
Abstract
The mitochondrion is the most important organelle in determining continued cell survival and cell death. Mitochondrial dysfunction leads to many human maladies, including cardiovascular diseases, neurodegenerative disease, and cancer. These mitochondria-related pathologies range from early infancy to senescence. The central premise of this review is that if mitochondrial abnormalities contribute to the pathological state, alleviating the mitochondrial dysfunction would contribute to attenuating the severity or progression of the disease. Therefore, this review will examine the role of mitochondria in the etiology and progression of several diseases and explore potential therapeutic benefits of targeting mitochondria in mitigating the disease processes. Indeed, recent advances in mitochondrial biology have led to selective targeting of drugs designed to modulate and manipulate mitochondrial function and genomics for therapeutic benefit. These approaches to treat mitochondrial dysfunction rationally could lead to selective protection of cells in different tissues and various disease states. However, most of these approaches are in their infancy. Antioxid. Redox Signal. 13, 279–347.
Mitochondrial ROS Scavenging and Its Potential Therapeutic Value
Mitochondrial DNA-Related Pathologies and a Potential Therapeutic Target
Mitochondrial Interaction with other Organelles: Therapeutic Implications
Mitochondria-Related Diseases and Cell Injury
Caveats and Potential Limitations in Mitochondrial Drug Targeting
I. Introduction and Topics Reviewed
A thorough understanding of mitochondrial function in normal and pathological states is critical in developing the full therapeutic potential of the organelle in mitigating or preventing a given disease. Mitochondrial-related diseases are vastly different and much of the science linking mitochondria to different disease states is still being intensively studied. The central premise of this review simply is that if mitochondrial abnormalities contribute to a pathological state (directly or indirectly), then alleviating the mitochondrial dysfunction should attenuate the severity or progression of the disease. Hence, the main objective of this review is to present the concept that mitochondria of varying cell types can be potentially targeted for therapeutic intervention in mitochondria-associated diseases. That is, the review will focus on how the mitochondrion has become a potential therapeutic target in disease management.
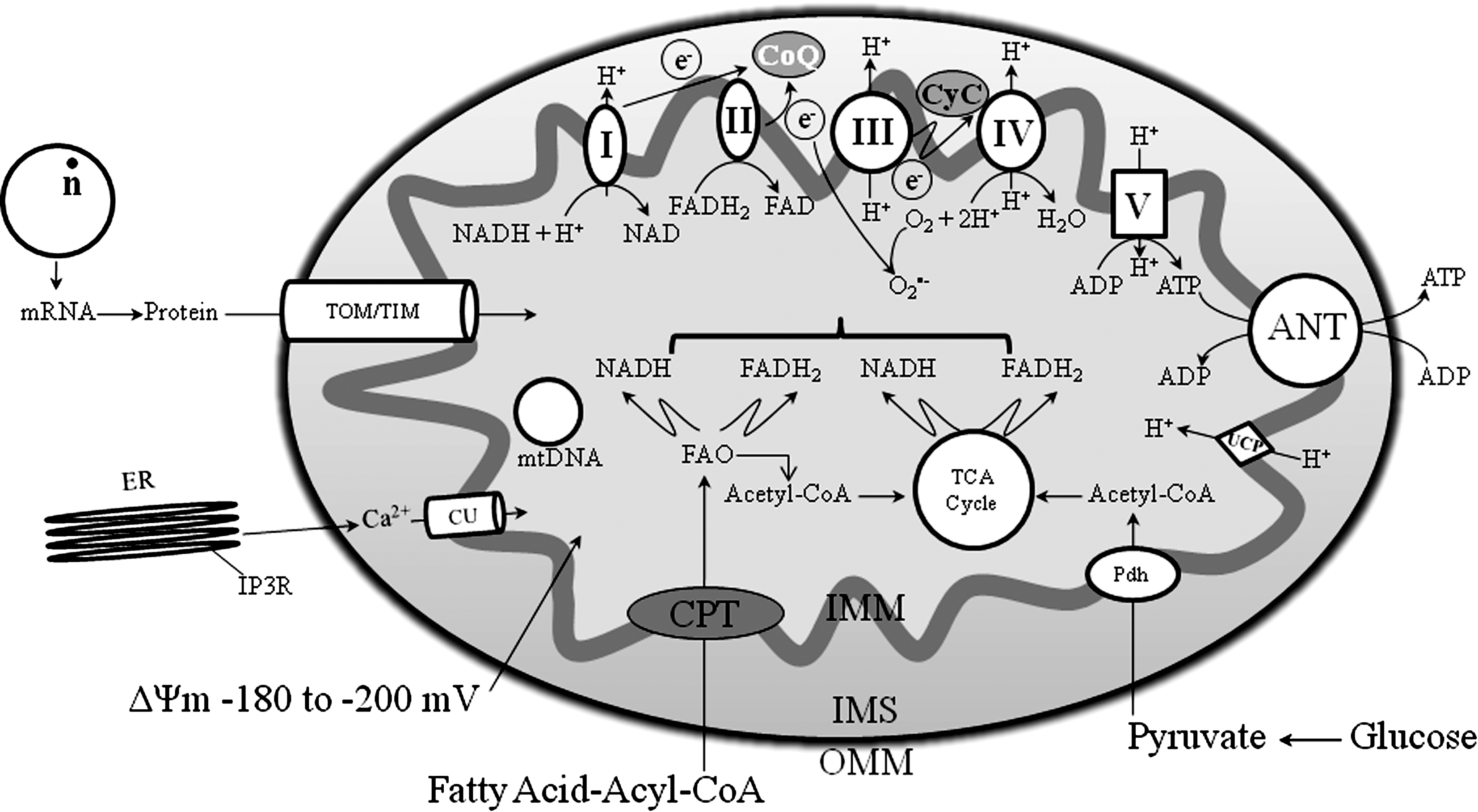
Sections II and III describe the primary function of mitochondria, ATP synthesis through oxidative phosphorylation (OXPHOS), and how this energy production is carried out on a folded inner mitochondrial membrane (IMM) with protein complexes that transfer electrons from one protein complex to another at different redox potentials. Figure 1 provides a simple scheme of mitochondrial function. Figures 2 and 3 illustrate the interactions of some IMM and outer mitochondrial membrane (OMM) proteins important in the regulation of cell survival and cell death as a consequence of oxidative stress, how regulation of transmembrane ion fluxes maintains cell homeostasis, and how perturbing this anatomical and functional link can lead to pathological conditions.


Section III explains one of the side effects of electron transport, generation of O2 •− and its products, and Section IV summarizes the roles of ROS and reactive nitrogen species (RNS) as both regulators of cell function and cell death. Section V describes the elaborate free radical scavenging system that regulates ROS within physiologically acceptable values and the critical importance of balance in the production and scavenging of O2 •− in normal cellular physiology. These sections summarize how mitochondria modulate bioenergetics and serve as an effector of cell viability and how an imbalance in the rate of ROS production and the rate of ROS scavenging lead to oxidative stress, a marked contributor of mitochondrial mediated pathology. Section VI discusses how uncoupling proteins and drugs enhance mitochondrial respiration, but not ATP synthesis, as a way to paradoxically protect the organelle and cell.
Mitochondrial dysfunction and dysregulation may have their genesis in mtDNA mutations (e.g., colon and prostate cancer) and/or impairment or reversal of mitochondrial electron transport chain (ETC). Section VII notes that many human diseases are linked to mtDNA mutations and mitochondrial dysfunction and describes how mitochondria have emerged as central foci in the investigation of the etiology of numerous cardiovascular, metabolic, neurological diseases, cancer, psychiatric disorders, and migraine. Mitochondrial membrane integrity and mitochondrial functional and morphological connectivity with each other and with other organelles, for example, the nucleus or the endoplasmic reticulum (ER) are critical in maintaining cellular integrity and they also provide continuity in cellular function. Most mitochondrial proteins are encoded by the nuclear genome and complexes are encoded by both mitochondrial and nuclear genomes. Consequently, any defects in the production of these proteins could induce mitochondrial cytopathies that underlie a multitude of diseases or pathological conditions. Thus Section VIII explores the interaction of mitochondria with themselves (the mitochondrial reticular network) and with the nucleus and ER. In this section it is discussed how mutations in the genes for nuclear-encoded mitochondrial proteins, the so-called nuclear–mitochondrial crosstalk, are implicated in a number of tissue degenerative disorders. Section IX summarizes how mitochondrial dysfunction underlies a number of diseases including cardiac ischemia and heart failure, diabetes, hypertension, as well as neurologic and neoplastic diseases and other lesser-known mitochondria-related diseases. For example, myocardial ischemia causes damage to the mitochondrial distal ETC that could be an important link between ischemia and the mitochondrial-induced myocyte damage that occurs on reperfusion. Section X explores known strategies for delivering drugs to the mitochondrion and discusses some mitochondria-targeted procedures and drugs that appear useful in treating some disease states, especially cardiac ischemia and reperfusion (I/R) injury. Other mitochondrial therapeutic approaches are presented in Section XI. Examples are lipid replacement therapy (LRT), transactivator of transcription (TAT) protein delivery, novel genetic approaches and the potential benefits of caloric restriction, and nutritional supplements. Section XII explores the role of mitochondria in the aging process and the role of the mitochondrial adapter protein, p66shc in lifespan determination. Finally, Sections XIII and XIV bring up the shortcomings and limitations of mitochondria-targeted drug delivery and the authors' conclusions and perspectives.
II. Anatomy and Function of Mitochondrial Membranes
A. Outer mitochondrial membrane and its potential role as a therapeutic target
The elaborate structure of a mitochondrion is important for the normal functioning of the organelle and therefore as a potential therapeutic target. Two specialized membranes encircle each mitochondrion, dividing the organelle into a narrow intermembrane space (IMS) bordered by the OMM and the inner IMM (Figs. 1 and 2). The OMM contains many channels formed by the protein porin that makes the membrane relatively permeable. One of the membrane proteins is the peripheral benzodiazepine receptor (PBR). PBR is a small evolutionarily conserved protein involved in cholesterol transport and steroid synthesis; it is also a regulator of apoptosis (177, 194, 217, 412). The PBR is also involved in OMM permeabilization by interaction with the pro-apoptotic Bcl family of proteins. However, OMM permeability may be independent of mitochondrial permeability transition pore (mPTP) opening because blocking PBR with 4′-chlorodiazepam (CDZ) protects against ischemia-induced cytochrome c release independent of damage to the IMM (74, 194, 411); CDZ also reduces ischemia-induced arrhythmias (74). PBR is found in close association with the voltage dependent anion channel (VDAC) and additional components of the mitochondrial contact site (194,412) as shown in Figure 2. This close association also suggests that PBR-VDAC may serve as a target for modulating apoptosis and may have implications for drug design to treat such disorders as cancer and neurodegenerative diseases (194, 412).
During the activation of cell death programs, permeation of the OMM occurs through the unopposed activation of effector proteins Bcl-2-associated X protein (Bax) and Bcl-2 homologous antagonist/killer (Bak) (3). These proteins are located in the cytosol but oligomerize and translocate to the OMM as a consequence of oxidative stress. They are activated by interaction with activator peptides including truncated-bid (t-bid) or Bim. The activator proteins are initially sequestered and inhibited by the anti-apoptotic proteins Bcl-2 and Bcl-Xl (3, 229, 311, 620). Bcl-2 and Bcl-Xl also interact with sensitizer BH3-only domain peptides including Bcl-2-associated death proteins (Bad), Bcl-2-interacting killer (Bik) protein, and perhaps Bcl-2/adenovirus E1B 19 kd-interacting protein (Bnip). These peptides preferentially interact with and sequester B-cell lymphoma (Bcl-2) and Bcl-2 X protein (Bcl-Xl), tilting the balance toward unopposed action of the activator peptides (229). When activated, Bak and Bax homo-oligomerize at the OMM and promote the release of apoptotic factors cytochrome c, AIF, HtrA22/Omi and other factors (Section II.C). The ‘BH3 only’ promotes the oligomerization of Bax and Bak to the OMM. The anti-apoptotic proteins Bcl-2 and Bcl-XL, located on the OMM, antagonize these effects. It is understood that the role of Bcl-2 and Bcl-Xl in inhibiting OMM permeation to cytochrome c is focused on direct interactions of Bcl-2 and Bcl-Xl with effectors Bax and Bak.
The VDAC is a mitochondrial protein synthesized by the nuclear genome. It is considered the principal site for exchange of metabolites between the IMS and the cytosol. VDAC has a large hydrophilic pore capable of translocating ions and a variety of metabolites such as ATP and ADP (420). As a major gateway in and out of the mitochondrion, VDAC mediates an intimate dichotomy between metabolism and death in all cells (583). The mitochondrial mediated cell death involves an array of OMM and cytosolic proteins that include the hexokinases (I and II) and the Bcl family of proteins that alternatively promote or prevent cell injury (78, 443, 444, 583).
Accumulating evidence has shown that hexokinases (HK) (Fig. 2) play a crucial role in promoting cell survival, when needed, and to instigate its removal if cell death is required. HK I, and II in particular, mediate cytoprotection by binding specifically to the VDAC (31, 195, 583), in part via the hydrophobic N terminus specific residues of the VDAC in the presence of Mg2+ (583). It is postulated that Mg2+ facilitates binding in part by charge shielding or specific bridging effects to facilitate the apposition of negatively charged amino acids on the enzymes and the phospholipids of the OMM (443). HK III and IV lack the terminal hydrophobic region, thus they cannot bind to the mitochondria (457). Like other cytoprotective maneuvers, HK is translocated from the cytosol to the OMM (74, 78, 194, 411, 414, 443, 444, 583) and the interaction with VDAC is believed to prevent mPTP formation. Indeed, in malignant tumor cells, unlike normal cells, the enhanced association between HK and VDAC provides extra protection against permeabilization of the OMM and resistance to apoptosis (195). Recent evidence shows that the interaction between HKs and VDAC is regulated by many factors, including protein kinases (443). However, it is worth noting that the binding to the VDAC is not a prerequisite for HK binding to OMM, because the enzyme binds to OMM in yeasts, which are known to be deficient in VDACs.
In recent preliminary studies, Cheng et al. (119) showed that HKs could also modulate VDAC activity via protein kinase-mediated interaction. Specifically, this study showed that human VDACs incorporated into lipid bilayers are phosphorylated under basal conditions because treatment with phosphatases increases channel conductance, and HK binding to the channel decreases conductance of the channel. The HK–VDAC complex significantly decreased VDAC conductance, and this effect was reversed by addition of phosphatase. Thus, it was concluded from these observations that at the functional level basal phosphorylation of cardiac VDAC may be required for modulation by HK. This observation is consistent with the notion that HK promotes VDAC closure. In a somewhat related study from Ardehali's group (549), a hypothesis was tested that HK overexpression increases VDAC phosphorylation and that this effect may autoregulate the binding of these proteins to mitochondria. They showed that full length HKs (I and II) expression resulted in a significant increase in VDAC phosphorylation and since HKs do not directly phosphorylate proteins, they proposed that the increased phosphorylation was via PKCɛ. Akt, an oncogenic protein kinase activated by PI3K, mediates HK binding to VDAC by affecting the phosphorylation state of VDAC and/or hexokinase. Indeed, Akt can directly phosphorylate HKII and this has been associated with protection against I/R injury (381, 443). Pastorino et al. (443) showed that GSK3-β, a kinase inhibited by Akt and located in mitochondria and nuclei, can phosphorylate VDAC, and that this phosphorylation affects HKII binding to mitochondria. Activation of GSK3-β has been linked to phosphorylation of Bax, which upon translocation to mitochondria, makes the cell more susceptible to apoptosis. In addition, a disruption of the HK–VDAC interaction, as in the phosphorylation of the HK docking site to VDAC by GSK3-β, facilitates the induction of OMM permeabilization and subsequent apoptosis (120, 195, 447).
Insofar as the GSK3-β is implicated in apoptosis, inhibiting the kinase can be cytoprotective. Yazlovitskaya et al. (637) showed that inhibition of GSK3-β with LiCl prevented radiation-induced damage to cultured hippocampal neurons. However, in some circumstances, GSK3-β can exert an anti-apoptotic effect, as evidenced by recent observation that certain GSK3-β inhibitors are able to induce apoptosis in tumor cells through a p53-dependent mechanism (210). Furthermore, GSK3-β expression in melanoma cell line has been shown to protect against the apoptotic effect of the chemotherapeutic agent sorafenib by increasing the basal levels of the Bcl-2 proteins. Thus there is a strong rationale for the use of GSK3-β inhibitors (e.g., GSK3-IX) as adjuncts in the treatment of cancer (437).
HK binding promotes oligomerization of the VDAC (443) and impedes cytochrome c release (2, 523). Neoplastic cells also resist death in part by increasing the interaction between mitochondria and HK; this could be prevented, as a therapeutic approach, by adding 3-bromopyruvate (3BrPA), an inhibitor of HK to VDAC, in order to kill a hepatoma cell line characterized by overexpression of HKI and HKII (447). Transfection of leukemia-derived U-937 cells with HK significantly reduced staurosporine-induced apoptosis when compared to GFP transfected cells (31). Taken together, these studies suggest that interference of the binding of HK to mitochondria by VDAC-derived peptides (2) and peptide targeting of the N-terminal of the HK protein (120, 443) may offer a novel strategy to potentiate the efficacy of other modes of conventional chemotherapy (2). However, recent evidence also suggests that HK-mediated protection against apoptotic signals can occur independently of VDAC (443). For instance, HK is known to prevent apoptosis by interfering with Bax binding to mitochondria to induce cytochrome c release (644). Thus, in normal cells, preservation of the OMM with HKs is accompanied by increased retention of cytochrome c and improved electron transfer and more effective OXPHOS.
