found to correlate with the occurrence of EMT (56), in human patients affected by CKDs a positive correlation with proteinuria and markers of EMT was found only for increased tubular expression of vimentin (216) or decreased cytokeratins levels (267), whereas no other marker of EMT was found to correlate with the degree of urinary protein excretion.
According to published evidence (reviewed in 148, 242, 297 and references therein), tubular epithelial cells may undergo EMT as triggered by four major stimuli: a) TGFβ1, which is known to be highly overexpressed in CKDs of different etiology; the relevance of TGFβ1 and downstream signaling pathway, including involvement of ROS generation (218), has been remarked by the results of Zeisberg and coworkers (299) that, by employing a mouse model of chronic renal injury, were able to reverse TGFβ1-induced EMT by administration of recombinant human BMP-7; the use of BMP-7 was applied also to other models (242, 296) as a putative example of a rather selective experimental therapeutic strategy based on the cross talk between BMP-7 and TGFβ1 in the regulation of EMT, with BMP-7 being potentially able to induce reversal of renal fibrosis; in addition, another example of putative therapeutic interference has been shown by data indicating that HGF suppresses TGFβ1-mediated renal interstitial myofibroblastic activation, an action of HGF that is likely to be related to a mitogen-activated and protein kinase-dependent blockade of Smad nuclear translocation (284). b) Changes in ECM composition, such as an increase in fibrillar collagen type I, operating through ILK (144) that, however, can also operate as a critical mediator of TGFβ1-triggered EMT. c) Activation of the plasmin system, where plasmin derived from proteolytic cleavage of plasminogen is able to induce ERK phosphorylation and EMT in tubular epithelia (303); this evidence has been confirmed by studies performed in mice lacking tissue plasminogen activator or tPA (148): in these mice a very significant prevention of interstitial fibrosis after obstructive injury was observed, possibly related to a decrease in MMP-9 expression and EMT triggering. d) Conditions of hypoxia, as will be detailed in a later section, are able to induce EMT in tubular cells through HIF-1α-mediated signaling and processes (98, 122).
A role for EMT has been proposed also for the response to injury and pathogenesis of fibrosis in the adult lung (47, 271, 272). Pulmonary fibrosis can be triggered by several etiological agents or conditions, including allergens, chemicals, radiation, and environmental particles, although the cause of idiopathic pulmonary fibrosis (IPF ), one of the most common pulmonary fibrotic clinical condition, is still unclear and possibly again related to multiple causes and mechanisms. From a historical point of view, IPF has been viewed as the result of persisting cellular injury and inflammation, leading to activation and proliferation of resident lung interstitial fibroblasts (63). This canonical view fits with the general three phase model of pulmonary wound repair and fibrosis that has been very recently outlined (see for more details Ref. 274 and refs. therein), including: a) Phase 1 or the phase of injury, which is dominated by epithelial and endothelial cell damage and death (by apoptosis), platelet activation, and formation of fibrin-rich clots, as well as increased expression of MMP-2 and MMP-9 that favor basement membrane and ECM degradation and allow egression or clearance of inflammatory cells out of the inflamed tissue and into the airspaces. b) Phase 2 or the phase of inflammation, in which chemokine gradients can lead to the recruitment of circulating inflammatory cells and of fibrocytes to the site of injury that, in turn, will supply cytokines and growth factors able to support activation of fibroblasts, events favored by concomitant angiogenesis. c) Phase 3 or the phase of repair, characterized by wound contraction operated by fibroblasts and MFs, by apoptosis and phagocytic removal of inflammatory cells and MFs, as well as by re-epithelialization and regeneration.
Recent findings from either experimental models of lung fibrosis and human patients affected particularly by IPF has led some authors to a reconsideration of inflammatory response as the major pathogenic event (271, 272) based on the two following points: a) inflammation is not usually prominent in IPF biopsies; b) anti-inflammatory therapy has offered little benefit in the treatment of IPF. This has led to the hypothesis that IPF progression may also rely on abnormal wound healing with alveolar epithelial cells (AECs) being not simply the target of injury (leading to cell death, usually by apoptosis) but also cells with a considerable plasticity, being able to contribute either to re-epithelialization and restoring of alveolar architecture as well as to fibrogenesis by acquiring mesenchymal-like phenotype and becoming a source of fibroblasts and MFs.
The problem in lung fibrosis, or at least in IPF and some interstitial lung diseases, is similar to the one mentioned for kidney fibrosis: the ECM-producing cells (the effectors of fibrosis) may have a multiple origin and again resident fibroblasts as well as either bone marrow- or AECs-derived cells are indicated as sources of α-SMA positive MFs (47, 63, 115, 271, 272, 279). The following points should be underlined. First, the most prevalent hypothesis still postulates that MFs originate from resident fibroblast as a response to several fibrogenic stimuli, with a major role for TGFβ1. However, some studies have proposed that bone marrow-derived progenitors may contribute to MFs induction and proliferation during pulmonary fibrosis; as reviewed by Lama and Phan (136), studies tracking CD34 and/or CD45 markers of fibrocytes show their presence in injured murine lung tissue. Moreover, bone marrow chimeric mice with green fluorescence protein (GFP)-expressing marrow cells show abundant GFP-expressing fibroblasts in their lungs in response to lung injury, being recruited by several chemokines. However, the real relevance of fibrocytes for lung fibrosis is still controversial since there are studies suggesting a protective effect for bone marrow-derived cells (136). Moreover, these marrow-derived fibroblasts do not express α-SMA and are resistant to TGFβ1-induced conversion of fibroblasts to MFs.
Where the origin of lung MFs from AECs is concerned, a number of observations and concepts should be recalled that seem pertinent (more details in Ref. 272). First, sustained AECs injury and apoptosis as well as retarded wound repair are likely to be critical in the pathogenesis of pulmonary fibrosis; moreover, it is known that type 2 alveolar cells (AT-2) must proliferate and differentiate into type 1 cells (AT-1) in order to allow re-epithelialization. Second, it has long been known that epithelial cells overlying fibroblastic foci show abnormal morphology (hyperplastic and dysplastic) and can secrete pro-fibrogenic cytokines, then being able to cross-talk with neighboring fibroblasts/MFs; this is particularly true for IPF conditions where abnormal and hyperplastic AT-2 cells have been detected with an intermediate phenotype overlying fibroblastic foci. In addition, alveolar epithelium is considered a major source of TGFβ1 and other cytokines but there is evidence suggesting that AECs can also respond to TGFβ1.