These studies demonstrate that cellular injury could be ascribed to increased permeability of the OMM and that limiting the permeability of the OMM will protect against cell damage and cell death due to oxidative stress. Indeed, HKII detachment from the OMM with clotrimazole, or with designed peptide fragments that target the N-terminal (amino acid sequences) of the HKII domain for VDAC (120), leads to cell death. Therefore, a mitochondria-targeted therapy designed for the OMM as a potential therapeutic maneuver is highly relevant and is the subject of intense research. A better understanding of the interaction between the cytosolic proteins and OMM will greatly optimize therapies for treating ischemic heart disease, neurodegenerative diseases, and cancer. These different mitochondrial related diseases and the potential targeting of the organelle as a mitigating factor will be discussed in much detail in the following sections.
B. Inner mitochondrial membrane and its potential role as therapeutic target
The IMM, relative to the OMM, is highly impermeant and allows only certain small molecules to pass through. It is convoluted into a large number of infoldings called cristae. Cation permeation is regulated by ion channels and exchangers whose functions are governed by a high IMM potential (ΔΨm) (Fig. 3). The transmembrane cation fluxes through specialized cation transporters and exchangers are essential for mitochondrial bioenergetics (53, 379). The specialized coupling of OXPHOS requires a low permeability of the IMM not only to protons but also to other cations (53, 201). Mitochondrial cation anti-porters/exchangers (proton-linked) regulate any osmotic differential across the IMM that would result from the high proton motive force (ΔμH+). The chemiosmotic hypothesis of energy conservation indeed requires the presence of electroneutral cation anti-porters, for example, the Na+/H+ exchanger (NHE) and the K+/H+ exchanger (KHE), as well as a low permeability to the cations K+ and Na+ (Fig. 3). The requirement of low cationic permeability and a strong H+ electrochemical gradient ΔμH (provided by the substrates and stored in the Δψm and pH gradients) along with cation exchangers to prevent any osmotic overload, might seem to obviate the need for specific cation channels/uniporters. However, it is now evident that there are or need to be mitochondrial channels for K+, Ca2+, and perhaps Na+ (53). These channels are likely to modulate ΔμH.
Mitochondrial Ca2+ (mCa2+) uptake through the Ca2+ uniporter (CaU) is mainly dependent on Δψm and the Ca2+ gradient between the cytosol and the matrix. This uptake of Ca2+ into mitochondria helps to buffer cytosolic Ca2+, bringing it to levels where the ER can handle it. Mitochondrial Ca2+ loading may have profound consequences for mitochondrial function such as regulating cellular respiration and mediating cell death by apoptosis or necrosis. A small increase in mCa2+ during increased workload is thought to be necessary for activity of TCA cycle enzymes to furnish the reducing equivalents necessary to match energy demand with supply. The buffering capacity of the matrix proteins, adenine nucleotides, and phosphates modulate mCa2+ to maintain a physiologically relevant free Ca2+ (249). However, high m[Ca2+], as observed during cardiac I/R, can impair ATP synthesis and lead to a loss of ion homeostasis, opening of the mPTP, matrix swelling, and OMM rupture (277, 569). This irreversible mPTP opening is associated with collapse of ΔΨm, release of cytochrome c and perhaps more ROS production (Section III), resulting in the vicious cycle of further amplification of ROS production, mCa2+ overload, and increasing irreversible cell damage (71, 391).
The electrophoretic Ca2+ uptake through the CaU is matched by Ca2+ extrusion primarily via the Na+-dependent Na+/Ca2+ exchanger (NCE) (71, 96, 139, 411) and via a putative Na+-independent Ca2+ efflux mechanism (NICE), [e.g., a Ca2+/H+ exchanger (CHE)] (226, 621). The Ca2+ efflux can also be regulated by the mPTP, which is insensitive to ruthenium red (RuR) (53, 177, 227). Transient opening of the pore, perhaps in a low conductance state, will result in Ca2+ efflux without significant depolarization. This is only possible if the pore opening is brief so that the transient depolarization can recover.
Attenuation of mCa2+ overload and the subsequent reduction in the sensitivity of the mPTP opening can be accomplished in part by inhibiting NHE or the CaU. Indeed, studies have shown that NHE inhibition and Ca2+channel blockers preserve tissue ATP and creatine phosphate levels during cardiac I/R injury (279, 322), in part by improving mitochondrial state 3 respiration (270). It was concluded from this study that inhibition of NHE might be mediated in part via mitochondria to prevent Ca2+ overload, which could mitigate mPTP opening and reduce cell injury (270). However, these effects of NHE inhibition could also be attributed to delayed recovery of intracellular pH, which inhibits mPTP opening (270).
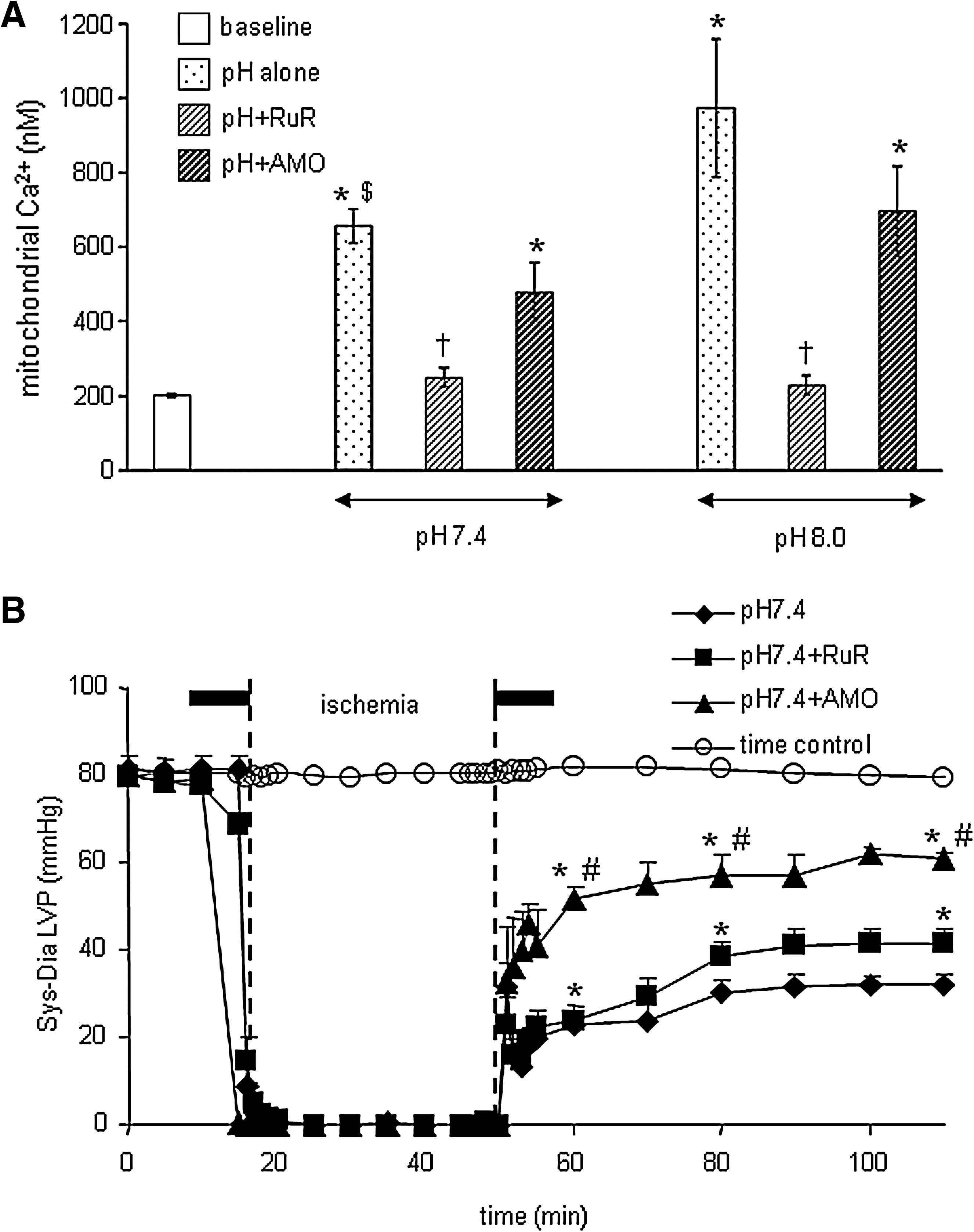
In a recent study we reported that activation or inhibition of NHE might impact mitochondrial bioenergetics directly as evidenced by changes in mitochondrial redox state, mCa2+ overload, and O2 •− production in isolated hearts. Oxidation of the mitochondrial redox state, increased O2 •− generation, and increased m[Ca2+] in hearts where NHE was activated were associated with compromised functional recovery. Blocking activation of NHE with a NHE inhibitor or by reperfusing with acidic buffer to reduce the pH gradient minimized the mitochondrial dysfunction (10). We suggested that the protection afforded by NHE inhibition is due to a direct impact on mitochondrial NHE, as well as on sarcolemmal NHE. Our interpretation was based on an observation that cariporide, an NHE-1 inhibitor, blocked mitochondrial matrix acidification and ATP depletion during simulated ischemia in cardiac myocytes (486). In the presence of the respiratory inhibitors oligomycin and KCN, inhibition of mitochondrial NHE increased mitochondrial acidification in permeabilized myocytes (228). Based on this scenario, a decrease in matrix pH and the concomitant depolarization of ΔΨm (228) should in turn reduce the driving force for mCa2+uptake and minimize mitochondrial damage. Furthermore, in preliminary studies we showed that RuR given in combination with perfusate buffer at pH 7.4 (Fig. 4B) and 8 (data not shown), 10 min before and after warm ischemia only improved cardiac function slightly, but with a marked reduction of mCa2+ (Fig. 4A). In the same study using a similar protocol, we showed that amobarbital, a complex I blocker, markedly reduced mCa2+ load and improved functional recovery (Figs. 4A and 4B) (11), consistent with another study (380).
The respiratory chain and the ion channel pumps are necessary to maintain the substantial electrical potential across the IMM (Fig. 3). This potential is about two times larger than the sarcolemmal membrane potential and therefore provides a unique chemical opportunity for selectively targeting the mitochondrion. This unique attribute of the mitochondrion, coupled with recognized peptide signal sequences following post-translational modification of nuclear encoded polypeptides, has been employed to direct the so-called “mitochondriotropic” drugs, where they accumulate in the matrix (71). For example, the cationic metalloporphyrin superoxide dismutase mimetic Mn (III) meso-tetrakis (N-ethylpyridium-2-yl) porphyrin (MnIII TE-2-Pyp5+) has been shown to accumulate in mitochondria derived from mice cardiac myocytes following systemic injection (534). Some of the mitochondrial antioxidants, for example, vitamin E and coenzyme Q, have been structurally modified to target the mitochondrion (218). Smith et al. (532) reported that complexing vitamin E with the triphenyl-phosphonium (TPP+) cation augmented mitochondrial uptake of the complex. Similarly, the scavenging capability and anti-apoptotic efficacy of ubiquinone was enhanced by complexing the protein with spin traps (286). The cell membrane permeable amphilite tempol (4-hydroxy-2,2,6,6, -tetramethylpiperidine) can be partitioned to the mitochondria (mito-tempol) by coupling it to TPP+ (619). Table 1 shows examples of mitochondria-targeted drugs or agents that are hitched to the carrier molecules that permeate the mitochondrion.
Mitochondria-targeted peptides could also be recognized by unique amino acid sequences that enable translocation of a peptide to a mitochondrion. However, other mitochondrial proteins translocate to the matrix without the targeting peptide sequences. These proteins interact with and bind to sites present on the OMM or IMM. For example, PKCɛ interacts with and phosphorylates its target proteins in the IMM by binding to sites/adapter proteins on the IMM (150, 417). This translocation of PKCɛ results in mitochondrial hyperpolarization and this may reduce depolarization of ΔΨm during ischemia and increase ATP synthesis on reperfusion, which in turn may increase the energy-dependent processes that are involved in establishing the ion gradient across the sarcolemma and mitochondrial membranes (150).
It is important that mitochondrial ion channels and exchangers are controlled in order to provide the balance between energy supply and demand that is crucial for normal cell function. Attempts to characterize the molecular structures of these channels remain elusive, however. Achieving this goal from a pharmacological standpoint could “spur the development of novel and specific therapeutic agents targeted to the mitochondria” (411).
C. Mitochondrial permeability transition pore
Crompton et al. (140, 141) were the first to demonstrate that the mPTP plays a crucial role in cardiac myocyte injury following oxidative stress. The mPTP, a large, nonspecific channel protein aggregate known to span the OMM and the IMM (Fig. 2), mediates the lethal permeability changes of the mitochondrial membranes leading to mitochondria-mediated death. It has been described as the rate-limiting step in the mitochondria-mediated cell death pathway (391). However, recent studies have implicated the pore as a “physiological valve”, which alleviates mCa2+ overload as a consequence of a brief surge of Ca2+ in a localized microdomain involving the ER (Section VIII,C) (530). This transient brief opening of the pore (flickering) has also been implicated in providing protection against cellular injury (301). This dual role of the mPTP in the survival and death of the cell is therefore critical in selective targeting of the pore for therapeutic interventions. Similarly, an understanding of the constituents of the pore and its molecular structure are paramount in this therapeutic goal.
A model structure of the mPTP put forward by Halestrap et al. (236, 237) and Di Lisa and Bernadi (161) involves the combination of the adenine nucleotide translocase (ANT) in the IMM, the VDAC of the OMM, and a regulatory protein cyclophilin D (CypD) in the matrix (Fig. 2). Other variants of the ANT, ANT2, which are overexpressed in cancer cells, help to stabilize mitochondrial membranes (195). Indeed, it was suggested that in cancer cells, small interfering RNA (siRNA) that down regulate ANT2 may constitute a valid strategy for the selective induction of tumor cell death (195, 269). Associated with the outer leaflet of the IMM, in the IMS, is the mitochondrial creatine kinase (CK). CK, under physiological conditions, is crucial in catalyzing the transphosphorylation of creatine by ATP to phosphocreatine and ADP. In apoptotic-induced cell death, mitochondrial CK may facilitate contact between the VDAC and ANT to form the pore at the contact site of the IMM and OMM (391) (Fig. 2). However, recent studies have demonstrated that the VDAC and ANT act more as regulatory proteins of the mPTP (37, 120).
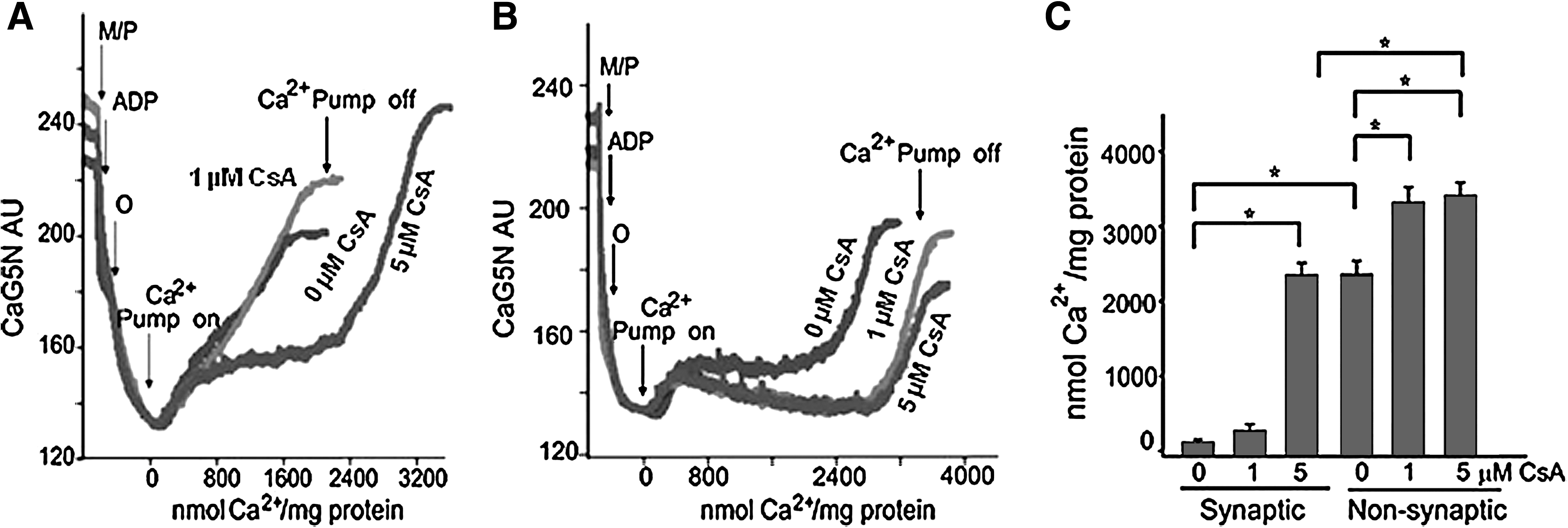
The physiological role of CypD is not known, but its pathological role as a component of the pore is widely accepted (37). CypD, a peptidylprolyl cis-trans isomerase, facilitates a conformational change in the ANT, converting it to an “open” pore (397). The role of CypD in the regulation of cell survival or death is evidenced by the finding that cells from mice lacking the Ppif gene that encodes the protein are protected from necrotic caspase-independent cell death but not from caspase-dependent apoptosis (36, 43, 297). In a related study Naga et al. (397) reported that synaptic mitochondria show greater vulnerability to Ca2+ overload when compared to nonsynaptic mitochondria. This differential sensitivity was attributed to higher levels of CypD in the synaptic mitochondria when compared to the nonsynaptic mitochondria. The differences in Ca2+ handling between the synaptic and nonsynaptic mitochondria were greatly reduced in CypD null mice, and a higher concentration of CsA was necessary to increase the Ca2+ retention capacity (Fig. 5) in the synaptic mitochondria (397). Interestingly, the levels of VDAC and ANT were not significantly different between synaptic versus nonsynaptic mitochondria (397). The authors postulated that the greater amount of CsA needed to block mPTP opening in the brain synaptic mitochondria, compared with the nonsynaptic mitochondria, has important implications with regard to the use of this compound and its derivatives as neuroprotective agents (397). Other OMM proteins, including anti-apoptotic proteins, are known to be associated with mPTP, but may mediate cell death independently of the megapore opening (71, 74, 194, 443, 550).