According to these concepts, the hypothesis is then that AT-2 cells, and possibly (see later) also some airway cells may be able, in defined conditions of chronic injury, to transdifferentiate into fibroblasts/MFs. As recently reviewed, in vitro and in vivo evidence supporting this hypothesis can be summarized as follows (272 and ref. therein): a) Prolonged exposure of primary AECs, as well as of AEC line (for example, RLE-6TN) or primary rat AT-2 cells in culture to TGFβ1 is known to be followed by classic EMT changes (273). This also applies to mouse AT-2 cells that, after exposure to both TGFβ1 and EGF, also expressed FSP-1. This is relevant for the well-known pro-fibrogenic role of this cytokine and because TGFβ1 is expressed at sites of epithelial injury and adjacent fibrosis in vivo. b) Morphological studies indicate that epithelial cells overlying fibroblastic foci in IPF have a fibroblast-like phenotype, change their pattern of cytokeratins expression and have transcriptional profile suggestive of EMT; moreover, fibroblasts adjacent to area of epithelial hyperplasia express a significant number of epithelial markers. c) There are convincing experimental studies, performed using sophisticated models of transgenic mice submitted to either bleomycin-induced chronic lung injury or intranasal instillation of TGFβ1, supporting the concept that both AECs and bronchial epithelial cells can undergo EMT in vivo, then contributing significantly to experimental pulmonary fibrosis (119, 120, 278). In these models, EMT seem indeed to represent a major event. d) Less detailed evidence for EMT in human lung biopsies has been provided until now, with co-expression of myofibroblast and AEC markers in AECs reported in biopsies from patients affected by IPF (273) or, again in IPF patients, with signs of EMT consisting in the detection of cells co-expressing the pro-surfactant protein and the mesenchymal marker N-cadherin (119). However, it is correct to note that little evidence for EMT has been reported in a more recent morphological study performed on 15 lung biopsies obtained from IPF patients and 10 biopsies from patients with a diagnosis of nonspecific interstitial pneumonia (NSIP) (282).
Where liver fibrogenesis and fibrosis are concerned, EMT of cultured neonatal hepatocytes has been described for the first time in 1995 (188) but only recently has EMT emerged as a possible contributing pathogenic mechanism sustaining progressive fibrogenesis in chronic liver diseases (CLDs). CLDs are due to chronic infection by hepatotropic virus (mainly hepatitis B and C viruses) or to metabolic, toxic/drug-induced (with alcohol being predominant) and autoimmune causes. Whatever the etiology, fibrotic progression of CLDs to the common advanced stage of cirrhosis can be envisaged as a dynamic and highly integrated cellular response to chronic liver injury. Liver fibrosis is accompanied by perpetuation of liver injury, chronic hepatitis, and persisting activation of tissue repair mechanisms, leading eventually to excess deposition of ECM components, liver cirrhosis, and hepatic failure. Liver fibrogenesis (i.e., the process) is sustained by heterogeneous populations of highly proliferative, profibrogenic, and contractile α-SMA positive MFs that, according to current literature, can originate from different cellular sources with a relative contribution that may vary depending on the specific etiology of the CLDs, the pattern of fibrosis and the prevailing fibrogenic mechanism (76, 108, 196).
Current literature indicates that the major contribution to liver fibrogenesis is from perisinusoidal hepatic stellate cells (HSCs), also known as liver-specific pericytes, that undergo a process of activation and trans-differentiation into a MF-like phenotype (HSC/MFs) that is believed to be regulated by cytokines, chemokines, growth factors, ROS, and several other mediators (75, 76, 108, 179, 196, and references therein). Available data indicate that HSC-MFs are involved in most of clinical conditions of CLDs, with a prevailing involvement in the pattern of fibrosis progression defined as “perisinusoidal/pericellular fibrosis,” recognizing a metabolic or alcoholic etiology (196). HSC are believed to significantly contribute also to the origin of the so-called interface MFs and then also to the pattern of “bridging fibrosis” found in patients affected by chronic viral hepatitis (196).
Before the proposal of a role of EMT (particularly concerning EMT of cholangiocytes, as we will discuss later) in the classic view, a second major contribution to fibrogenesis was attributed to portal fibroblasts. These cells, located in the connective tissue of portal areas, have been reported to undergo a process of activation/transdifferentiation into MFs that are considered relevant in ischemic conditions and in obstructive cholestatic diseases (pattern of biliary fibrosis, 76, 108, 196).
Under conditions of chronic liver injury, pro-fibrogenic MFs (mainly interface MFs and some portal MFs) have been described also to originate from progressive recruitment of bone marrow-derived cells, including mesenchymal stem cells or MSC (221, 262) and circulating fibrocytes (124).
As an essential basis of knowledge, the reader should be aware that cells displaying EMT (i.e., expressing both mesenchymal and epithelial markers) have been detected in fetal liver stroma during the hematopoietic phase but not in nonhematopoietic liver by the end of gestation and in the normal adult liver (36). Moreover, cells resulting from early hepatic differentiation of endoderm, usually defined as hepatoblasts, as well as a numerically limited population of hepatic progenitor cells (HPCs) or oval cells that proliferate in the adult injured liver, share the capacity to give rise to either mature hepatocytes or bile duct epithelial cells (BDEC, also defined as cholangiocytes) (70, 252) that are then the two epithelial hepatic lineages that may potentially undergo EMT in CLDs. According to these premises, we will consider first in vivo and in vitro evidence supporting the occurrence of EMT in hepatocytes. As previously mentioned, but sometimes neglected, typical EMT changes in vitro were first reported by Vilaró et al. (188, 189) in cultured rat neonatal hepatocytes, and later on by other groups using cultured primary mouse hepatocytes (113, 300) or in different nontumorigenic hepatocytic cell lines (42, 113). In these studies, several growth factors and cytokines were described to have a role in EMT (42, 113, 189, 300), with TGFβ1 being able to induce, through Smad2/3 signaling, all the canonical EMT-related changes, including SNAI1 induction, E-cadherin delocalization and downregulation, downregulation of the hepatocyte transcriptional factor HNF4, and upregulation of mesenchymal and invasiveness markers (42, 113, 300). In one of these latter studies (300), the authors described a progressive appearance in the injured livers of FSP-1 positive cells, although less than 10% of FSP-1+ cells were shown to co-express the typical and widely accepted MF marker α-SMA. These authors also performed lineage-tracing experiments using AlbCre.R26RstoplacZ double transgenic mice in order to investigate whether hepatocytes undergoing EMT may contribute significantly to fibrosis induced by chronic treatment with the hepatotoxin CCl4. They reported that approx. 15% of hepatic cells were FSP-1 positive at the time of severe fibrosis and that approx. 5% of the hepatic cells were co-expressing either FSP-1 and albumin or FSP-1 and β-gal, suggestive of EMT. The authors also performed experiments showing that BMP-7, which is known to antagonize TGFβ1 signaling, significantly inhibited progression of fibrosis and almost abolished putative EMT-derived fibroblasts. Similar results, pointing more specifically to a hepatocyte contribution to fibrosis by a TGFβ1-dependent induction of EMT, were also described by another group that employed a transgenic mouse model of Smad7 overexpression in hepatocytes to counteract CCl4-induced fibrosis (60). In the latter study, the authors also reported preliminary morphological evidence suggesting that signs of activation of TGFβ1 signaling (i.e., detected by immunoistochemistry for phospho-Smad2) are expressed in the nuclei of hepatocytes in biopsies from patients affected by chronic HBV infection or schistosomiasis. In HBV biopsies they also showed by immunohistochemistry the presence of nuclear staining for SNAI1 that was, however, found in both transferrin positive and negative cells.