The mPTP allows passage of electrolytes and solutes and metabolites up to 1.5 KDa. In addition to excess mCa2+ load and ROS production, mPTP opening can also be promoted by ΔΨm depolarization, Pi, and thiol modification of specific mitochondrial proteins. Adenine nucleotides, Mg2+, and matrix H+ inhibit the pore (71, 177, 236, 237, 411). Pore opening causes dissipation of ΔΨm and is exemplified by equilibration of H+ across the IMM, which leads to inhibition of ATP production, further generation of ROS and ultimately to colloid osmotic swelling (514) and rupture of the OMM (514, 572, 573). Depletion of intracellular ATP in turn leads to derangement of ionic homeostasis and prolonged pore opening; this could lead to irreversible cellular damage and necrosis. Caspase-dependent apoptotic cell death on the other hand is dependent on residual ATP production from “stunned” mitochondria (573).
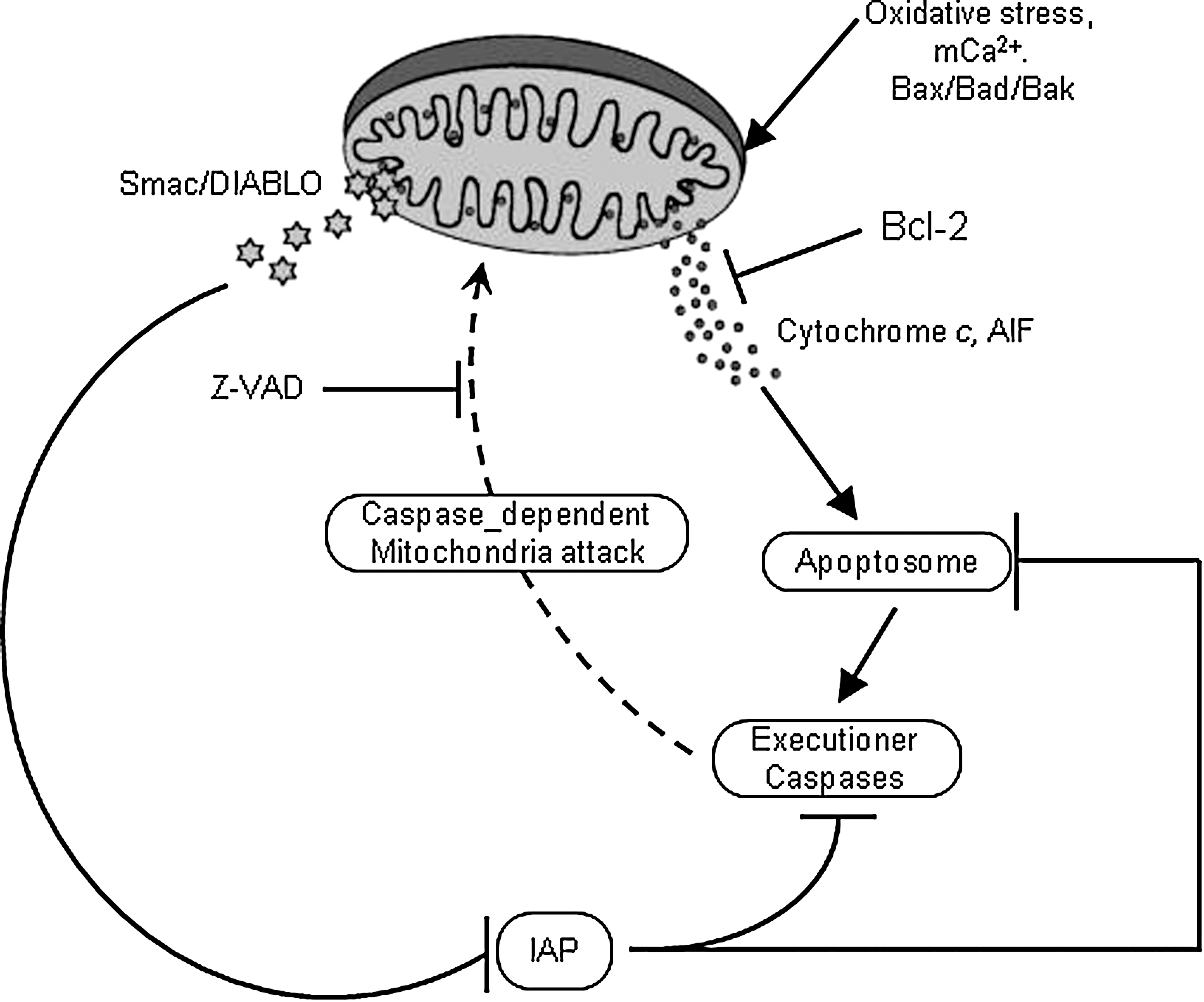
The most basic function of the mPTP is thought to be initiation of mitochondrial turnover in instances where individual mitochondria are dysfunctional because of accumulating mutations in mtDNA and oxidative damage to membranes and proteins. Moreover, the opening of the pore has been associated with numerous pathological conditions (e.g., stroke accompanied by brain ischemia). In this case, prolonged pore opening led to loss of mitochondrial proteins, most notably cytochrome c, second mitochondria-derived activator caspase/direct inhibitors of apoptosis protein (IAP)-binding protein (Smac/Diablo), apoptosis-inducing factor (AIF), endonuclease G (Endo G), and HtrA2/Omi (295, 363, 546). Once released into the cytosol, these mitochondrial proteins trigger both caspase-dependent (by cytochrome c, Smac/DIABLO, or HtrA2/Omi), and caspase-independent (by AIF, Endo G, or HtrA2/Omi) apoptosis (110, 268).
AIF is a phylogenetically old protein confined in the IMS in the healthy cell. Upon lethal signaling, AIF is released to the cytosol and is then translocated to the nucleus where it binds to DNA to instigate caspase-independent chromatin condensation (93). Endo G is a mitochondria-specific nuclease that once released from mitochondria translocates to the nucleus and cleaves DNA into nucleosomal fragments independent of caspases (336). Smac/Diablo and HtrA2/Omi are two IAP antagonists identified in mammals. They are both nuclear encoded mitochondrial proteins and cleavage of their mitochondria-targeting sequence generates active Smac and Omi. Released in the cytosol these peptides bind to and cleave IAPs and thereby induce apoptosis (177, 642). For example, increased expression of HtrA2/Omi in cells increases cleavage of XIAP, while suppression of HtrA2/Omi by siRNA has the opposite effect (535). Therefore, release of these pro-apoptotic peptides could initiate and/or amplify cell death that occurs via apoptosis (177, 550). Conversely, IAPs block the enzymatic actions of the caspase proteins that mediate cell death (177, 550) (Fig. 6). Cancer cells are able to utilize the IAP proteins by overexpression to confer chemoresistance. Recent studies have also shown that the mitochondrial apoptotic protein Smac can abrogate the protective function of IAPs (134, 144, 546). These findings suggest the potential clinical utility of Smac mimetics to trigger apoptosis and overcome drug resistance conferred by IAPs.
mPTP opening can also mediate fast Ca2+ release from Ca2+ loaded mitochondria (Fig. 2), perhaps through a Ca2+ induced release mechanism (177, 550). These actions of the mPTP apply in most instances of cell damage and suggest that pharmacological agents or any other maneuvers that influence the pore could minimize the extent of permanent damage that arises. In this regard, protective strategies directed to mitochondria might be beneficial (572, 573). It is therefore significant that achieving control of the mPTP during the disease process is an important goal from the perspective of minimizing the early loss of function, and maximizing salvage as time proceeds following initial injury.
Despite considerable effort, however, the molecular identity of the mPTP remains controversial and uncertain. This uncertainty in the pore structure, its constituents or secondary targets (ROS production, Ca2+ uniporter), have complicated drug development directed at influencing pore opening. Furthermore, the dubious role of the mPTP constituents in the preservation of cell life or mediation of mitochondrial injury complicates the therapeutic goal of targeting the pore. For example, one therapeutic strategy targeting the mPTP is overexpression of CypD, which has been shown to paradoxically increase resistance of cells to oxidative stress-induced cell damage. In this case, CypD acts both as an instigator of cell death on the one hand, and on the other hand as a chaperone-like protein to protect against oxidative stress (341, 602). Nonetheless, several pharmaceutical agents targeting the mPTP have been successfully employed in numerous models of cellular injury to mitigate damage (572, 573, 602). This is evident by the recent human clinical trial that shows CsA reduced infarct size and improved recovery of contractile function on reperfusion. The authors proclaimed that large-scale trials are ongoing to address if these treatments might improve clinical outcome in patients after acute myocardial infarction (213).
These studies do provide some hopes for a clinical possibility of targeting the mPTP. But other models of the pore maintain that activation of the pore with ROS would impair the antioxidant effect of CypD. Overall, the major goal is development of novel, specific, and potent inhibitors that target both the primary constituents, which still remains elusive, and secondary targets (e.g., CaU) whose activities directly or indirectly modulate pore activity. An example in this case is the use of the antidepressant drug nortriptyline, which exerts its neuroprotective effects against cerebral I/R injury in part via delayed mPTP opening by resisting Ca2+ overload (646). Therefore, a complete molecular characterization of the CaU could lead to better therapeutic targets that could minimize matrix Ca2+ uptake and indirectly mitigate mPTP-mediated cellular damage.
Other therapeutic strategies involve the direct targeting of constituents of the pore. For example, ANT inhibitors have been used to block mPTP, but their use in the heart is of limited values because the heart stops beating (74). CsA, and its nonimmunosuppresive derivative NIM811, prevent mPTP activation in part by blocking CypD binding to ANT and thus prevent mitochondrial depolarization (573). Sanglifehrin, a novel immunosuppressive natural product that also binds to CypD and inhibits its peptidyl-prolyl isomerase, is effective in protecting against pore opening and minimizing I/R-mediated cellular injury. The translation of these agents from experimental studies to clinical trials is hampered, however, by their undesirable side effects. CsA is known to exert unwanted side effects on the heart by inhibiting calcineurin (138, 236). It is reported that CsA has a narrow window of activity; the optimal concentration is approximately 200 nM for optimal protection but it declines as a protective agent at higher concentrations. Other mPTP inhibitors whose mode of actions are not well known include trifluoperazine, which is only active in energized mitochondria, and ubiquinone analogues which modulate pore opening by interacting with complex I (236).
Knowledge of the structural constituents of the mPTP and how agents modulate the dynamic function and structure of the mPTP is essential to understand the role of mitochondria as a therapeutic target for human diseases in which apoptotic and anti-apoptotic mechanisms are directly implicated in the etiology. The goal here would be to selectively manipulate mPTP protein function by therapeutic intervention, either to activate it to induce apoptosis for cancer therapy, or to inhibit it to protect against cell death during cardiac or cerebral ischemia.
III. Electron Transport Chain and Oxidative Phosphorylation: Modulation by Mitochondrial Ion Channels and Exchangers
Mitochondria are the primary organelles for the generation of ATP under normal aerobic conditions. They contain the terminal oxidative pathway (TCA cycle) for carbohydrate and fat oxidations that produce the reducing equivalents NADH and FADH2 (H+ and electron pairs). In OXPHOS, electrons are transferred from NADH and FADH2 to molecular O2 through the ETC complexes I–IV until two electrons and two protons combine with ½O2 to produce H2O at complex IV (respiration). Concomitantly, protons are pumped from the mitochondrial matrix into the IMS. This generates a pH gradient and an electrostatic potential, ΔΨm, across the IMM. Under normal physiological conditions, ΔΨm contributes most of the ΔμH, which drives the protons back into the mitochondrial matrix down their electrochemical gradient through the F1F0–ATPase (ATP synthase) to synthesize ATP (phosphorylation). Both ΔΨm and ΔμH tend to decrease if the supply of NADH and FADH2 through the TCA cycle does not match the increased flux through the ETC during mitochondrial respiration. Together, the various compartments of mitochondria are able to work in harmony to generate ATP in a complex multistep process.
ATP is involved in a myriad of cellular processes that are essential for cell survival such as maintaining ionic homeostasis, cell proliferation, and gene regulation. The dependence of cells on mitochondrial ATP varies. For example, cancer cells and astrocytes can survive well on ATP generated from glycolysis and are much less dependent on mitochondrial OXPHOS to generate ATP. Other cells such as neurons and cardiomyocytes depend almost entirely on mitochondrial OXPHOS for their function. Preservation of the constituents of the mitochondrial ETC is paramount in maintaining the bioenergetics status of the mitochondrion and the cell homeostasis. Indeed, mitochondrial defects encompassing complex I–IV of the ETC characterize a large number of neurodegenerative diseases (124, 125).
Mitochondrial ETC complexes are involved in cytoprotection. Studies have shown that amobarbital and volatile anesthetics block complex I, diazoxide blocks complex II, and hydrogen sulfide blocks complex IV. Although these drugs have additional effects, they emerge as potential means to protect against cellular injury following I/R (9, 113, 244 –246, 324, 325). The targeting of mitochondrial complexes for a therapeutic purpose is in part ascribed to their vulnerability to oxidative stress. Therefore, a limitation of electron transfer during ischemia to complex III, a major site for electron leak and ROS production, is a new concept to limit mitochondrial damage specifically during ischemia (9, 116, 117, 330). Mitochondria sustain progressive damage to the ETC during the course of myocardial ischemia; 10–20 min of ischemia decreased complex I activity and caused damage to the OXPHOS apparatus, including complex V and the ANT (330). As ischemia time lengthens (30–45 min), damage to complex III and IV becomes evident.
Hence, while a complex I defect occurs early in ischemia, damage continues to progress to involve complexes III and IV. Complex I activity will go down due to a decrease in the NADH dehydrogenase component, possibly the loss of the FMN coenzyme; complex I activity is also modulated by post-translational modifications including S-nitrosylation and phosphorylation. These peptide alterations are amenable to pharmacologic manipulation, as in the use of S-nitroso-2-mercaptopropionyl glycine (SNO-MPG) in providing protection against ischemic damage (396). SNO-MPG inhibits complex I during the critical late ischemia and early reperfusion stage (59, 66) and in that way provides protection from ROS generated at complex III (117) (Section III).
The above observations are consistent with our recent studies showing that blocking complex I with the reversible inhibitor amobarbital protected the heart, and its mitochondria in particular, from I/R injury when the blocker was present only during ischemia. Amobarbital, a short-acting barbiturate, inhibits complex I at the rotenone site at concentrations of 1–3 mM. At higher concentrations (5 mM), amobarbital also inhibits succinate respiration and complex V (113, 116). We have furnished recent additional data for the specificity of amobarbital treatment in intact isolated hearts that gives further insight into its mechanism of protection (9). This mechanism of protection was similar to that of ranolazine, a late sodium channel blocker and anti-angina drug that is also known to block complex I (82) and to inhibit β-oxidation enzymes (68, 538).
In a preliminary study, guinea pig hearts were perfused with either ranolazine (5 μM) or vehicle for 1 min up to the onset of 30 min no flow global ischemia; ischemia was followed by 120 min reperfusion without ranolazine. Mitochondrial NADH and FAD, ROS, and Ca2+ were monitored online continuously using a fiberoptic probe placed against the left ventricular (LV) free wall and connected to a fluorescence spectrophotometry (9). Ranolazine was present only during the 1 min period prior to ischemia and during ischemia, and was completely absent during reperfusion, and so did not contribute to reperfusion function. We found that ranolazine treatment resulted in a more normalized mitochondrial redox state (NADH, FAD) (Figs. 7A and 7B), less O2 •− production during I/R, a higher phasic LV pressure (Figs. 7C and 7D), and a reduction in the incidence of ventricular fibrillation (data not shown) (8). Mitochondria isolated from hearts treated with ranolazine just before the index ischemia showed a greater propensity for Ca2+ retention than the control untreated hearts when challenged with pulses of increasing concentrations buffer CaCl2 (Camara AKS and Stowe DF, unpublished observations). In a related study (324), we showed that a limitation of electron transfer by the irreversible complex I inhibitor rotenone did not provide as much protection as when the drug was given before ischemia and was present during the ischemic episode. Thus, blockade of ETC during ischemia preserves respiratory function in isolated mitochondria (116) and in the intact heart (9, 117); this is accompanied by decreased mCa2+ overload and less O2 •− generation in the isolated beating heart (9).