At present, only two experimental studies have provided some in vivo evidence for hepatocellular EMT and we still lack clinical studies properly designed in order to ascertain the real contribution of such a process in progressive fibrogenesis associated to the most common forms of human CLDs where the major role of HSC/MFs has extensively described (76, 108, 196).
More convincing should be considered published in vivo and in vitro data related to EMT of BDEC in either experimental or clinical conditions associated with a form of biliary fibrosis, which represent approx. less than 10% of CLDs in western countries. The first report suggesting a BDEC origin of portal MFs was published in 2006 (280): this study described that BDEC undergoing the well-known process of bile ductular reaction in the rat model of secondary biliary fibrosis of bile duct ligation (BDL, resulting in obstruction of biliary tree) were co-expressing α-SMA and cytokeratin 19 (CK-19, a BDEC and HPCs marker), a EMT scenario fully reproduced by treating in vitro BDEC with TGFβ1 (followed by upregulation of α-SMA and fibronectin, with downregulation of CK19) and prevented, both in vivo and in vitro, by pretreatment with HGF. The experimental scenario was confirmed in the same model of BDL (rat and murine) by a series of elegant studies from the group of Anna Mae Diehl, the most relevant (184, 185, and reference therein) being able to describe a clear cause–effect relationships between EMT of BDEC, appearance of portal MFs and biliary fibrosis, as well as the closely related major involvement of Hedgehog signaling pathway (Fig. 15). The most relevant point, in our view, is that the same group as well as other groups were able to convincingly describe evidence for EMT of BDEC (monitored by following co-expression of several markers including CK19, α-SMA, HSP-47, phospho-Smad2/3, FSP-1, etc) not only in experimental conditions but also in biopsies obtained from human patients affected by primary biliary cirrhosis (PBC, 112, 185, 219), primary sclerosing cholangitis (123), and biliary atresia (54). Moreover, EMT of BDEC was also described in post-transplantation recurrence of PBC (219). Again, the relevance of Hedgehog and TGFβ1-Smad2/3 signaling was reported also in human patients (112, 185, 219, 222). Indeed, these studies seem to support the view that EMT of BDEC undergoing bile ductular reaction may significantly contribute to the genesis of portal MFs in those conditions collectively described as of biliary fibrosis (76, 108, 196). At present, two reports (111, 222) have also provided the first evidence for EMT in BDEC and/or HPCs in biopsies from patients affected by alcoholic liver disease, with again Hedgehog and TGFβ1-Smad2/3 signaling having a major role. Although more studies are needed, these latter findings may suggest that in a rather advanced phase of ALD (in the early phase fibrosis by chronic alcohol consumption is peri-sinusoidal and sustained by activated HSC) (76, 108, 196) this process may contribute to increased ECM deposition in alcoholic patients.
D. EMT in cancer progression and metastasis or Type 3 EMT: The initiation of invasive and metastatic behavior of epithelial cancer cells
Accumulating data from both experimental animal models and clinical studies have provided convincing evidence for the relevance of a deregulated form of EMT, now also indicated as Type 3 EMT (114, 298), in cancer progression (88, 104, 114, 126, 138, 139, 168, 201, 207, 250, 251, 283, 298), although this is still somewhat controversial (246). Where carcinomas (i.e., malignant tumors of epithelial origin that represent the most prevalent malignancies in humans) are concerned, it is well known that progression is associated with the loss of epithelial features and progressive acquisition of a motile phenotype as well as increased ability to degrade ECM and to survive outside the epithelium. Indeed, similar to embryonic development, EMT and the reverse process of MET (35) have been proposed to play a critical role in cancer, with EMT being recognized as a mechanism contributing to increased invasiveness, metastatic dissemination, and resistance to therapy, whereas MET is likely to occur following dissemination and favoring formation of distant metastasis (Fig. 16).

Since we have already described in detail stimuli, signaling, and transcriptional mechanisms able to trigger and modulate EMT, in this section we will intentionally focus attention on those EMT-related concepts and mechanisms that are likely to have a significant impact and clinical relevance for tumor progression and cancer-related mortality, rather than to cite the now impressive literature available. A number of relevant points should be underlined.
A first point is that many of the pathways that the literature associates with tumor progression seem to converge on E-cadherin downregulation, considered as a critical event in the development of metastasis in most if not all epithelial tumours (15, 20, 201); accordingly, the loss of E-cadherin expression in sporadic tumors is associated with a poor clinical prognosis (90).
As a second issue, genetic as well as epigenetic changes have been suggested to promote or favour EMT in tumors (15, 207, 283), including mutations within E-cadherin gene (as in familial gastric cancer, with a subset of these patients being also more susceptible to develop lobular breast carcinomas) and polymorphism of E-cadherin promoter, although these changes may not unavoidably lead to EMT. By contrast, hypermethylation of CpG islands in the promoter of E-cadherin has been shown to preceed cancer cell invasion and to persist in invasive and metastatic lesions (15). In addition, sustained activation of EMT can lead mammalian cells to epigenetic alterations such as methylation of CpG sites and of several other gene promoters, including those of the estrogen receptor and Twist, then inducing heritable effects that may maintain the mesenchymal phenotype even after EMT-initiating signals are no longer present in the environment (64). Moreover, a recent report suggests that genes encoding for transcription factors implicated both in the induction of EMT and stem cells functions are hypomethylated and highly expressed in cancer cells, with the degree of expression being correlated with a poorly differentiated phenotype and increased risk of metastasis (22).