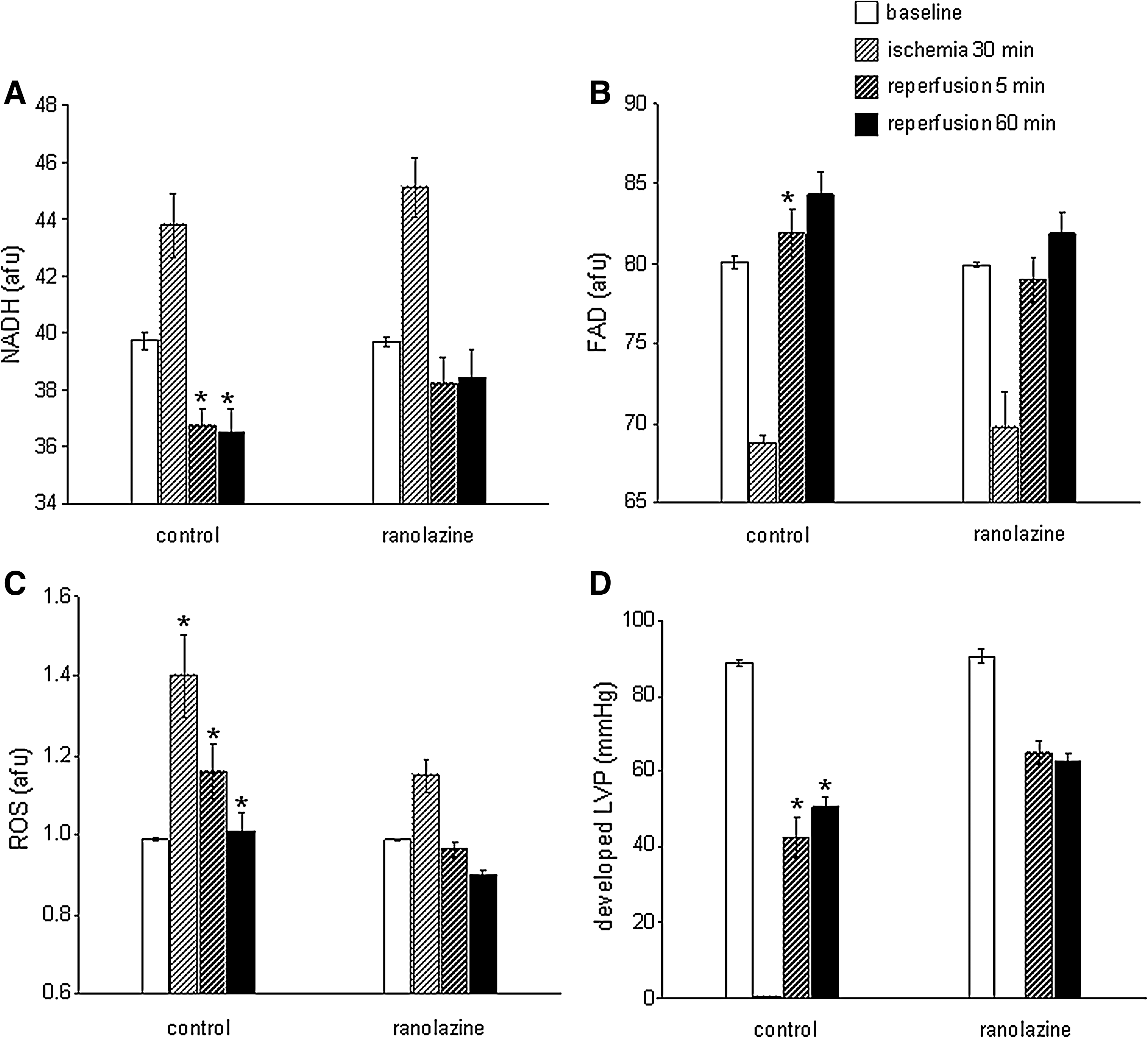
Alleviating mitochondrial dysfunction is not limited to targeting complex I of the ETC. Hanley et al. (243 –245) reported that the putative mKATP channel opener diazoxide inhibited complex II of the ETC and suggested this could provide protection in part by inhibiting electron transfer to complex III and in the process minimize O2 •− generation. The benefit of targeting mitochondrial ETC is that it provides an alternative approach to cardioprotection against I/R injury when ischemic or pharmacological preconditioning is impaired (113, 396). The structural integrity of the IMM is equally important in preserving the mitochondrion for normal and efficient OXPHOS. The IMM contains cardiolipin, a special phospholipid that is rich in linoleic acyl-groups that are highly susceptible to ROS produced during oxidative stress (29). Preservation of IMM was also observed with rotenone (113, 324). Loss of cardiolipin results in dysfunction of complex V, impaired ATP levels, and subsequent derangement of cellular ion homeostasis and cell death. Overall, these results highlight an emerging paradigm that reversible metabolic inhibition may be a common pathway leading to cellular protection and that the ETC regulates apoptosis.
Mitochondrial ETC function is modulated by several trans-matrix ions that enter and exit via several mitochondrial ion channels, exchangers, and symports (Sections IIB and IIC). In the mitochondrion, a principal cation uptake pathway is via K+ channels. There is a concerted interplay between K+ uptake, via one or more K+ channels, and the primary K+ efflux route via the K+/H+ exchanger (KHE), which controls mitochondrial volume homeostasis (200, 202). The existence of regulated pathways for both K+ uptake and K+ efflux may allow for a very fine-tuning of mitochondrial volume, and thus the rate of respiration. Changes in mitochondrial volume regulate mitochondrial energy metabolism through their effects on the TCA cycle enzymes and respiratory chain (356, 555). During the steady state, respiration is balanced by K+ influx into mitochondria through K+ channels and efflux through the KHE. An imbalance in this dynamic relation could lead to matrix swelling and on to cellular damage by apoptosis or necrosis.
Indeed, ischemic damage has been associated with derangements in mitochondrial ion flux regulation and matrix volume (9, 10, 90, 173, 201, 307, 474, 475). Decreased Δψm during ischemia may lead to a contraction of matrix volume and result in decreased and less efficient OXPHOS. Increased K+ flux via the putative mitochondrial KATP (mKATP) channel may counteract this effect with a concomitant increase in volume that may improve the mitochondrial redox state (173, 201, 307, 474, 475) and allow for more efficient ATP synthesis (201) and cellular preservation. However, Shalbuyeva et al. (514) reported that Ca2+-sensitive K+ channel (KCa) and KATP channel blockers (e.g. charybdotoxin and 5-HD, respectively) did not suppress Ca2+-induced swelling in mitochondria isolated from brain cells, and inhibitors of the mitochondrial KHE (e.g., quinine, dicyclohexylcarbodimiide) inhibited the recovery phase of the reversible mitochondrial swelling. In addition, CsA supplemented with cytochrome c did not reverse mitochondrial swelling in both liver and heart mitochondria (503, 514). It was proposed that Ca2+-induced K+ influx leading to swelling causes activation of KHE to extrude K+ and thus reduce mitochondrial swelling.
We have recently provided novel evidence for a regulatory role of the putative mKCa on ΔμH. We inferred that opening mKCa channel allows a small H+ influx (“leak”) via the KHE (253). When the leak is small (instigated with <30 μM NS1619), H+ pumping may increase to drive respiration and ATP synthesis without changing the Δψm. If the leak is large (instigated with >30 μM NS1619), Δψm will decrease and ultimately dissipate Δψm (253). This scenario is supported by our recent experiments showing that NS1619-induced matrix K+ uptake and mitochondrial swelling were observed only when quinine (KHE inhibition) was present in the buffer (7). The implications of these channels and/or exchangers in mitochondria-mediated cellular damage have been proposed in numerous studies and their potential therapeutic utility at the mitochondrial level is currently being pursued.
IV. Mitochondrial ROS and RNS
A. Mitochondria and reactive oxygen species
In this and the subsequent section, ROS (O2 •−, H2O2 and OH•, etc) and RNS (NO• and ONOO-, etc) generation and scavengers will be discussed. Even though ROS and RNS are discussed separately, the two subjects are tightly intertwined and as such, the discussions will overlap in several sections. ROS scavengers and antioxidants are used interchangeably.
In excitable tissue, especially cardiac and neuronal, mitochondria represent a major source of O2 •− as a consequence of mitochondrial respiration, which generates unpaired electrons that interact with molecular O2 to produce O2 •− (20, 84, 85). These O2 •− anions are readily interconverted to other radical species, such as H2O2 and ferryl radicals (Fe(VI) = O•) and perhaps OH• (487, 488). H2O2 is a relatively inactive compound. However, if reduced iron (Fe2+) is abundantly present, as in I/R, as a result of increased release of Fe from ferritin or aconitase (TCA cycle enzyme), the highly reactive [Fe = O]• radical will be formed (80, 137, 230). In support of this it was found that the Fe chelator desferrioxamine, when administered upon reperfusion, improved function following I/R (169, 331, 343).
Beyond their roles in aerobic energy metabolism and maintenance of ionic (e.g., Ca2+) homeostasis, mitochondria have other important physiological and pathophysiological processes. Mitochondrial ROS are involved in cell signal pathways as noted for ischemic and pharmacological pre- and postconditioning (Sections X.D and X.E). ROS are also involved in transcriptional regulation and normal cell proliferation. Indeed, ROS are important in normal cellular development and a limited amount of ROS in specific cells is necessary to mediate the programmed cell death that is required for cell elimination and mitochondrial autophagy during development and elimination of injured mitochondria or poorly performing cells. So, one would assume that teleologically mitochondria produce some ROS that are important for normal cellular function and survival despite the elaborate scavenging system (Section V). Indeed, overexpression of matrix scavenger proteins (e.g., manganese superoxide dismutase (MnSOD), the mitochondrial variant of SOD), could provide effective scavenging, but because O2 •− plays an important physiological role, excess scavenging may be deleterious.
Mice with overexpressed MnSOD exhibit reduced fertility and abnormal development (338, 473, 478). On the other hand, an MnSOD gene knockout was shown to be lethal (321, 340), whereas the CuZnSOD (extra-matrix SOD) gene knockout was not, though lifespan was shortened (183) and oxidative stress was elevated (389). These findings indicate that the targeting of mitochondrial O2 •− as a therapeutic goal during I/R would require careful balancing of the “good” (physiological) and “bad” (pathological) O2 •−. Deviation from a tight regulation of ROS is likely to contribute to mitochondrial O2 •− damage leading to numerous degenerative diseases and promotion of the aging process (Sections IX and XII).
Under physiological conditions, a net amount of O2 •− is produced (i.e., O2 •− emission), as determined by the rate of O2 •− generated minus the rate of O2 •− scavenged. To maintain this delicate balance, mitochondria are equipped with a variety of endogenous antioxidant defenses that regulate O2 •− within a physiological range. However, under pathological conditions, as in cardiac I/R and in the aging process, the delicate balance (generation—scavenging) that keeps the level of O2 •− to a minimum is altered so that the rate of O2 •− generation exceeds the rate of scavenging. This can result in further damage to mitochondria and may exacerbate ROS-induced ROS damage (66, 657).
Superoxide anions are generated from numerous sources in the mitochondrion. These include monoamine oxidase and cytochrome b5 reductase in the OMM (85), in the IMM along the ETC (63, 64, 316, 347, 536, 580, 656), in the TCA cycle from α-ketoglutarate dehydrogenase and aconitase, and from non-TCA cycle enzymes, pyruvate dehydrogenase and glycerol-3-phosphate dehydrogenase (GPDH) (335). For example, isolated mitochondria supplemented with GPDH can produce O2 •− from complex I to the matrix side and some from the cytosolic side (20, 345) (Fig. 8). However, a majority of mitochondrial O2 •− is generated within the IMM of the ETC, in particular at complexes I and III (85, 113, 118, 581). The superoxide anions are generated by the ETC directed vectorially into the IMS and the matrix. O2 •− generated in the OMM and from the TCA cycle will not be discussed further in this review.

In metabolically active cells such as cardiomyocytes and neurons, mitochondrial ETC is a key contributor in cellular O2 •− production under normal and pathophysiological conditions. In the absence of electron transport through the ETC, these cells cannot consume O2 or generate O2 •− from mitochondria (235, 578). But the specific sites and mechanisms of O2 •− generation along the ETC remain controversial (14, 34, 118, 580). For example, the precise site for ROS production from complex I is uncertain. It is believed that FMN in NADH dehydrogenase (220, 580), Fe–S cluster N2, and the two tightly bound ubiquinones located distal in the path of electron transfer through complex I (206, 316), are all potential sites for O2 •− generation.
It is widely acknowledged that complex III is the dominant site for the net production of ROS from intact mitochondria in the baseline state (118, 240, 407, 536). Experiments show that complex III generates O2 •− through the oxidation of ubisemiquinone, a radical intermediate formed through the Q-cycle of the complex, particularly at the Qo site which faces the IMS, while complex I-mediated ROS is directed mostly toward the matrix side (113, 117, 118). Inhibition of forward electron flow at upstream or downstream sites of complex I (NADH dehydrogenase) decreased and increased O2 •− generation from complex I, respectively, whereas inhibition of reverse electron flow at the upstream site of complex I enhanced O2 •−; this suggested that the site for O2 •− generation in complex I is distal to the FMN center at the FeS cluster N2 (418, 512). However, many inhibitors of complex I may share common binding domains (422).
In normal healthy aerobic cells, oxidation and the generation of O2 •− occur at a controlled rate. But under high stress conditions or in disease states including cancer, nervous system disorders such as Parkinson's disease (PD) and Alzheimer's disease (AD), or cardiovascular disorders, ROS production is greatly increased, causing peroxidative changes of many proteins and lipids (421, 638, 639). Mitochondrial DNA is also one of the main cellular targets of ROS-induced oxidative damage due to their lack of histone protein protection (334) (Section VII). Increased mitochondrial ROS production, for example during hyperglycemia, may be a major factor in the pathology of diabetes. Glucose-stimulated insulin secretion by isolated islet cells can be used as an index for oxidative stress and/or impaired oxidative metabolism (51).
In cardiac I/R injury, impaired complex I (113, 327) can enhance O2 •− formation as a result of increased electron leak as electron transfer is impeded. When FADH2-related substrates are used and electrons enter the ETC at complex II, O2 •− may be generated by reverse electron transfer to the FMN site of complex I (113, 543, 656). Although complex I is a site for O2 •− generation in cardiac cells under ischemic conditions, complex III is also a major site for ROS production (117). Ischemia damages complex III by a functional alteration in the Fe–S protein subunit (113). Regardless of the source of O2 •− production, the mechanism and quantity of O2 •− produced in vitro is dependent on the experimental substrate, the energetic conditions, and the trans-matrix pH gradient (316, 317).
An increase in O2 •− production under pathological conditions can also occur as a consequence of depletion or a defect in the mitochondrial antioxidant system. Increased ROS production under such conditions has been ascribed to a self-regenerating ROS production facilitated by ROS-induced ROS release (66, 543, 657). This increase in oxidative stress results in further damage of OMM, IMM, and matrix proteins that are highly sensitive to oxidative stress. A point is reached where the scavenging system almost completely collapses and generation of ROS is perpetuated in a vicious cycle. The association of ROS generation and various pathological conditions has made development of the ideal antioxidant therapy to target the mitochondrion a pre-eminent goal. The therapeutic strategy to limit mitochondrial O2 •− production during hyperglycemia, for example, counteracts their damaging effects and may be a useful complement to conventional therapies designed to normalize blood glucose (218).
However, targeting O2 •− emission during I/R could be problematic because recent evidence shows that O2 •− production occurs in heart cells not only during reperfusion but also during ischemia with a surge during late ischemia (9, 10, 90, 289, 290, 591, 592). Thus during ischemia, O2 •− generation sets the stage for an increase in O2 •− emission during reperfusion as a mechanism of cellular injury (9, 113, 117). Clinically, this is a relevant area of research because patients with active myocardial ischemia could theoretically receive pharmacologic therapies that target the spatial and temporal aspects of ROS generation. Pharmaceutical agents that provide ROS scavenging systems are most effective if they address the problem at its source, in this case in the IMM (262).
Overall, a better understanding of the sources and direction of O2 •− generation from the mitochondrion is crucial to an understanding of the potential of particular antioxidants used to mitigate oxidative stress and cellular damage. However, effective delivery of these antioxidants into the cytosol or matrix as a therapy is quite problematic. Attempts to boost antioxidants by dietary supplements do not help, probably because they cannot permeate the mitochondrial membrane into the matrix where some free radicals are produced. To address these physical limitations, therapeutic antioxidants have been reformulated based on the strong negativity of the matrix membrane potential (Table 1). This will be addressed further in Sections X and XI.
B. Mitochondria and reactive nitrogen species
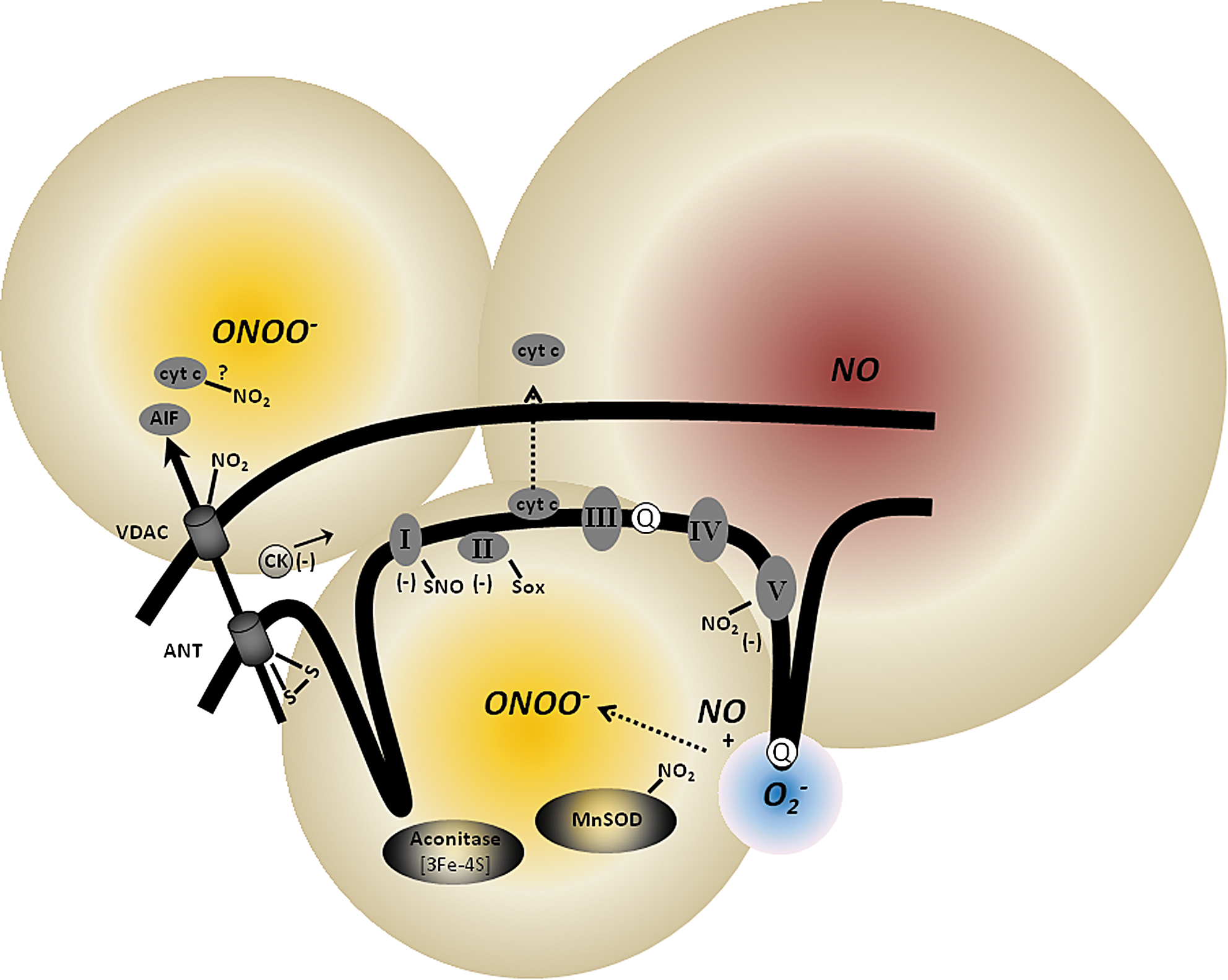
The free radical NO• is an endogenous mediator of numerous vital physiological processes, including cytoprotection. NO• can also mediate cell injury (533). NO• has emerged as a crucial and potential player in the control of mitochondrial function: it modulates mitochondrial activity at complex IV; it generates peroxynitrite (ONOO-) when it reacts with O2 •− (Fig. 9; and it regulates mitochondrial biogenesis via activation of guanylate cyclase. NO• is a major target for numerous signaling pathways, and it in turn can trigger the release of factors/proteins that initiate cellular events critical in cell survival or death. These roles of NO• are determined by a delicate balance between physiologically relevant levels and pathological concentrations (298).