As already mentioned previously in this review, one of the main mechanisms underlying the downregulation of E-cadherin in both normal development and cancer is represented by direct transcriptional regulation of the gene, including mutations and deregulation of oncogenic signal transduction pathways (15, 198, 207, 283). A first relevant example is represented by the notion that SNAI1 is overexpressed at the invasive front of tumors, where it is inversely correlated with E-cadherin expression, in lymph node metastases of breast cancer, and has been suggested as a putative prognostic marker of malignancy (21). Similar considerations apply also to upregulation of TWIST, ZEB1, and ZEB2 in malignant and/or metastatic tumors (199) as well as to other transcription factors involved in EMT triggering. Where deregulation of signal transduction pathways is concerned, carcinoma cells that arrive at the stroma/tumor interface deregulate signaling cascades induced by TGFβ, HGF, and Wnt ligands (including in the scenario also the established interactions between the Wnt/β–catenin pathway, GSK3β, and SNAI1 and SNAI2) in order to promote cell survival and EMT (135, 220).
As a further preliminary message, as recently reviewed (207), hypoxia and miRNAs are emerging as relevant conditions able to induce (hypoxia) or modulate EMT in tumors (see sections IV.F and II.F, respectively, for more details).
The previous considerations should be implemented by other messages and findings that should in the end identify which is the clinical relevance of EMT in cancer progression, invasiveness, and metastasis (or Type 3 EMT).
A first concept is that, as nicely suggested very recently by Polyak and Weinberg (207), the overall EMT-triggering scenario and related morphological and functional changes may significantly contribute to the well-known phenotypical heterogeneity observed within large solid tumors, in addition to those changes that are known to be related to the typical genetic and epigenetic instability of cancer cells.
If we decide to go further in this direction, most authors (15, 20, 21, 53, 126, 164, 168, 198, 199, 201, 207, 250, 251, 281, 283) consider that EMT, by either favoring the appearance/selection of a more invasive phenotype and/or sustaining epigenetic changes in cancer cells (see Fig. 17 for a synthesis of major evidence),is of course implicated in tumor progression and metastasis, as well as in the acquisition of therapeutic resistance, two processes primarily responsible for cancer-related mortality. However, as the most recent and authoritative reviews suggest (15, 28, 168, 198, 207, 228, 283), these two processes may be strictly linked to a third one: EMT may favor/induce the generation of cancer cells expressing stem cell features (i.e., cancer stem cells or CSCs), as revealed mainly by studies performed on mammary epithelial cells and breast cancer as well as on colorectal cancer.
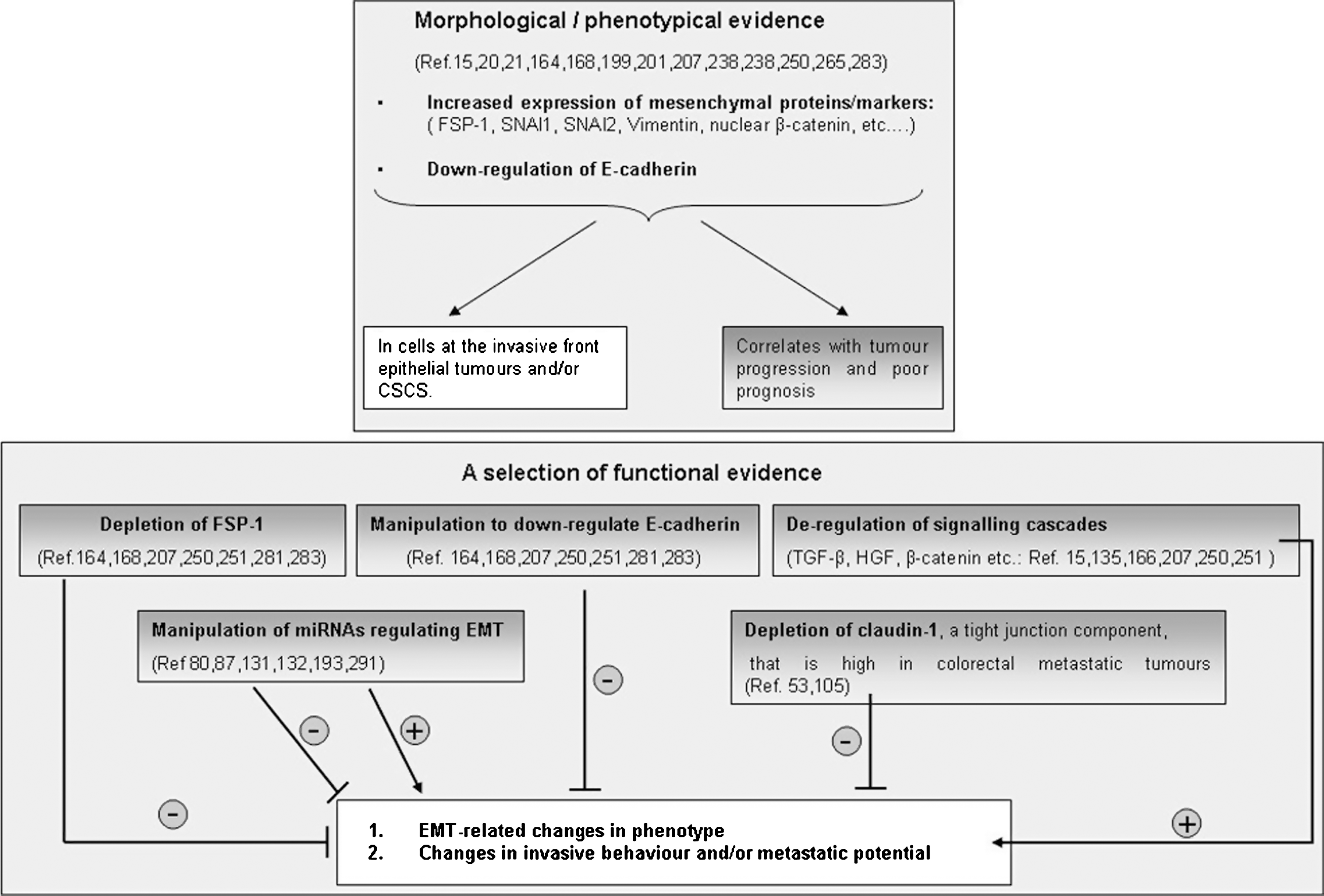
In the last 2 years, different laboratories have published studies on mammary epithelial cells indicating that expression of EMT-triggering transcription factors like SNAI1 or TWIST or treatment with TGFβ can result in an increase in the number of cells showing characteristics of CSCs, as suggested by their antigenic profile as well as by the ability to form mammospheres, soft agar colonies, and tumors more efficiently (154, 165). Moreover, a series of studies performed on breast cancer (207) has proposed that CD44+ CD24− stem cell-like cells, that express high levels of EMT markers, are mostly related to the basal-like subtype of human breast cancer. Accordingly, in a tissue microarray-based immunohistochemical study performed on 479 biopsies of invasive breast carcinoma, it has been shown that the basal-like subtype of human breast cancer is indeed characterized by high expression of EMT-induced markers (vimentin, α-SMA, N-cadherin), SPARC, laminin, and fascin, as well as by low expression of E-cadherin, and have an especially poor prognosis being associated with an increased attitude to form metastasis (225).