The source of mitochondrial NO• may be vascular endothelium, nerve terminals, or other cytosolic sources, as the activity of mitochondrial NO• is low (533) or nonexistent. Some have proposed that mitochondria contain nitric oxide synthase (NOS), which can be source of mitochondrial NO• (44, 209). However, the notion of mitochondrial NOS remains controversial with major contentions surrounding the purity of mitochondria and several experimental artifacts in NO•-measuring systems (81). For example, in a recent study, Venkatakrishnan et al. (596), using HPLC-mass spectroscopy, found no evidence for NOS derived peptides, calmodulin (needed for NOS activity), or NOS activity measured as conversion of arginine to citrulline in highly purified liver mitochondria. The controversy over this subject will not be discussed any further in this review.
NO• has multiple targets in mitochondria including hemoproteins such as cytochrome oxidase, proteins, and lipid thiols. The role of NO• and its products in the cell is akin to a double-edged sword. NO• can act both as a scavenger and as a facilitator of cellular injury depending on the concentration and the conditions of the lipid environment (81). NO• can act directly on mitochondria to protect tissue/organs, or it can provide protection by way of a NO•-mediated signaling cascade. Recent data support strongly the role of NO• as a key mitochondrial regulator. Mitochondria can be considered a cellular “hub” for NO• signaling, as evidenced by the presence of many metal clusters and thiols; mitochondria also generate secondary intermediates crucial for other NO• mediated functions (81). One of the most important functions of NO• in mitochondria, and its most characterized effect, is the competitive reversible inhibition of O2 binding at the binuclear site of complex IV, the terminal component of the ETC where electrons are transferred to O2. NO• inhibition of O2 binding is reversible as this depends on the concentration of the two gases in the mitochondrion. Thus the relative concentration of NO• is crucial in the mechanism for controlling respiration, and if ONOO- is produced, in cell death (71, 298).
At a low concentration, NO• provides protection against I/R injury, but at higher concentrations, or in the presence of increased matrix Ca2+, NO• increases the apoptotogenic effect of O2 •− (391). Inhibition of complex IV by NO• is thought to elicit cardioprotection by preserving the limited supply of O2. By binding to complex IV, NO• may provide protection to mitochondria by indirectly reducing mCa2+ overload. Some of these effects are sensitive to the NO• inhibitor cPTIO. A study on the interaction of NO• with complex IV suggested that NO• interacts with either oxidized Fe2+ or Cu2+ so that NO• is reduced to nitrite. Nitrites have been used to treat angina in patients for a very long time (576). In isolated rat heart experiments it was shown that reduction of nitrite to NO• during ischemia protects against myocardial damage (611). In addition, NO• is strongly implicated in the mechanisms underlying IPC, including nitroalkanes such as nitro-linoleate and S-nitrosation of complex I (395). NO• is also known to preserve cardiolipin, which helps maintain IMM integrity and complex IV activity, and to minimize the release of cytochrome c release and other apoptotic factors (215, 425, 601). NO• may decrease mCa2+ overload by binding to complex I and this appears to cause a decrease in Δψm (81, 533). It is possible that some of the protective effects of NO• can be attributed to a coordinated series of responses by modest levels of NO•, which indirectly enhances mitochondrial function by increasing local blood supply to enhance O2 and substrate delivery to mitochondria.
The effects of NO• are mediated in part via signaling molecules including a guanylate cyclase-dependent pathway. NO• also appear to be involved in regulation of mitochondrial biogenesis. The cGMP-dependent pathways are thought to activate PGC-1α and PGC-1β, which lead to expression of the nuclear respiration factors NRF-1 and NRF-2. The transcription factors transcribe the nuclear genes that encode subunits of the ETC complexes and the mitochondrial transcription factor A (496) resulting in increased mitochondrial biogenesis.
At higher rates of NO• production, NO• overwhelms the cellular protective mechanisms and shifts the balance towards apoptosis. In mitochondria, NO• is present at fairly high concentrations under pathological conditions such as ischemia and displays a broad chemical reactivity with oxidative inflammatory mediators. In this way higher concentrations of NO• could induce cell death by necrosis and apoptosis via mPTP opening (533). Under disease conditions the excess NO• in the presence of diminished O2 tension may inhibit mitochondrial respiration and lead to increased O2 •− production. The O2 •− in turn reacts with the NO• to produce the potent ONOO- compound, a highly reactive non-free radical (Fig. 9). The reaction between NO• and O2 •− is limited by the diffusion rate of NO• (630), with NO• being much more permeable than O2 •−.
Like NO•, ONOO- has been reported to be cytoprotective when administered in small concentrations (81). However, at higher concentrations ONOO- is highly toxic to mitochondrial membrane and proteins. It is believed that the most damaging effects of NO• activity on mitochondria are attributed in large part to ONOO- (177). ONOO- may reduce the NO• half-life under conditions in which mitochondrial O2 •− formation is stimulated, reducing the effects of NO• on complex IV activity, and reducing NO•-dependent mitochondrial signaling. ONOO- can be derived from extra- or intra-mitochondria sources (Fig. 9). Extra-mitochondrial ONOO- might diffuse into mitochondria to exert its effect but intra-mitochondrial ONOO- has a short half-life due to large abundance of metalloproteins and fast acting thiols (461). Cardiolipin is most susceptible to ONOO- damage (113) and lipid peroxidation can damage mitochondria and lead to apoptosis.
It is important to note that while NO• mainly reacts reversibly at complex IV, ONOO- is known to block complexes I, II, and V by irreversible nitration of the tyrosine residues and transition metal centers in these proteins (177). ONOO- also nitrates VDAC, ANT, MnSOD, and aconitase, and it is also involved in oxidative damage of other complexes (Fig. 9). ONOO- is not believed to exert effects at Complex IV, however, because of its remarkable resistant to oxidative damage (648). Thus, conditions leading to excess ONOO- will promote enhanced mitochondrial lipid/protein oxidation, swelling, and rupture of mitochondrial membranes, Ca2+ loading, and cytochrome c release.
Ubiquinol, which can reduce ONOO-, has been proposed to protect mitochondria against damage (64, 85, 584). Expansion of the ubiquinol pool either by pharmacological manipulation of the ETC or its exogenous addition, correlates significantly with decreased mitochondrial nitration and attenuated ONOO--dependent damage (461). Glutathione, reduced cytochrome c oxidase, and perhaps NADH also minimize ONOO- levels (462). Given that ONOO- is formed from NO• and O2 •−, inhibition of either O2 •− or NO• generation will attenuate the levels of ONOO- and minimize mitochondrial damage. For example, overexpression of MnSOD has been shown to protect neuronal-like cells against NO•-dependent cell injury, tyrosine nitration, and lipid peroxidation. In addition, overexpression of nNOS instigates NO•-dependent neuronal cell death, which is alleviated in cells enriched in MnSOD (462). However, the chemical reaction between NO• and O2 •− is faster than the reaction with MnSOD (187) and ONOO- mediated inactivation of the enzyme serves to amplify NO• and ONOO--dependent mitochondrial oxidative stress (462). Therefore attempts to scavenge O2 •− to reduce ONOO- might not be a feasible approach in mitigating ONOO--induced damage. However, targeted approach toward excess ONOO- and NO• or use of “small” concentrations of the NO• as a signaling mediator for cytoprotection appears as a novel and important potential strategy against cell injury.
V. Mitochondrial ROS Scavenging and Its Potential Therapeutic Value
ROS and RNS are clearly involved in normal cellular functions because they act as signaling agents in cellular protection, such as in cardiac preconditioning (135, 289, 377, 408, 409, 428, 429, 476, 477, 649), postconditioning (135, 223, 382, 429, 431), and cold preservation (90, 475). But ROS and RNS, as described above, can induce cell damage if their levels are not controlled within acceptable physiological limits. With this dual role, can modulation of ROS be an effective therapeutic tool? To address this question, the need to effectively detoxify pathologic ROS has to be balanced with the need to maintain physiological ROS. It is this delicate balance that is used to control and manage cancer. Increased generation of ROS, which challenges ROS scavenging systems, can lead to increased apoptosis of tumor cells, or alternatively increase the scavenging capability to reduce ROS needed for tumor growth, in this case a desirable effect (193).
During pathological stress with a sustained increase in ROS levels, an ideal strategy would be to boost O2 •− scavenging by using nontoxic catalytic antioxidants that are either delivered tissue-specifically or produced where needed from inactive precursors. Another strategy would be to decrease the primary O2 •− production by preventing the over-reduction of intra-mitochondrial NADH (556) or by using mild uncouplers, that is, decrease ΔΨm (529, 556) (Section V), or to pharmacologically stimulate the expression of endogenous mitochondrial and intracellular antioxidant systems (20). But these strategies are limited in their capability to mitigate ROS-induced damage if ROS inflicted damage results in further mitochondrial damage and leads to additional activation of O2 •− generation in a vicious positive feedback loop that results in increased ROS production. In this case, the most efficient way to reduce mitochondrial O2 •− production may be to prevent O2 •− generation rather than to scavenge the emitted O2 •− (20).
The endogenous scavenging system of mitochondria has been widely covered by others (20, 23, 258, 259, 450, 543, 625). So only a brief discussion will be presented here. Discussion on the use of antioxidants is only presented where they are relevant in ameliorating mitochondrial related diseases. These antioxidants are mainly synthetic agents, for example MitoQ, α-tocopherol, vitamin-based antioxidants, and genetic maneuvers, such as overexpression of MnSOD.
Mitochondria possess an elaborate and well-defined multileveled antioxidant defense system of enzymes and nonenzymes to scavenge mitochondrial O2 •− (345). The scavenging system includes the matrix MnSOD and the glutathione (GSH) (Fig. 8) (20, 543) and thioredoxin (TRXSH2) (266, 571) systems, cytochrome c, peroxidase, and catalase (543). The counterpart to the matrix-bound MnSOD is the Cu-Zn SOD (SOD1) found predominantly in the cytosol, although recent studies have reported the presence of SOD1 in the IMS (337); extracellular SOD is also found in interstitial fluid, plasma, lymph, and synovial fluid (285).
A. Manganese superoxide dismutase
MnSOD (SOD2) is a metallo-enzyme located primarily in the mitochondrial matrix at levels of 10–20 μM (461) (Fig. 8) that does not require co-factors to detoxify O2 •− radicals. MnSOD plays an essential role in protecting against oxidative stress and the assembly of this tetrameric peptide into the active manganese-based enzyme is key for survival. Its only known function is to detoxify O2 •− to H2O2, thereby protecting mitochondrial Fe–S cluster containing enzymes from oxidative damage (20, 199, 543). A defect in genes encoding SOD1 or SOD3 (extracellular Cu/Zn SOD) results in a mild, nonlethal phenotypic enzyme expression (98, 470). In marked contrast, a SOD2 (MnSOD) knockout results in neonatal lethality (263, 264) due to dilated ventricular cardiomyopathy, fibrosis, and other complications (20, 145, 262 –264). Overexpression of the MnSOD gene on the other hand has been associated with protection against oxidative stress-mediated cell death and cellular injury (145). For example, overexpression of MnSOD has been shown to ameliorate the expression of proteins implicated in retinal impairment in a mouse diabetic model (498).
A number of studies show that cells respond to oxidative stress, like lipopolysaccharide treatment, by increasing the level of MnSOD (142, 479). In contrast, in neoplastic cells where MnSOD is normally low, induction of the scavenger has been implicated in the suppression of tumor growth. This suppressive effect in tumor cells is believed to be due to changes in the activity and expression of transcription factors including NF-κB (nuclear factor kappa of activated B cells) and NFAT (nuclear factor activated-T cells) (415).
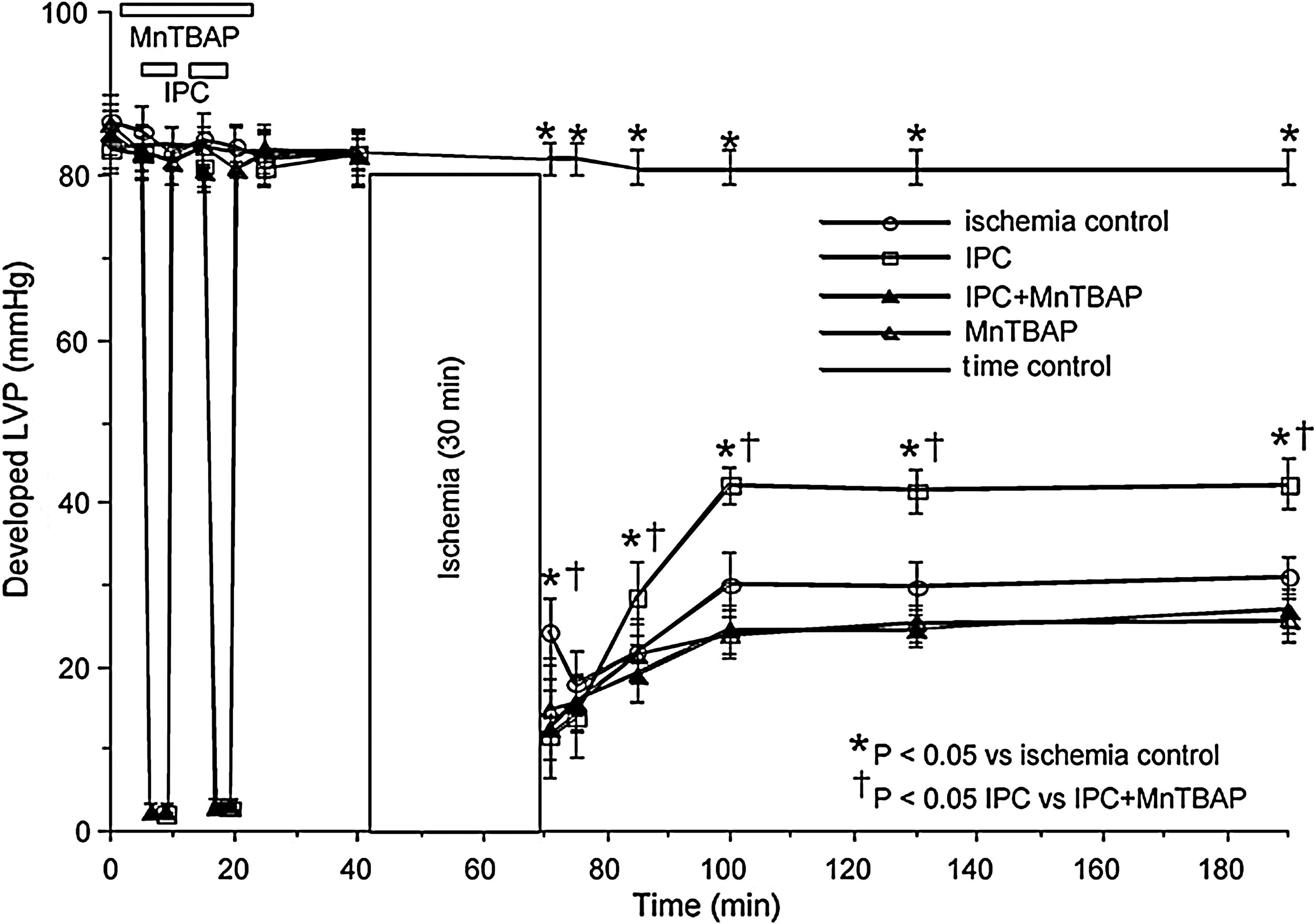
Mitochondrial SOD mimetics (nonprotein) have been developed to allow uptake into the mitochondrion to scavenge ROS. The mitochondrial superoxide dismutase mimetics MnTBAP, and Mn(III) meso-tetrakis (N-methylpryidinium-2-yl) porphyrin (MnTE-2-Py5+) are permeable to the IMM. MnTE-2-Py5+ was shown to accumulate in heart mitochondria following intraperitoneal injection and MnTBAP was reported to improve survival in MnSOD knockout mice (49). Since O2 •− is involved in signal transduction of IPC, we showed that MnTBAP given alone abolished the cardioprotection afforded by preconditioning (289) (Fig. 10). MnTBAP was also shown to scavenge ROS generated during cold perfusion (91), which confirms uptake into mitochondria. Genetically altered mice deficient in the sod2 gene die with heart failure in conjunction with other severe complications (264, 340, 369). Treating mice with MnTBAP ameliorated the complications and greatly increased lifespan (370).