Even more consistent and worth mentioning is the body of evidence relating EMT and stemness to the generation of gastrointestinal cancer, with a major focus on colorectal cancer (CRC), a scenario which has received major contributions and has been nicely reviewed by Thomas Brabletz and coworkers (28, 228, and references therein). A critical concept in the genesis of these tumors is represented by the knowledge that regulation of intestinal stem cells (ISCs) is based on a permanent cross-talk between epithelial cells and the underlying mesenchymal/stromal cells in the intestinal stem cell niche. This cross-talk is mediated by a defined number of signal transduction pathways that are all critically involved in EMT, including Wnt/β–catenin, Notch, PI-3K/c-Akt, Hedgehog, and BMP pathways. Literature suggests (28, 228) that any significant disturbance of such a delicate cross-talk can initiate intestinal cancers and that these disturbances, in association with other additional genetic alterations or environmental activation of EMT, can lead to cancer progression, invasion, and metastasis. A major role is attributed to deregulation of Wnt/β–catenin pathways for both initiation and malignant progression of CRC and, indeed, aberrant Wnt signaling, under the form of APC mutations or loss of function as well as of β-catenin activating mutations, has been detected in putative familial adenomatous polyposis coli (FAP), adenomas, as well as almost all human colorectal adenocarcinomas and carcinomas. Where initiation of CRC is concerned, the idea is that initiating APC or β-catenin mutations, alone or associated to alterations of other regulatory pathways (PI3K, BMP, or Notch pathways), will result in a deregulation of ISCs leading to a weak activation of Wnt signaling sufficient to lead these stem cells to increased proliferation and survival in either FAP and adenoma, then initiating the carcinogenic process.
Where malignant progression towards invasive carcinomas and metastases is concerned, the involvement of EMT and its links with stemness are strongly suggested by a number of related observations that eventually led Brabletz and his colleagues to propose the model of migrating cancer stem (MCS) cells (28, 29, 228). In synthesis, it has been shown that the more undifferentiated cancer cells at the invasive front of typical colorectal adenocarcinomas, but significantly not those of the more differentiated central tumor area or those of metastases, were characterized by strong β-catenin nuclear staining as well as by loss of E-cadherin. These phenotypic changes, related to aberrant Wnt signaling, suggested that invasive cancer cells were then likely to undergo first an EMT to leave the primary tumor and then a MET process in the metastases.
In the model of MCS cells, Brabletz and coworkers propose that due to additional somatic mutations as well as environmental factors and signals, tumor cells, and particularly the cells recently defined as stationary cancer stem cells (SCS-cells, 28), usually embedded in benign adenomas and found also in the cental areas of carcinoma and metastases, may undergo a more significant activation of Wnt signaling, resulting in induction of EMT. These cells, expressing nuclear β-catenin and downregulation of E-cadherin, then become mobile and have been referred to as MCS-cells that, according to their ability to divide asymmetrically, have been proposed to be able to both contribute to the increase of the mass of the primary tumor or to the dissemination through blood and lymphatic vessels that leads to metastases.
Another message to be recalled is that EMT may be relevant in resistance to cancer chemotherapy. As a matter of fact, CD44+CD24− stem cell-like cells expressing EMT markers have been found to be enriched in the residual tumors following standard antracyclin/taxane chemotherapy protocol for breast cancer (143). Similarly, epithelial tumor cells have been shown to be significantly more sensitive to treatment with EGFR inhibitors than tumor cells that have undergone an EMT-like transition and acquired mesenchymal characteristics, including non-small cell lung and colorectal carcinomas, as well as carcinomas of head and neck, bladder, breast, and pancreas (13).
E. Hypoxia as an emerging and independent master condition able to trigger EMT in human diseases
Hypoxia, with its related signaling and transcriptional mechanisms, is emerging as one of the most relevant conditions able to induce EMT in embryogenesis, organ fibrosis, and tumors (71, 99, 100, 207, 234). According to present knowledge, hypoxia can be defined as an oxygenation state that is below the norm for a particular tissue, with the average oxygen partial pressure (pO2) of normal tissues usually exceeding 20 mm Hg (52, 231, and ref. therein). The transition between normal and hypoxic conditions for a defined tissue usually requires that pO2 falls to levels lower than 10 mm Hg and is unequivocally marked by a transcriptional response of the exposed cells that result in increased expression of several genes that are regulated by a family of transcription factors known as hypoxia-inducible factors (HIFs). An extensive analysis of HIFs and HIFs-dependent mechanisms is out of the scope of the present review, and the interested reader can found more details in authoritative reviews (52, 215, 231 and ref. therein). Here only major concepts will be briefly recalled.