Another cell permeable SOD2 mimetic, Mn(III)tetrakis[(1-methyl-4-pyridyl)-porphyrin] (MnTPyP), has both MnSOD and catalase mimetic effects (607). We observed that MnTPyP, given before prolonged cold ischemia, protected the isolated heart from cold-induced ROS damage during ischemia better than MnTBAP (Camara and Stowe, unpublished observation). The protection afforded by MnTPyP was similar to that provided by an MnTBAP, catalase, glutathione cocktail (90). These actions of MnTPyP may be similar to the actions of a class of small molecular weight catalytic scavengers of ROS, the salen-manganese complexes. These scavengers act as SOD and catalase mimetics, catalytically eliminating O2 •− and H2O2 (45, 170). They have also been found effective against some mouse models of Parkinson's disease (PD) as they protect the dopaminergic neurons of the substantia nigra from damage induced by mitochondrial-targeted toxins (Section IX,E,2) or 6-hydroxydopamine; they have also shown efficiency against the cytotoxicity of β-amyloid peptide associated with Alzheimer's disease (AD) (76) (Section IX,E,1).
Ischemia and reperfusion injury is not infrequent as a result of vascular surgery, organ procurement, or transplantation. For example, renal ischemia is an unavoidable complication of cross-clamping the donor kidney for removal during transplant (492). Human heart allografts show oxidative stress that is associated with time-dependent changes in endogenous SOD levels (500). Previous attempts to improve organ graft survival by means of exogenous administration of SOD proteins have proven ineffective (403). Studies have shown that oxidative stress resulting from I/R during coronary and lower limb arterial reconstruction can cause endothelial dysfunction in the vein graft with the possibility of graft failure. The addition of MnTBAP was shown to improve endothelium-dependent vasorelaxation in harvested saphenous vein (516). In another study, Nilakantan et al. (403) showed that continuous treatment with MnTPyP had beneficial effects on graft function in a rat model of acute cardiac transplantation. The improvement of function in the allografts by MnTPyP was attributed to attenuated ROS in cardiac allografts, reduction of genes involved in early stages of organ rejection, and decreased apoptosis. MnTPyP was reported also to potentially counteract nitration of MnSOD in allografts by preventing ONOO- levels via scavenging of O2 •−. Similarly, Mn-containing metalloporphyrin can attenuate liver damage, lipid peroxidation, and protein nitration from I/R injury in isolated perfused rat liver (629).
These studies clearly demonstrate the potential beneficial role for these antioxidants in organ and tissue transplantation. However, attempts to incorporate these therapies in the realm of clinical translation for human organ transplantation remain unresolved. To date, only experimental studies in animals have been attempted and there are no known recent clinical trials on ROS scavenging during organ transplantation. One would hope in time that with increased oxidative stress due to reduction in endogenous scavengers in allografts (500), the potential beneficial role of synthetic MnSOD mimetics would become clinically applicable.
B. Glutathione thioredoxin, and peroxiredoxin systems
Redox homeostasis in mitochondria is regulated by the coordinated activity of various antioxidant mechanisms including GSH, thioredoxin (TRX) and peroxiredoxins (PRX). These thiol-reducing agents are critically involved in defense against large increases in O2 •− production as well as in redox regulation of signaling processes ranging from cell division to cell death by apoptosis (345, 510, 543, 582). Cellular antioxidant defenses also depend on the reduction potential of the electron carriers and the reducing capacity of linked redox couples in the matrix (NADH/NAD+ and FADH2/FAD) and cytoplasm that are required to restore the antioxidant activity of the redox systems (25) (Fig. 8).
GSH is a tripeptide with the thiol (-SH) residue of cysteine as its active site. It provides protection for mitochondria against endogenous ROS. Matrix GSH, at 5–10 mM (461), is highly regulated by cytosolic redox state and it is rapidly taken up from the cytosol via the decarboxylate and 2-oxoglutarate transporters (20, 25, 430). In this way it effectively links changes in the cellular redox state (25). Indeed, one defense against I/R-induced ROS accumulation and damage may involve preservation of the NAD(P)H pool. The NADH/NAD+ level through the NADH kinase and transhydrogenase-dependent mechanism (248, 426) maintains the mitochondrial NAD(P)H pool required to maintain the redox status necessary for effective scavenging. This NADH is generated from the TCA cycle and during β-oxidation; thus an increase in NADH would correlate with increased NAD(P)H dependent redox scavenging. The IMM nicotinamide nucleotide transhydrogenase, with binding domains for both NADH and NADP(H), uses the transmembrane proton gradient in the presence of NADH and NADP+ to generate NADPH (461). Cytosolic NAD(P)H is derived mainly from the pentose pathway (20, 543). The enzyme glutathione reductase (GR) uses NAD(P)H as its source of electrons to regenerate GSH. Hence an increased concentration of NAD(P)H relative to NADP+ promotes the production of GSH, which is a substrate for peroxidase (Fig. 8).
GSH, as an abundant source of reducing equivalents, provides the substrates necessary for the proper functioning of mitochondrial proteins containing critical sulphydryl residues, such as the dehydrogenases and ATPase. GSH provides the substrate for glutathione-S-transferase and glutathione reductase to scavenge H2O2. In so doing, GSH protects mitochondria from lipid peroxidation by reducing phospholipid hydroperoxides (PHP) and H2O2, among other peroxides, via PHP GSH-peroxidase, an enzyme essential for life (20). GSH protects mitochondria from extra-mitochondrial ROS and detoxifies O2 •− and Fe = O• in a nonenzymatic fashion. The high reducing power of GSH makes it a major contributor to the recycling of other oxidants that have become oxidized and could be a basis by which GSH helps conserve lipid-phase antioxidants like α-tocopherol (vitamin E) (367).
Mitochondria appear to be the most susceptible foci in the GSH-depleted state. Changes in mitochondrial GSH status have been associated with activation of signaling pathways and expression of genes that regulate apoptosis, cell death, and differentiation (430). In recent studies, Aon and colleagues (25) reported that GSH depletion may be the ultimate factor determining the vulnerability to oxidant attack. Indeed, depletion of GSH has been associated with many degenerative diseases. This is evident by high levels of ROS production and activation of the mPTP that is independent of CsA, but is sensitive to CDZ, the PBR antagonist (25). These findings suggest the involvement of OMM proteins mediating the GSH depleted cellular damage, and highlight the significance of targeting the OMM as a way to mitigate mitochondria-related cellular injury. Inhibition of GSH transport with butyl malonate led to substantial depletion of mitochondrial GSH levels and could therefore render the mitochondrion more susceptible to oxidative stress (25) and subsequent cellular injury if there is an imbalance in the rate of ROS scavenging to the rate of O2 •− generation.
GSH deficiency has been linked to widespread mitochondrial damage, which is lethal in neonatal rats and guinea pigs deficient in ascorbate (368). The combination of GSH and ascorbate to remedy these defects is ascribed to the evidence that they spare one another. Ascorbate increases mitochondrial GSH in GSH-deficient animals and GSH delays the onset of ascorbate deficiency-related pathologies such as scurvy. In this way, GSH and ascorbate function together to protect against oxidative stress. Diabetic nephropathy is another example of the importance of GSH. The disease is characterized by mitochondrial dysfunction and decreased rates of GSH transport. In this case, a potential remedy is overexpression of mitochondrial GSH carriers to revert the phenotype to normal (149, 318). Specifically, overexpression of the GSH carrier in renal proximal tubular cells from diabetic rats improved mitochondrial function and redox state and resulted in normal mitochondrial function and reversal of diabetic nephropathy and other models of renal injury (318).
TRX has multiple actions; it is a small redox protein containing a thioredoxin active site localized in mitochondria and encoded by the nuclear genome (252, 575). Mitochondrial thioredoxin reductase-2 (TrxR-2) is ubiquitously expressed, but is present in larger concentrations in the brain, heart, and liver (491). TRX is reduced by TrxR-2, which utilizes the mitochondrial NADH/NAD redox state, similar to GR, as a source of reducing equivalents (Fig. 8). TrxR-2 protects against H2O2-induced cytotoxicity and regulates hemeoxygenase, a protein that prevents accumulation of heme and thus reduces oxidative stress associated with heme buildup. The mitochondrial TRX system appears essential during development because disruption of the Trx2 gene in the mouse results in massive apoptosis during early embryogenesis and embryonic lethality. Overexpression of cardiac TRX has been associated with improved post-ischemic ventricular recovery and reduced infarct size when compared to hearts from wild-type mice (180, 181). The TRX system has also been shown to have a potential therapeutic value against cardiac hypertrophy and cardiac failure, and as an antioxidant; it can also protect cells from oxidative stress by inducing MnSOD (19, 147, 294). Induction of MnSOD leads to increase H2O2 generation, which is converted by H2O2 detoxifying enzymes to H2O, and hence maintains oxidative stress within the physiological range.
Peroxiredoxins (PRX) are the recently discovered thioredoxin-dependent peroxide reductases, which reduce H2O2 and lipid hydroperoxides (102, 103, 345, 625). There are various isoforms of PRX in mammalian mitochondria including Prx3 and Prx5. A part of the mitochondrial TRX system is involved in maintaining the PRX proteins. Indeed, it has been reported that overexpression of CypD, a strategy targeting the mPTP, increases cells' resistance to oxidative stress-induced damage. This paradoxical effect can be explained in part by the fact that CypD can be an activator of TRX-mediated conversion of H2O2 via peroxidase (236).
The contribution to scavenging potential of mitochondria by these redox proteins is therefore dependent on the mitochondrial bioenergetic function. If the redox balance is compromised, increased ROS production could lead to detrimental oxidative injury to proteins and lipids, which are highly susceptible to oxidative damage. Thus, maintaining a large pool of reductants like GSH and TRX requires mitochondria to regenerate the reduced state via the NADH/NAD+ redox pair after detoxifying the ROS. Hence, supplying exogenous GSH or TRX-2 may be protective, but only if there are also sufficient reducing equivalents.
It is also important to note that, under certain pathological conditions, understanding the balance between the O2 •− producing side and its scavenging side is crucial for an effective therapy. In ischemia the excess O2 •− appears to be confined to the production side and less to the detoxifying side. This is evidenced by the apparent intactness of the scavenging capability of glutaredoxin or glutathione peroxidase (GPx) and MnSOD in isolated mitochondria during the ischemic insult (275, 541). Thus, targeting the ETC to attenuate electron transfer, and consequently to decrease the electron leak, may represent a better strategy than changing the matrix antioxidant capacity during ischemia.
C. Catalase and glutathione peroxidase
Catalase is present in cardiac cell mitochondria where it is thought to comprise about 0.025% of the total mitochondrial protein (20). Mitochondrial fractionation studies and quantitative electron microscopic immunocytochemistry showed that most of the catalase is located in the matrix. In myocardial tissue, catalase activity is lower and mitochondrial H2O2 production/g tissue is greater than in most organs (463). Catalase protects the organelle against intra- and extra-mitochondrial generated H2O2 (20). GPx, ubiquitously expressed in mammalian tissues and present in the mitochondrial matrix, seems to be the predominant H2O2 detoxifying agent in the heart. Catalase or GPx coupled to GR converts H2O2 to H2O (193) (Fig. 8). It is noteworthy that in mice, an increase in lifespan was found to be due to decreased mitochondrial ROS via greater expression of catalase in mitochondria (504). Recent studies have shown that targeting catalase to the mitochondria can reduce cardiac pathology (e.g., cardiomyopathy with age, and cardiac amyloidosis) consistent with the effect of endogenous ROS to decrease heart functional capacity with aging (577).
D. Cytochrome c
Cytochrome c is present at high concentrations in the IMS (>1 mM) (461) and it is one of the mediators of apoptosis. Cytochrome c can accept or donate an electron depending on the redox state of its heme (Fe). Thus, it is also a scavenger of O2 •− through its capacity to be reduced alternatively by the transfer of electrons or by O2 •−, thereby reducing ROS emission (20, 543). The reduced cytochrome c is reinstituted by donating its electron to cytochrome c oxidase. In I/R and neurodegenerative diseases, loss of cytochrome c inhibits respiration, which leads to increased electron leak (337, 652); the outcome is more O2 •− production and more cell damage. Another possible reason for an increase in O2 •− after mPTP opening is the loss of cytochrome c. It was shown that addition of exogenous cytochrome c to cytochrome c-depleted mitochondria reduced O2 •− levels by 7–8-fold (609, 652). In the cytochrome c-deficient Keilin–Hartree heart muscle model, Zhao et al. (652) showed that electron transfer through the ETC was attenuated and O2 •− generation was significantly greater in the mutant than in the wild type. Reconstituting cytochrome c in the cytochrome c-depleted hearts resulted in less O2 •− accumulation. Therefore, an adequate concentration of cytochrome c in the ETC is necessary to maintain ROS at physiological levels (652). In the I/R model, blocking electron transfer prevents O2 •− formation and preserves the integrity of the mitochondrial membrane including cardiolipin that functions to retain cytochrome c content (114, 115, 262, 324). Hence, maintaining the integrity of cytochrome c could represent a potential strategy for mitigating mitochondria-related cellular injury.
E. Mitochondria as scavengers of cytosolic O2 •−
Mitochondria are major scavengers of O2 •− produced from extra-mitochondrial sources. Mitochondria scavenge cytosolic O2 •− radicals by maintaining a polarized IMM that is positively charged during respiration. The [H+] in the IMS attracts the cellular O2 •−. Here, O2 •− radicals are protonated to form hydroperoxyl radicals (224, 495), which can diffuse into the matrix and become deprotonated so that the O2 •− is dismutated by matrix MnSOD. Net O2 •− consumption in mitochondria creates a gradient for O2 •−, which favors diffusion from the cytosol to the IMS. Therefore, increased MnSOD in mitochondria augments O2 •− removal from the cytosol as well as from the mitochondria. In this regard, it may not be a coincidence that mitochondria play a central role in cell death and that a lack of MnSOD in the mitochondria leads to cardiac and neuronal lethality.
VI. Uncoupling Proteins in Modulation of Mitochondrial Function: Physiological and Pharmacologic Relevance
Mitochondrial uncoupling is an important physiological regulator of its function and redox potential and thus, is also a regulator of O2 •− production. One of the control systems that regulate mitochondrial function and the flux of protons back across the IMM to maintain a suitable Δψm (379) is a group of uncoupling proteins (UCPs). The UCPs (1 –4) and fatty acids are believed to induce an inward H+ “leak” in energized mitochondria (70, 274, 543, 544). A key role for UCPs in regulating mitochondrial metabolism is supported by the presence of different isoforms in various mammalian tissues. UCPs prevent excessive ROS accumulation by maximizing respiration rate (401). The obscure roles of UCPs in normal physiology, and their emerging role in pathology, provide exciting potential for further investigation. However, neither the exact physiological nor biochemical roles of UCP homologues are well understood (342, 626).
Natural uncouplers like fatty acids and proteins, and artificial uncouplers like carbonyl cyanide m-chlorophenylhydrazone (CCCP), inhibit ROS production by decreasing Δψm (253, 536). UCPs are IMM proteins that dissipate the mitochondrial H+ gradient. Other biological effects of UCPs are their ability to attenuate mitochondrial ROS production (537) and to reduce the damaging effects of ROS during cardiac I/R or hypoxia injury (42, 188). In IPC and PPC, activation of putative mKATP channels may lead to mild uncoupling of mitochondrial respiration via mild “proton leak” with a concomitant increase in O2 •− generation that provides the signal for protection against ischemic damage (261). This form of “proton leak” is attenuated by GDP, a UCP inhibitor, suggesting that the H+ leak is mediated via UCPs.
In a recent study (253), we showed that a H+ leak instigated by the putative mitochondrial big KCa (mBKCa) channel in cardiomyocytes opening led to ROS production without disturbing Δψm. This is possible because the small H+ leak and the concomitant small increase in H+ pumping by ETC complexes increases the respiration rate without decreasing Δψm while increasing the rate of H2O2 production (253). It is possible that this “mild” uncoupling may contribute to the protection of the heart against I/R injury as shown by the BKCa channel agonist NS1619 (542). We showed that NS1619-induced protection was mediated by preservation of mitochondrial redox state, reduced mCa2+ loading, and better functional recovery; this protection was abolished when hearts were treated with MnTBAP and paxilline, the BKCa channel antagonist.
It has also been reported that overexpression of UCP2 protein in mice protects brain from severe infarction in I/R (154). UCP2 is thought to mediate such protection, possibly by causing mild mitochondrial depolarization that could limit mCa2+ uptake, reduce ROS production, and so protect cells from damage (177). It is feasible that UCPs could alleviate damage from I/R injury if they could be activated or stimulated before the insult to reduce Δψm, and before the redox increases because of inhibited electron transfer (543). Moreover, it is possible that shifting from state 4 to state 3 respiration could be sufficient to lower Δψm enough to have the same effects as uncoupling agents on ROS production. Thus maintaining a depolarized Δψm state is crucial in mitigating ROS-mediated injury.
Recent studies have shown that UCPs play an important role in the pathogenesis of obesity, type-2 diabetes, aging, and tumor progression (626). It was reported that the expression of UCPs increased in response to increased mitochondrial oxidative stress and that they serve as the link between diabetes and mitochondrial ROS (449). In endothelial cells, high glucose levels increase mitochondrial ROS, and the normalization of mitochondrial ROS production by inhibitors of mitochondrial metabolism, or by the overexpression of UCP-1, prevent the glucose-induced formation of advanced glycation end products that are believed to underlie major molecular diabetic complications (405). In contrast, in ob/ob mice that lack a functional UCP-2 gene, or when UCP-2-deficient mice were fed high fatty diets, glucose stimulated insulin secretion was enhanced compared to the wild type (342). These studies imply that UCPs may be important in insulin-glucose homeostasis and may contribute to impaired glucose-stimulated insulin secretion in diabetes. In human islet cells, chronic glucose or free fatty acid concentrations increase UCP-2 expression (342). These results are consistent with the finding that UCP-2 overexpression impairs β-cell function (342). Thus, minimizing UCP-2 activity in pancreatic β-cells could represent a valid and viable approach to improve β-cell function and to treat diabetes. However, such a conclusion is considered tenuous or “precocious” according to Anetor et al. (22), who reported an absence of significant oxidative stress in mitochondria, so that it was thought less likely that the UCP-2-superoxide pathway was involved in the inhibition of glucose-stimulated insulin release.
Another possible alternative approach to ameliorate ROS production is to modulate mitochondrial bioenergetics with low doses of artificial uncouplers. As discussed previously (543), ROS production is more likely when the Δψm is highly polarized. Therefore, any slight decrease in Δψm, as has been reported by most studies (309,317,543), or trans-matrix ΔpH (317), results in a marked reduction or cessation of ROS production. In this case, temporary partial uncoupling of respiration from phosphorylation by inducing a mild H+ “leak” has potential therapeutic benefit. For example, the use of dinitrophenol (DNP), which increases permeability of the IMM to protons, has been proposed and even attempted in treating obesity. However DNP has a low therapeutic index (narrow concentration difference between therapeutic potential and toxicity) that limits its utility as a viable therapeutic option (Section VI) (58). An alternative approach to using artificial uncouplers is the use of endogenous uncouplers that induce H+ leak (449). However, the development of this approach is dependent on unraveling the mechanisms involved in activating the uncoupling proteins.
Some lipid peroxidation products, such as 4-hydroxy-trans-2-noneal (HNE) may induce partial uncoupling of mitochondria through UCPs and are thought to initiate protective mechanisms (60, 182). HNE can also induce uncoupling of OXPHOS by enhancing H+ leak through other membrane proteins such as ANT if Δψm is high (32). It has been proposed recently by Brookes (69, 70) that ROS and H+ leak comprise a loop not requiring UCPs to operate, but rather is dependent on Δψm alone. A high Δψm would generate ROS and the ROS would, in turn, cause a H+ leak to reduce the Δψm in a feedback manner. The ROS so generated could induce H+ leak in part by lipid peroxidation or possibly by protonation of O2 •− in the acidic IMS to HO2 • (346, 543), which is membrane permeable and is deprotonated by the alkaline pH in the matrix (153, 543). A decrease in OXPHOS as a result of uncoupling and the less efficient metabolic rate associated with a H+ leak may be linked to the aging process (70) (Section XII). Taken together, these findings suggest a better understanding of the physiological roles and molecular mechanisms of uncoupling proteins and the consequence of H+ leak or H+ “slip” (70) to better understand the role of this phenomenon in mitochondrial function and dysfunction (449). A better knowledge of the mechanism of H+ leak by UCPs and synthetic uncouplers could provide the basis for better design of drugs that target this aspect of mitochondrial biology involving compromised mitochondrial function in the disease state.
VII. Mitochondrial DNA-Related Pathologies and a Potential Therapeutic Target
Mitochondria are inherited through the maternal lineage, though some recent evidence suggests that in rare instances mitochondria may also be inherited via a paternal route (506). Mitochondrial DNA (mtDNA) exists in hundreds of identical copies/cell (homoplasmy) but can also exist in multiple nonidentical copies within individual cells (heteroplasmy). Unlike nuclear DNA (nDNA), mitochondrial nonchromosomal DNA (mtDNA) does not get shuffled every generation, so it is presumed to change at a much slower rate (75). It is believed that mtDNA, organized in nucleoprotein complex (nucleoids), are particularly sensitive to oxidative stress due to its proximity to the ETC and the lack of histones (521, 634). Mitochondrial DNA also lacks introns, and as a result, mutations in the genome occur primarily in the coding sequence, consistent with the notion that mtDNA has a higher mutation rate (10–20 times) than nDNA (50, 221). For these reasons, accumulation of mtDNA damage plays a causative role in various disorders that are associated with aging, cancer, neurodegenerative diseases, and other diseases (634).
However, based on the organization of mtDNA into nucleoids and their proximity to the ETC, nucleoids may have an integrated antioxidant system to protect mtDNA from oxidative stress (293). Indeed, Kienhofer et al. (293) recently confirmed the presence of SOD2 in the nucleoid and noted that it binds directly to mtDNA through ionic binding forces. This mtDNA–SOD2 interaction in the nucleoid protected against oxidative damage as evidenced by the increased vulnerability of free mtDNA to X-ray and H2O2 than mtDNA in a nucleoid complex. The mtDNA–SOD2 interaction may also explain in part the enhanced ROS levels, the increased mtDNA damage, the reduced activities of the ETC complexes and TCA cycle enzymes, and endothelial dysfunction observed in MnSOD+/− mice (614).
Much of the mtDNA is used to code the manufacture of proteins that are key components of the energy production system. All 13 proteins encoded by mtDNA contribute subunit components to most of the respiratory complexes. mtDNA encodes 7 subunits of complex I, 1 subunit of complex III, 3 subunits of complex IV, and 2 subunits of complex V. Cells depleted of mtDNA, ρ0-cells, lack some of the critical subunits of the ETC complexes, which result in a defective respiratory system evidenced by a reduced Δψm. These cells are resistant to anoxia-induced cell death (lack of ROS) and they are dependent exclusively on anaerobic energy production via glycolysis (312, 524), much like tumor cells.
Most mitochondrial proteins that regulate replication and repair are encoded by the nuclear genome (540). Complex I, the largest holoenzyme in the ETC, has 45 nuclear-encoded and 7 mitochondria-derived subunits (438). Complex II consists only of nuclear-encoded subunits, and complex III consists of 10 nuclear-encoded and 1 mitochondria-derived subunits. Complex IV has 7–11 nuclear-encoded subunits, depending on the tissue type, and 3 subunits are mitochondria derived (323, 598) as noted above.
Currently, it is believed that various mutations are responsible for more than 120 syndromes associated with mitochondrial proteins (357, 433) (Section IX). There are also a number of mitochondrial diseases associated with specific mutations in mtDNA or in nDNA coding for mitochondrial proteins (165, 176, 515) (Table 2). In other diseases like PD, type 2 diabetes, and cancer, the disease process is more complex and involves multiple genetic and environmental causes (178, 423, 439, 458). In all these diseases, the genetic mutations lead to impaired mitochondrial energy-generating machinery. In this scenario, disabled mitochondria with impaired ETC would cause electron leak and increased O2 •− production. Indeed, the most common source of somatic mutation of mtDNA is O2 •− generated from the ETC. The O2 •− produced in the mitochondrion can continue in a self-perpetuating process leading to even more damage and more O2 •− generation (Section IV,A). It is likely that gene replacement has potential to be used to correct a mutant mitochondrial genome similar to classical gene transfer therapies that have replaced defective nuclear genes (509). Indeed, genetic maneuvers of different sorts have been employed to reverse mitochondrial related diseases. These include, but are by no means limited to: a) DNA coupled covalently to mitochondrial leader peptides (chimeras) that enter mitochondria through protein import pathways; b) manipulating mtDNA replication by import into mitochondria endonucleases that might selectively destroy a specific mutant sequence; and c) suppression of mutant mtDNA expansion to alternatively salvage OXPHOS (498, 655).
Mitochondrial DNA has the capacity to form a mixture of both wild-type and mutant DNA genotypes within a cell (heteroplasmy). Cellular dysfunction usually occurs when the ratio of mutated to wild-type mtDNA exceeds a threshold level (56). Mitochondrial disorders of the heteroplasmic type and the associated disease could be remedied by selectively blocking the replication of mutant DNA molecules to allow repopulation of the wild-type mtDNA (567). On the other hand, a potential therapeutic maneuver for destroying fast growing tumor cells involves the use of ethidium bromide to deplete mtDNA and the antibiotic ciprofloxacin to block OXPHOS and deplete ATP needed for cell proliferation (28, 29, 100, 320).
Mitochondria have developed a complex system to import proteins. The import of nuclear encoded mitochondrial proteins is based on specific targeting sequences (498) that could be altered by mutation of the nucleotide base in the nuclear genome. The protein import pathway has been used to direct the import of chimeric proteins into mitochondria. It could also be used to direct the appropriate mitochondrial gene sequences to mitochondria to correct for a defective protein (28, 29). In this case, the use of mitochondrial protein importation machinery may lead to novel mtDNA delivery strategies (Section VIII). A number of mitochondria-related diseases could be linked to faults in the transcription-translational process, in the mitochondrial importation of proteins following post-translational modification, or simply due to mutation of the mitochondrial genome. Defects in the import system are rare, but mutations of genes involved in the mitochondrial import machinery have been shown to be the cause of several maladies, including the Mohr–Tranebjaerg syndrome (sensorineural deafness, dystonia, dysphagia, cortical blindness, and paranoia) (498). Abnormalities in mitochondrial protein import have also been implicated in neurological disorders such as AD (159). A detailed description of mitochondrial and nuclear genomic related diseases is provided in the literature and will not be described here beyond cursory observations.
VIII. Mitochondrial Interaction with Other Organelles: Therapeutic Implications
Mitochondria often form a 3-D branching network inside cells along the cytoskeleton with tight connections to other organelles. This association with the cytoskeleton determines mitochondrial shape and function (466) and ultimately proper cellular function (21, 26). This anatomical connection provides for a dynamic feature in mitochondrial biology. Mitochondria in the cell tend to have anatomical and functional connectivity with each other and with other organelles such as the nucleus and the endoplasmic reticulum (ER). These dynamic functional interconnections are essential for normal function of mitochondria and play a vital role in preserving cellular function and integrity. It is therefore anticipated that derangement of this link could be involved in pathologic states associated with mitochondrial dysfunction. Thus, exploitation of this dynamic anatomic relationship could represent a new target for potential therapy in altering mitochondrial-mediated cell death.
A. Mitochondrion–mitochondrion interaction
Mitochondrial shape, size, and number vary from organ to organ, tissue-to-tissue, and cell-to-cell, and they vary under physiological and pathological conditions. In vivo, mitochondria often merge to form a reticulated network that under physiological conditions are governed by the dynamic equilibrium between fusion and fission. Indeed, the entire mitochondrial population is in constant flux and the identity of an individual mitochondrion is influenced by a nearby mitochondrion with its potentially different mitochondrial genome (heteroplasmy) (105). Interestingly, proteins such as GTPases, kinases, and phosphatases are involved in bi-directional communication between the mitochondrial reticular network and other organelles and the rest of the cell (365).
In recent years this new emerging concept has provided increased molecular definition to the mitochondrial network as a central platform in the execution of diverse cellular functions (i.e., from maintaining normal cell function to initiating cell damage). With this comes a new concept focused on the idea that mitochondrial dynamics (membrane fusion/fission) is a potential target in mitigating mitochondrial related diseases. The ΔΨm is essential in preserving and maintaining the dynamic process involved in establishing the mitochondrial network, which in turn is important for unifying function and responses to intracellular signals (314). This dynamic relationship is maintained by specific proteins whose presence is important in maintaining and regulating the integrity of the IMM and cytochrome c, both of which are necessary to maintain mitochondrial respiration by generation of the H+ gradient necessary to establish ΔΨm.
Mitochondrial fusion and fission genes have been described recently (105). The mitochondrial fusion proteins, encoded by mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2), help to regulate mitochondrial function and to maintain OMM and IMM fusion between mitochondria. To achieve fusion, the proton gradient of the IMM, but not the ΔΨm, is required for the connection of the OMM and to achieve IMM fusion; a large ΔΨm and GTP must be available (29, 105). The dynamins mitofusin and optic atrophy 1 (OPA-1) are GTPase proteins associated with the OMM and IMM, respectively, and are essential for mitochondrial fusion (562). The morphological orientation of mitochondria is relevant in regulating cellular apoptosis with increased fragmentation of the network leading to an increased tendency for cell death (365, 562). In normal cells where ΔΨm is maintained, mitochondrial fusion in a tubular network is maintained. In cells derived from disease states that show compromise in the ΔΨm as a result of a defect in the ETC complexes and reduced respiration, the cells show a degree of mitochondrial network disintegration with increased fragmentation leading to increased cell death (365, 562). Loss of function of Mfn2 results in decreased substrate oxidation and ΔΨm as a result of repression of nuclear encoded proteins involved in OXPHOS. Thus, the machinery that governs mitochondrial dynamics also participates in the temporal regulation of metabolism (365).
Defects in mitochondrial fusion proteins and subsequent fragmentation of the mitochondrial tubular network are implicated in numerous mitochondrial myopathies, including neurodegenerative diseases (314) and I/R injury (24, 65). For example, a set of mutations in Mfn2 leads to Charcot–Marie–Tooth type 2A, a peripheral neuropathy characterized by axonal degeneration (372 –374). In mouse embryonic fibroblasts, a deficiency in Mfn1 or 2 leads to accumulation of fragmented mitochondria and embryonic lethality, whereas overexpression of either one of the mitofusins restores the mitochondrial tubular network (151, 152, 373). In another study it was shown that a muscle-specific knockdown of the Drosophila homologue of Mfn (Marf ) or OPA-1 results in significant mitochondrial fragmentation and damaged cristae (155). In all these abnormalities, the common denominator is alteration of ΔΨm because of reduced activity of the complexes. The process is reversed by OPA-1, which prevents release of cytochrome c and maintains IMM integrity.
Mitochondrial fission (fragmentation) depends on the mitochondrial fission proteins (Fis1) and another GTPase dynamin family protein, DRP-1. These mitochondrial membrane proteins, when present, mark the spot where fragmentation is initiated. Overexpression of Fis1 proteins in normal wild-type cells resulted in increased fragmentation and increased apoptosis as a result of increased Bax and Bak on the OMM and subsequent cytochrome c release (105, 314, 498). Overexpression of DRP-1 has been associated with significant fragmentation and disruption of IMM and matrix cristae (155). These proteins provide an intriguing link between pro- and anti-apoptotic proteins and permeabilization of the OMM. Mitochondrial fission also plays an important role in normal mitochondrial function. For example, it has been reported that disruption of fission proteins might lead to abnormal mitochondrial function (105, 314, 498). Clearly, normal Δψm and electron transfer have local effects on mitochondrial structure, networking design, and normal function. Thus, membrane dynamic proteins of the IMM are also intimately involved in apoptotic regulation and represent potentially novel targets for therapeutic intervention.
In many diseases, in which a less polarized Δψm may contribute to mitochondrial integrity, a strategy to maintain integrity of the ETC with normal electron flux is a therapeutic option. Moreover, the site within the ETC where the defect lies may be critical in the design of drugs to target mitochondria. For example, in PD defects in complexes I and III are associated with compromised Δψm and may contribute to subsequent fragmentation of mitochondrial connectivity (40, 304, 497, 557). On the other hand, a defect in complex V in the disease NARP (neuropathy, ataxia, and retinitis pigmentosa) does not alter Δψm. Impaired levels of Bcl-2 anti-apoptotic proteins are also characteristic of NARP (314). A possible therapeutic approach in the situation, where Δψm instability is a contributing factor in mitochondrial fission, is to provide substrates like N,N,N′,N′-tetramethyl p-phenylenediamine to restore electron flux and thereby reestablish membrane functional integrity and Δψm. A note of caution is that this type of pharmacological maneuver may not be suitable for diseases that are not linked to specific defects in the ETC. It is therefore essential to understand the specific underlying cause of the defect to properly design the appropriate therapy. Another approach to reduce cytotoxicity involves overexpression of the fusion genes and inhibition of the fission genes in a replicative cell line (606). However, this approach will not likely work in mature post mitotic cells.
B. Mitochondrion–nucleus interaction
The existence of two spatially separated genomes, each contributing, albeit asymmetrically, to the biogenesis of mitochondria, has led to the suggestion that the two organelles interact to provide a coordinated cellular response to intracellular changes. How the nucleus and mitochondria interact, how mtDNA and nDNA gene expression is coordinated, or how mtDNA is maintained within the cell (a process strictly driven by nuclear factors), are key factors necessary for understanding the molecular mechanisms underlying many disease states. The nucleus regulates numerous mitochondrial functions because a majority of mitochondrial proteins are encoded by the nuclear genome. Thus, disruption in the line of communication between the two organelles can lead to initiation of adaptive responses to reorganize cellular metabolism. The mitochondrial proteins encoded by the nuclear genome are synthesized on cytoplasmic ribosomes and the peptides are imported into the matrix after specific post-translational modifications. Import of nuclear encoded proteins or precursor peptides relies on a proper function of the mitochondrial protein uptake machinery, including chaperones and transmembrane peptide import systems.
Mitochondrial oxidative stress that leads to the accumulation of unfolded proteins in the matrix subsequently leads to the upregulation of nuclear-encoded chaperone genes that are transcribed and imported into the mitochondria. Mitochondrial chaperones, chaperonin 60 and chaperonin 10, are upregulated via activation of the transcription factor CHOP, a CEBP-β analogue (650). The mitochondrial factor that leads to CHOP transcription remains unclear. The response is specific since chaperonin 10 and 60 are induced, but HSP70, another mitochondrial chaperone is not. Thus, mitochondria can signal the nucleus in a specific fashion in order to mount a stress response that is specific to the mitochondrial compartment.
The transmembrane peptide import systems (i.e., the outer and inner membrane translocases (TOM and TIM, respectively)), import proteins determined by the amino terminal sequences of the pre-protein. The direction of proteins to specific mitochondrial compartments contributes to regulation of mitochondrial metabolism (305, 330, 392). Thus, a coordinated protein import system is essential for mitochondrial maintenance and biogenesis; a disturbance of this communication between these organelles may underlie a disease process.