HIFs are members of the PAS (PER-ARNT (arylhydrocarbon receptor nuclear translocator)-SIM) family of basic helix–loop–helix (bHLH) transcription factors that bind to DNA as heterodimers composed of an oxygen sensitive α subunit (three subunit described and designated as HIF-1α, HIF-2α, and HIF-3α) and a constitutively expressed β subunit (two subunit described, ARNT or HIF-β and ARNT2) which heterodimerize and bind to DNA at hypoxia response elements (HREs) in promoter or enhancer regions of several hypoxia target genes to upregulate their expression. At present, three HIFs have been described (HIF-1, -2, and -3). HIF-1 is the best characterized regulator of the cellular response to hypoxia, usually formed by HIF-1α and ARNT, being ubiquitously expressed in most organ and tissues and able to upregulate many hypoxia-inducible genes. HIF-2 is more specific with HIF-2α being expressed, under hypoxic conditions, only in some specific cell types including endothelial cells, glial cells, type II pneumocytes, cardiomyocytes, fibroblasts of the kidney, interstitial cells of the pancreas and duodenum, hepatocytes, and likely, as recently suggested, cancer stem cells (100, 207).
Under normoxic conditions, HIF-1α is continuously modified by a number of enzymes (proline hydroxylases or PHs; the asparaginyl hydroxylase HIF-1 α subunit inhibitor or HIF1AN, also known as FIH1) whose activity is dependent on oxygen levels. Two major mechanisms are known that prevent the formation of transcriptional complexes able to translocate into the nuclei and bind HRE sequences: a) HIF-1α, when hydroxylated on proline residues and/or acetylated on lysine residue in normoxic conditions, binds to the von Hippel –Lindau protein and other polypeptides forming a multisubunit complex that is ubiquitylated and continuously degraded via proteasomes; b) alternatively, HIF1AN can hydroxylate an asparagine residue of HIF-1α preventing the formation of the final transcriptional complex.
Under hypoxic conditions, PHs and/or HIF1AN are progressively inactivated and HIF-1α can form the heterodimer with ARNT; HIF1 is then phosphorylated/stabilized by intervention of kinases and can form the ultimate transcriptional complex able to bind HRE sequences. Several sets of genes are activated in a HIF-1-dependent manner, including those involved in vasomotor control and erithropoiesis (inducible NO synthase, endothelin 1, and erythropoietin), energy metabolism (such as glucose transporters like GLUT-1 and -3 and several glycolytic enzymes), cell survival (ion transporters and exchangers able to regulate intracellular pH), cell proliferation, and cell cycle, angiogenesis, and ECM degradation, and remodelling (members of the VEGF family and related receptors, angiopoietins and related receptors, collagen prolyl-4-hydroxylase, the receptor for urokinase–plasminogen activator or uPA-R and the plasminogen activator inhibitor type 1 or PAI-1).
In some tissues or tissue areas, pO2 is low even in physiological conditions, including the normal liver, where the first rim of perivenular hepatocytes has been described to be under conditions of partial hypoxia, as well as renal tubular epithelium, myocardium (particularly during exercise), and subregions of the bone marrow that are enriched for stem cells.
Pertinent to this review, although under hypoxia HIF-1α activation is mostly obtained by post-translational mechanisms, there are conditions able either to lead to increased HIF-1α mRNA transcription (including cytokines, growth factors, oncogenes, metabolic stress, and reactive oxygen species or ROS, by operating through activation of PI3K and Ras/Erk signaling pathways) or to HIF-1α increased stabilization (for example, following direct interaction with ROS). This scenario, in which HIF-1α may operate also independently on hypoxia, is likely to play an additional role in conditions of chronic injury and organ fibrosis (71, 99) as well as of cancer progression (52, 100, 215, 231).
Where relationships between hypoxia and EMT in human diseases and related experimental animal models are concerned, according to available literature we should focus the attention to their relevance mainly in conditions of organ fibrosis and cancer, although theoretically these relationships should be relevant also for embryogenesis. Along these lines, it is indeed worth mentioning that, as nicely and extensively reviewed by Simon and Keith (234 and ref. therein) low levels of oxygen are also occurring in developing embryos and available data suggest that cells can respond to the hypoxic microenvironment during embryogenesis through HIFs, then coordinating the development of the different structures, organs, and tissues (including blood, vasculature, nervous system, organs, placenta, etc.). Moreover, embryonic stem cells as well as progenitor cells are believed to be located in hypoxic niches, with low pO2 levels being suggested as critical for the regulation of their differentiation and fate. Surprisingly, hypoxia-related EMT involvement in embryogenesis is still essentially unexplored and the few available data are then to be referred to conditions of chronic injury and organ fibrosis as well as of cancer progression and metastasis.
The relationships between hypoxia, fibrogenesis, and fibrotic progression of diseases under conditions of chronic injury are well established as the following messages from CKDs and CLDs can recall (71, 99, and ref. therein): a) areas of hypoxia are present in both clinical conditions and experimental models of CKDs and CLDs; b) hypoxia is of course considered as a major inducer of angiogenesis (52, 215, 231) and, in turn, angiogenesis is currently seen, at least for CLDs, as a major event favoring fibrogenesis and fibrotic progression of the disease, unrespective of etiology (71); c) hypoxia, through HIF and/or proangiogenic cytokines as well as through cross-talk with TGFβ is likely to concur in regulating ECM turnover and synthesis of collagen in ECM-producing cells (71, 99), migration of MFs (71, 180), as well as in sustaining inflammatory response (71, 99). However, at present the most pertinent and convincing results directly relating hypoxia, EMT and fibrosis are those published on CKDs (98, 99, 122): a) Hypoxia alone is able to induce EMT in primary renal epithelial cells in a HIF-1α–dependent manner; lysyl oxidases isoforms (LOX and LOXL2) have been suggested as critical mediators of hypoxia-induced EMT, likely by functional interacting with transcriptional repressors that regulate epithelial cell plasticity, although HIF-1α-dependent upregulation of the Notch target gene HEY1 in the same cells may also play a role. b) Conditional genetic inactivation of HIF-1α in epithelial cells of proximal tubules of murine kidneys subjected to unilateral ureteral obstruction revealed that hypoxia, through HIF-1α, was able to induce migration of epithelial cells through upregulation of lysyl oxidase genes. c) Genetic ablation of HIF-1α also resulted in a significant prevention of tubulo-interstitial fibrosis, as shown by decreased deposition of fibrillar collagen, decreased infiltration of inflammatory cells, and decreased number of FSP-1 positive cells in the injured murine kidney. d) Prominent expression of HIF-1α was described as associated to tubulo-interstitial injury in 13 of 21 biopsies from patients affected by diabetic nephropathy, as well as in biopsies from patients affected by IgA nephropathy; moreover, microarray analysis of the same human material disclosed upregulation of HIF-1α-dependent genes, including LOXL2 and CXCR4.
Where the relationships between hypoxia, EMT and cancer are concerned, it is well known that hypoxia is a major and common feature of almost any clinically relevant human solid malignant tumor, including cancer of the breast, prostate, lung, pancreas, rectum, brain, uterine cervix, melanomas, liver, soft tissue sarcomas, and renal cell cancer, with hypoxic or anoxic areas being usually heterogeneously distributed within neoplastic mass (30, 52, 215, 231, 264). Moreover, a significant decrease in oxygen tension is currently believed to provide a strong selective pressure able to positively modulate the growth of tumor cells as well as to eventually favor survival of the most aggressive malignant cells (30, 52, 215, 231, 264). Clinical and experimental evidence also indicate that neoplastic cells surviving to hypoxia can exhibit enhanced invasive propensity, suggesting that hypoxia may favor cancer progression and indeed, the detection of hypoxic areas within a neoplastic mass is considered as an independent prognostic indicator of poor outcome with a significant risk to develop metastasis that may escape therapy (30, 52, 215, 231, 264). Moreover, hypoxia should be considered as an unstable condition within a tumor mass and is likely to fluctuate in the tumor mass (the concept of cycling hypoxia, reviewed in ref. 52), leading to cycles of hypoxia/reoxygenation, increased generation of ROS and in the end to irregular and unsatisfactory response to radio- and chemotherapy. At present, different laboratories have suggested that hypoxia, as an independent condition, is able to induce in cancer cells of epithelial origin the process of EMT with its major features (including E-cadherin downregulation, upregulation of mesenchymal markers, nuclear translocation of SNAI1 and modulation of other transcription factors, increased migration and invasiveness) (33, 84, 100, 107, 141, 207, 223, 283) and a number of major EMT-triggering mechanisms have been suggested to play a role.
In breast cancer cells, hypoxia-induced EMT was accompanied by increased expression of the urokinase-type plasminogen activator receptor (uPAR), activation of signaling factors downstream of uPAR, including Akt and Rac1 and phosphorylation of GSK3β; hypoxia-induced EMT was reported to be blocked by uPAR gene silencing and mimicked by uPAR overexpression in normoxia. Moreover, procedures able to antagonize Rac1 or PI-3K also inhibit development of EMT associated feature under hypoxia (141).
As already detailed in a previous section, in a subsequent study (33) hypoxia, through an early switch requiring mitochondrial generation of ROS and a later HIF-1α and VEGF-dependent step, was found to stimulate EMT and increased invasiveness in several human cancer cell lines by determining PI-3K- and ERK-mediated phosphorylation/inactivation of GSK-3β, thereby allowing β-catenin and SNAI1 to translocate into the nucleus. In that study early events were prevented by blocking ROS generation whereas increased invasiveness was abolished by using siRNAs against HIF-1α and leading to VEGF downregulation.
It should be noted that hypoxia has been shown to upregulate not only HIF-1α but also several other factors related to the EMT process, including HGF, NF-κB, Notch signaling, Twist1, and DNA hypomethylation, as recently reviewed (84). Moreover, hypoxia-induced EMT led to increased motility, and invasiveness was abrogated by inhibition of Notch signaling and, conversely, mimicked by an activated form of Notch (223). Authors reported that Notch operated through two distinct mechanisms that acted in synergy to control the expression of SNAI1. Notch could either directly upregulate Snail-1 expression by recruitment of the Notch intracellular domain to the SNAI1 promoter or potentiate HIF-1α recruitment to LOX promoter, resulting in upregulation of LOX which, in turn, may stabilize the SNAI1 protein.
A final message is linking this section on the relationships between hypoxia, EMT, and cancer to a previously introduced concept (see section IV.D): EMT, based on available literature (15, 168, 198, 207, 283) may favor/induce the generation of cancer cells expressing stem cell features (i.e., cancer stem cells or CSCs). Indeed, increasing evidence (100, 234, and ref. therein) is suggesting that hypoxia, present in crucial areas defined as hypoxic niches, not only promotes the persistence of stem cells during embryogenesis and in the adult but may also concur to maintain the stem cell phenotype. As recently reviewed by Hill and coworkers (100), literature already suggests that (cancer) stem cell-related features may be sustained by either HIF-1α and HIF-2α-dependent transcriptional programs, with HIF-1α leading to Notch-1 activation as well as increased expression of CXCR4, MMPs, uPAR, and VEGFs genes (i.e., potentially relevant for motility, invasion, and metastasis) and HIF-2α being primarily involved in induction of genes related to embryonic staminality (like Octamer 4 or Oct-4) or proliferation (like c-myc). Whether this hypothesis is correct, remains, at present, an open question that will require further experimental and clinical experimentation.
F. Endothelial to mesenchymal transition: A peculiar form of EMT involved in pathophysiology
Endothelial to mesenchymal transition (EndMT) is a process that is usually classified as a peculiar form of EMT in which resident endothelial cells undergo a number of coordinated changes in morphology and behavior in order to abandon the organized endothelium and to acquire a mesenchymal-like phenotype. In order to offer a brief synopsis of the similarity of the two processes, the following major features can be recalled (208, 250, 293, 294, 295, and references therein): a) EndMT, similarly to what previously described for epithelial cells undergoing EMT, is characterized by loss of cell–cell contacts and junctions as well as of classic endothelial markers (CD31; vascular endothelial cadherin or VE-cadherin; receptors for pro-angiogenic cytokines like VEGF-R2, Tie1, and Tie2); the process then results in the progressive acquisition of typical mesenchymal markers (FSP-1, αSMA, fibronectin, the ability to synthetize components of ECM like collagen type I and III, etc) and of migratory and invasive properties. b) EndMT was originally proposed as a process mainly involved in development, with a major role ascertained for heart formation during embryogenesis, with endothelial cells of the primitive heart tube being able to invade the surrounding tissue, eventually leading to formation of valves and septa of the adult heart. However, as already established for EMT occurring in the adult, recent evidence is indicating that postnatal EndMT is likely to be involved also in different pathological conditions, once again, as detailed later, including organ fibrosis, wound healing and cancer progression. c) The large majority of data concerning EndMT come indeed from embryological studies that delineated a significant overlapping between EndMT and EMT also in terms of signaling pathways, with EndMT being triggered and regulated by signals like TGFβ, BMPs, and by Notch signaling pathway; additional signals involved in physiological EndMT have been reported, including wnt/β–catenin pathway and VEGF. However, apart from some evidence suggesting the involvement of SNAI1 and SNAI2 as repressor of VE-cadherin expression, most of the downstream transcriptional mechanisms mediating EndMT are still largely unknown; d) Where organ fibrosis and/or the response to either acute or chronic injury are concerned, expression of markers of EndMT has been shown particularly in cardiac fibrosis (295), where approx. one-third of fibroblasts in fibrotic heart were reported to be generated through EndMT; similarly to what was reported for EMT in CKDs, TGFβ and SMAD3-dependent signaling is likely to play a major role, as also confirmed by reduction of both EndMT and heart fibrosis obtained in experimental models by treating mice with BMP7. Kidney fibrosis is another example of chronic injury in which EndMT is likely contribute to fibrosis by originating (depending on the animal model of CKD employed) from 30% to 50% of fibroblasts/myofibroblast (293). EndMT has been also described in few other pathological settings, including atherosclerosis, chronic pulmonary hypertension, and wound healing. e) EndMT has been also involved in cancer progression in a study in which the use of Tie2-Cre;R26R-lox-STOP-lox-lacZ strain of transgenic mice (a strain that should trace only endothelial cell-derived lineages), has suggested that a consistent number of so-called cancer associated fibroblasts or CAFs (from 30% to 50%, depending on the animal model of experimental cancer), particularly at the invasive front of experimental tumors, may originate from EndMT that, in turn, may represent also a mechanism for their recruitment (294). In addition, the same authors also showed that TGFβ is an effective stimulus for the induction of EndMT in mouse endothelial cells. f) The same study (294) also provided evidence that EndMT may occur from endothelial cells belonging to neovessels (i.e., angiogenic vessels). Recently, the same research group has proposed that EndMT may enable the so-called tip cells that are the migrating endothelial-derived cells driving angiogenesis (i.e., cells that differ from proliferating stalk cells in neovessels) to migrate into the surrounding tissue; the suggestion is based on the peculiar phenotype of tip cells that is at least compatible with the one of cells that have undergone EndMT (208). The authors also suggested that EndMT may also play an additional role in angiogenesis by contributing to recruit pericytes for neovessel stabilization.
As a final comment and note of caution in interpretation of results that may apply particularly to the EndMT-related results reported by using Tie2-Cre;R26R-lox-STOP-lox-lacZ strain of transgenic mice for cancer studies (294) and the hypothesis suggesting a role of EndMT in tumor angiogenesis, one should remember that Tie2 can not be considered as an antigen restricted only to endothelial cells. As reviewed in Ref. 142, Tie2 is also expressed by a hematopoietic lineage of pro-angiogenic monocytes defined as Tie2-expressing monocytes or TEMs. Considering that bone marrow-derived cells are known to significantly contribute to tumor angiogenesis, these TEMs have been reported to be selectively recruited to spontaneous and orthotopic tumors and to account for most of the proangiogenic activity of myeloid cells in tumors. Not surprisingly, hypoxia has been found to represent a major stimulus for Tie-2 expression in these TEMs and, together with angiopoietin-2, has been reported to downregulate the antitumor activity of these cells. Moreover, genetic knockdown of TEMs has been described to dramatically prevent angiogenesis in mouse tumors and to induce significant tumor regression.
V. Final Comments
Epithelial to mesenchymal transition (EMT) has emerged in recent years as a fundamental biological process, paradigmatic of the concept of cell plasticity that leads epithelial cells to lose their polarization and specialized junctional structures, to undergo cytoskeleton reorganization, and to acquire morphological and functional features of mesenchymal-like cells. As described in the present review, recent literature has disclosed that EMT is not only involved in embryonic development but also in other physiological and pathological conditions, with a proposed role in sustaining organ fibrosis as well as, even more relevant, in affecting invasive and metastatic behavior of cancer cells, thereby modulating cancer progression. In order to conclude the present review we would like to focus the attention on the following points: a) Experimental work performed in vitro, due to the simplified context and the chance to carefully control conditions, has been instrumental in the identification of pathways and mechanisms involved in the induction and regulation/modulation of EMT. However, as elegantly reviewed by Thiery and Sleeman (250), in vitro studies have limitations, including the relatively few number of immortalized cell lines that can undergo full EMT, the fact that most available epithelial cell lines are not fully polarized and may lack tight junctions as well as the intrinsic, cell type- and context-dependent variability in the kinetics of EMT that may stand from few hours to several days. b) In vivo occurrence of EMT has been shown quite convincingly in defined animal models of disease, but properly designed corresponding human studies are still limited in number. Moreover, due to technical and ethical limitations, the correct identification of the “signature of EMT” is still a major concern; indeed, the reader should consider that EMT is a transient process that can be then temporally missed by the analysis, and that the relevance of EMT in a disease is likely to change significantly depending on the etiology, the context-dependent signals and stimuli able to trigger or modulate the process as well as differentiation events. c) Although several laboratories suggest the general relevance of EMT for fibrogenesis, relevant differences in cell lineages and tissue-specific conditions exists that are likely to significantly affect the overall relevance of EMT, as is the case for CLDs where the number of FSP-1 cells is much lower of that of α-SMA positive MFs and the prominent pro-fibrogenic role of HSC/MFs is widely accepted. d) An objective view of the role of EMT in cancer progression and metastasis suggests that EMT is likely to be just one of the mechanisms or steps required by malignant epithelial cancer cells to establish productive expansion via increased invasiveness and local intravasation (i.e., in the tumor vasculature). e) At present, the real role of ROS in mediating and/or modulating EMT is likely to be largely underscored and may emerge as very relevant under hypoxic conditions, particularly if one considers that significant fluctuation of pO2 values in organ fibrosis and in the mass of malignant tumors is a very common event. f) Technical improvements (from more powerful imaging techniques to specifically designed array analysis and much more) may favor an increasing comprehension of signaling pathways and mechanisms involved in EMT, potentially leading to the generation of new therapeutic approaches to counteract fibrosis and metastasis to be translated to clinical conditions.
Footnotes
Acknowledgments
The authors acknowledge financial support from the Italian Ministero dell'Università e della Ricerca (MIUR, Rome - PRIN Project 2006067527), the Regione Piemonte (Torino), the Fondazione CRT (Torino), and the Fondazione Bossolasco (Torino).
Abbreviations Used
Reviewing Editors: Philip Gregory, Fazlul H. Sarkar, Pierre Savagner, Hideyuki Saya, Guojun Sheng, Jian–Guo Song, and Jiri Zavadil