Mitochondria–nuclear communication has been implicated in modulating mitochondrial biogenesis and intra-mCa2+ distribution and swelling (550). A nuclear encoded protein, the peroxisome proliferator-activated receptor-gamma coactivator-1α (PGC-1α), is a transcriptional co-activator that modulates mitochondrial biogenesis and in this way regulates cellular metabolism. One of many functions of PGC-1α is regulation of nuclear transcription factors, NRF-1 and -2 and mitochondrial transcription factor-1, a protein that transfers from the nucleus to the mitochondria where it promotes an increased production of mitochondrially encoded proteins and the replication of mtDNA (177, 589). PGC-1α also enhances the activity of mitochondrial specific scavengers, including MnSOD and GPx1, during oxidative stress (537, 589). Indeed, increasing PGC-1α levels markedly enhances cell survival from oxidative stress. St-Pierre et al. (537) showed that PGC-1α levels are regulated by mitochondrial ROS which in turn activates the complex and multifaceted ROS scavenging system. The implication of PGC-1α in regulating mitochondrial function, while at the same time minimizing ROS production, makes it an ideal protein to control or to limit mitochondrial damage. A deficiency in PGC-1α in the brain renders it susceptible to neurodegeneration with apoptotic cell death and increased oxidative injury (537). On the other hand, endothelial cells with overexpressed PGC1-α exhibit reduced accumulation of ROS, increased Δψm, and reduced cell death (589). The role of PGC-1α as a regulatory function in lipid metabolism also makes the protein a target for pharmacological intervention in the treatment of type-2 diabetes (Section IX,C).
PGC-1α regulates nuclear gene transcription through a mCa2+ signal to the nucleus and conversely, PGC-1α levels are partly regulated by cytosolic Ca2+ (94, 550). This suggests that a feedback regulation operates between mitochondria and the nucleus through cellular and mCa2+ signaling. In addition, the release of cytochrome c, and the subsequent activation of caspase activity, ultimately lead to nuclear fragmentation, chromatin condensation, and the formation of apoptotic bodies and programmed cell death. Another signaling agent between mitochondria and the nucleus is H2O2, which can diffuse to the nucleus where it acts as a mitogen at low concentrations (602). Therefore a strategy involving overexpression of PGC-1α that is modulated by Ca2+ and other signaling pathways suggests a multifaceted therapeutic approach in managing cell death under oxidative stress. Targeting the mitochondria by inducing PGC-1α to increase ETC activity, and at the same time minimizing net ROS production, makes it an ideal protein to reduce damage in PD and AD. This strategy could be more relevant to the brain where increasing the levels of a specific protein is a challenge (537).
C. Mitochondria–endoplasmic/sarcoplasmic reticulum interaction
Mitochondria–endoplasmic reticulum (ER) communication is a vital component in the structure and function of the mitochondria. For example, many of the lipids of the IMM and OMM are not synthesized in the mitochondria but rather are imported from the ER to the OMM. Since mitochondria are not connected to other organelles through vesicular trafficking pathways, mitochondria–ER communication is via discrete sites of close apposition that would facilitate lipid and Ca2+ exchange between the organelles (308). A complex molecular tether that associates the OMM with the ER mediates the interorganelle communication. In a recent study, Kornmann et al. (308) developed an elegant genetic screen that identified the Mmm1/Mdm10/Mdm12/Mdm34 complex as the tether between ER and mitochondria. The Mmm1 (maintenance of mitochondrial morphology protein 1) of the complex connects the ER to mitochondria and the other core molecules maintain mitochondrial shape and structural framework to connect the two organelles. It is postulated that the ER–mitochondria junction may also influence the structural organization of the IMM, mtDNA, and the regulation of mitochondrial protein import (308, 617, 618).
Mitochondrial Ca2+ dynamics as well as generation of ROS are important events during the course of cellular injury and cell death. Mehrotra et al. (366) reported that oxidative stress potentiates membrane damage induced by Ca2+. The anatomical proximity between mitochondria and ER creates a local microdomain in which the local mitochondrial [ATP] within the microdomain is required for ER Ca2+ signaling (177, 550). This proximity of mitochondria to Ca2+ release sites has functional consequences for intracellular Ca2+ signaling. Mitochondrial uncouplers and the complex V blocker oligomycin are known to blunt ER Ca2+ uptake. In reverse order, ER supplies Ca2+ to mitochondria as needed for control of mitochondrial metabolism or to instigate apoptosis/necrosis (167, 550), depending on ER Ca2+ content. This “dynamic duo” is critical in preserving the functional integrity of the cell.
Despite the physiological significance of ER-mitochondria microdomain, little is known about the molecules that regulate Ca2+ dynamics between the two organelles. It has been proposed that these interactions are modulated in part by locally generated mitochondrial ROS, which are believed to act as the trigger to regulate the Ca2+ flux between the organelles (605, 610). It is believed that ROS increases the probability of Ca2+ release from the ER, probably through modulation of thiol groups on ryanodine, or via the ER IP3 receptor (IP3R) (177). Other possible molecules that regulate the mitochondria–ER contacts include the “sorting” protein, phosphofurin acidic cluster sorting protein-2 (PACS-2). PACS-2 helps mitochondria to maintain their membrane integrity and to translocate Bid from the cytosol to mitochondria in response to apoptotic inducers (526). A defect in PACS-2 results in uncoupling of mitochondria from ER and mitochondrial fragmentation (Section VIII,B). Recent studies also reveal that the mitochondrial VDAC, mitofusins, and the IP3R, provide specific interactions between the organelles (151, 152, 373, 550). Indeed, it was reported that after IP3-triggered Ca2+ release from the ER, the uptake into Mfn2-deficient mitochondria was markedly diminished (151, 152, 373).
ER–mitochondria apposition ultimately affects Ca2+ signaling and amplification of apoptotic signals (151). In the early apoptotic process, it is believed that Ca2+ release from the ER activates the mPTP and causes cytochrome c release; the cytochrome c then binds to the IP3R resulting in an unrestrained increase in cytosolic [Ca2+]; the subsequent increase in cytosolic [Ca2+] leads to mCa2+ uptake (151, 152, 550), which would lead to a further increase in cytochrome c release (204) and ultimately cell damage. It is therefore conceivable that interrupting this interaction between mitochondria and the ER could mitigate mitochondrial-mediated cell injury.
As an example, it has been shown that Bcl family of proteins, which localize to the ER, interact with the IP3R to modulate its phosphorylation state (204); this action could reduce the feed-forward system that amplifies the release of cytochrome c and mCa2+ load (167, 168). In this case, overexpression of anti-apoptotic Bcl-2 or Bcl-xl, or ablation of pro-apoptotic Bax and Bak, reduced ER calcium content by increasing the leak of Ca2+ through IP3 receptors and protected against cell death in vitro (615). Recent studies have shown that anti-apoptotic Bcl-2 and Bcl-Xl reduced ER Ca2+ content by binding to and sensitizing IP3 receptors (615). The increased Ca2+ leak may not alter m[Ca2+] but could lead to a moderate elevation in cytosolic [Ca2+], which unlike excessive cytosolic [Ca2+] may protect against cell injury. Conversely, Bax and Bak overexpression enhances the ER-mitochondria Ca2+ transfer and lowers the threshold for mitochondrial apoptosis (413). The chaperone protein HSP 70 appears to have a cytoprotective function by inhibiting the apoptosis induced by various insults. One possible anti-apoptotic mechanism suggests that it blocks Bax translocation (204) to the ER and reduces the initial cytochrome c -mediated Ca2+ release.
In a recent study, de Brito and Scorrano (151, 152) showed complementary mechanisms for the Bcl family protein NIX-mediated cardiac cell death that involve direct ER–mitochondrial disruption. Since it is believed that an important mechanism of apoptotic cell death involves the mPTP (167, 168), a strategy of combined protection of mitochondria and ER may represent an innovative therapeutic approach to enhance organ viability and functional integrity (176). This approach will likely involve pharmacological modulation and or genetic manipulation of anti-apoptotic proteins.
IX. Mitochondria-Related Diseases and Cell Injury
Studies in the past decade have identified a host of common maladies with apparent links to mitochondria. These diseases have been linked to defects in nuclear genes, mitochondrial genes, or potentially a combination of the two (121, 502). The mitochondrion is essential for ATP production; hence, when the production of ATP is impaired through mutation of a gene encoding a specific polypeptide involved in OXPHOS, tissues that rely heavily on high levels of ATP, such as the brain and heart, are most affected. Other organs beside the heart are highly susceptible to mitochondria-related dysfunction; these include liver, skeletal muscle, kidney, and the endocrine and respiratory systems. In the brain, this can be phenotypically observed as degeneration of motor neurons [e.g., amyotrophic lateral sclerosis (ALS) and Friedreich's ataxia (FA)], tremor [e.g., Parkinson's disease (PD)], and progressive dementia [e.g., Alzheimer's disease (AD)] (6, 39, 166, 225, 247, 254, 384, 502, 634) (Section IX,E). In heart failure, defects have been purported to occur in ETC complexes or in components of the OXPHOS machinery. These alterations are usually manifested frequently as cardiomyopathy (481). Concomitant with impaired OXPHOS is increased O2 •− generation and mCa2+ overload, which lead to the “vicious cycle” hypothesis of mitochondrial dysfunction (348) that may underlie the maladies associated with mitochondrial related disorders.
Mitochondrial metabolism and the OXPHOS cascade are emerging as key features in the generation of ROS associated with a large number of diseases. Thus, the basic role of mitochondria in sustaining the normal cellular function in every tissue and organ has made dysfunction of this organelle a central feature of numerous diseases in any organ system at any stage of life (391). In fact the term “mitochondrial medicine” has emerged recently as an active field of research and clinical trials (531). Derangement of mitochondrial function and loss of mitochondrial cell volume are now associated with several human disorders categorized as mitochondrial cytopathies (440, 501), of which mitochondrial encephalopathy, caused in part by a point mutation manifested as a defect in cytochrome c oxidase function, is a notable example. The phenotypic presentation of mitochondrial diseases includes a wide range of clinical manifestations that can affect all body tissues and the onset of these ailments varies from early infancy to senescence. However, the mystery remains as to how tissue or cell type specificity occurs, and how a systemic disorder of one mitochondrial complex can cause a selective disease phenotype while leaving other tissues intact. For example, why do only voluntary motor neurons die in ALS, or only basal ganglia neurons in PD?
It is believed that many of the mitochondria-related diseases might actually be an expression of progressive organ system failure due to disruption of specific aspects of mitochondrial function (391). Mitochondrial DNA is a particularly vulnerable target because of its proximity to the ETC and ROS. Thus, in most cases mitochondria-related diseases result from mutations in mtDNA (Section VII) due to oxidative stress. Most mitochondrial diseases manifest themselves primarily in young children, but the impaired metabolic function of mitochondria in adult onset diabetes has become much more commonly appreciated (232). In other mitochondrial diseases, the underlying mitochondrial mutations accumulate over time and the dysfunction that becomes evident correlate with the increasing fraction of mutated mtDNA (391). These diseases appear to be more prominent during later life in brain, skeletal muscle, and heart where defects accumulate more extensively than in rapidly dividing cells (391). In these postmitotic cells, mitochondria must remain in steady state, by either not replicating, or by at least maintaining a balance between degradation and replication.
Rare mutations in mitochondrial proteins can cause severe multisystem failure (540). A single mutation can lead to different syndromes, whereas the same phenotype can be caused by different mutations, so many aspects of mitochondrial diseases remain a mystery to date (540). Duchen (177) argues that the challenge facing experts engaged in the study of mitochondria-related diseases is to fully appreciate and understand the extent to which changes in mitochondrial function represent primary vs. secondary components of the pathological process, and to fully understand the processes that lead to manifestation of the disease. Each disease (e.g., cancer, type 2 diabetes, ischemic heart disease, and even aging) has unique triggers and symptoms, and establishing the causal link with mitochondria is still in its infancy. However, attempts to unravel this link, even though tenuous, have led to some drug designs that target the mitochondrion.
It suffices to say that the subject of mitochondrial diseases has been discussed extensively in the literature. In this review, we offer only a brief discussion of a few selected mitochondria-related diseases. Table 2 summarizes some of these diseases and their possible etiologies. Even though dysfunctional mitochondria appear to be a common underlying problem for all these diseases, they each exhibit unique triggers and symptoms. Some of the diseases to be discussed further in this review include cardiac I/R injury, heart failure, hypertension, neurodegenerative diseases (PD, AD, and ALS), diabetes, psychiatric disorders, and migraine headache; potential therapeutic approaches targeting mitochondria will also be discussed.
A. Mitochondria and cardiac ischemia and reperfusion injury
Mitochondria are crucial regulators of life and death in a variety of cells and play a pivotal role in cardiomyocyte death in response to cardiac I/R injury. During ischemia, ETC complex activity is depressed as a result of damage to cardiolipin, ETC complexes, and increased H+ leak in the IMM, thereby compromising its ability to maintain ΔΨm and to provide a sustained energy supply. This leads to impaired ATP-dependent ion pumps required to maintain ion homeostasis. Intracellular acidosis produced during ischemia quickly recovers on reperfusion and this leads to increases in intracellular [Na+] and [Ca2+] (Section II.B). The alteration of cytosolic Na+ and Ca2+ eventually predisposes mitochondria to dysregulation of ion homeostasis and increased mCa2+ load. A large increase in matrix [Ca2+] (Section II.B) and recovery of matrix pH (less acidic) might alter both the function and structure of mitochondria and contribute to opening of the mPTP (Section II.C) and subsequent loss of ΔΨm. Other alterations of mitochondrial function observed during I/R include changes in mitochondrial ROS production and redox state.
As noted in Section IV.A, ROS are a normal byproduct of mitochondrial respiration. Mitochondria generate cytotoxic amounts of ROS during cardiac ischemia mainly through the ETC (Section III), as shown by damage to the respiratory complexes (117, 327, 332, 407) and the use of mitochondrial inhibitors (83, 91, 113, 118). Extra-mitochondrial sources of ROS including NAD(P)H oxidase (219, 299, 624) and xanthine oxidase in vasculature (46, 571) are other likely sources of ROS. However, our recent experiments using MnTBAP (SOD2 mimetic) in isolated hearts indicates that cardiomyocyte mitochondria are likely the main source of ROS during cardiac ischemia (289). This subject is discussed in greater detail in our recent review article (543), and by others (14, 591, 592), and will not be covered in any more detail here.
It is often stressed that excess ROS and mCa2+ overload are the two major factors that are intertwined in the pathology of I/R injury. But how they are interrelated or how they influence each other is a subject of intense debate. It suffices to state here that mCa2+ overload leads to inhibition of the major matrix scavenging enzymes and in this way may increase net ROS production. Mitochondrial Ca2+ overload could also lead to mPTP opening and loss of GSH with dissipation of ΔΨm and NADH; these are all key factors involved in maintaining the redox balance in the GSH/GSSG system and efficient scavenging capacity (20, 391) (Fig. 8).
The association of O2 •− generation with I/R injury has made the development of antioxidants as therapeutic targets a pre-eminent goal (Section X). For example, it has been proposed that an ideal strategy would be to boost ROS scavenging by using nontoxic catalytic antioxidants that are delivered tissue-specifically or produced from inactive precursors (391) (Sections X and XI). Enhancing the endogenous levels of the GSH pool is a viable strategy to protect against mitochondria-related cellular injury. Indeed, we showed that administering a cocktail of mitochondrial scavengers (MnTBAP + glutathione + catalase) prior to 2 h cold cardiac ischemia followed by 2 h warm reperfusion provided better protection against mCa2+ overload and ROS production and better preservation of cardiac function than did MnTBAP alone, which only dismutates O2 •− to H2O2 (90). Other potentially beneficial strategies may involve decreasing the primary ROS production by preventing the overproduction of NADH, which could help minimize ROS production by the use of mild uncouplers (Section V) (20); or, pharmacologically directed attempts could be made to stimulate the expression of endogenous mitochondrial and intracellular antioxidants.
The limited O2 availability during ischemia or hypoxia leads to a shift of glucose metabolism from OXPHOS to substrate level phosphorylation (glycolysis), and prolonged ischemia leads to accumulation of lactic acid and depletion of NAD+. An insufficient O2 supply also results in decreased NADH dehydrogenase (160, 162), which appears to be associated with inhibited electron transfer at the mitochondrial cytochrome a,a3 complex of the ETC. This could lead to an increase in NADH and a decrease in FAD; that is, during cardiac ischemia, mitochondria are in a relatively reduced state. Consistent with this notion, we observed that during ischemia NADH increases (reduced mitochondria) but on reperfusion, we observed that NADH and FAD fluorescence signals pointed to more mitochondrial oxidation (9, 10, 90, 475, 542). The implications of our results are that a reduced redox state in the cardiac cell could be an important condition for efficient resumption of the H+ gradient for ATP synthesis and hence for better functional return. Prevention of ATP hydrolysis to maintain ΔΨm during ischemia is therefore a valid goal for preserving function.
B. Mitochondria and the failing heart
Substrate metabolism is dysfunctional in the failing heart. ATP synthesis in the healthy mammalian heart is primarily oxidative with greater than 95% of ATP synthesized in the mitochondria via OXPHOS (416, 539, 645). The major sources of mitochondrial fuel are illustrated in Figure 11; acetyl-CoA that supplies the TCA cycle is derived from β-oxidation of fatty acids and from dehydrogenation of pyruvate that is supplied by glycolysis, glycogenolysis, and lactate oxidation. Under normal conditions, between approximately 60% and 90% of the acetyl-CoA consumed by the heart is derived from fatty acids, with the remainder derived primarily from pyruvate (208, 538, 539). Under different stressful conditions, including ischemia and hypoxia, the heart switches from a dependence on fatty acid metabolism toward utilization of glycolytic metabolism (Fig. 11). This substrate switch makes adaptive sense because under stressful conditions, the limited O2 supply makes oxidation of glucose yielding glycolytic ATP more efficient than fat oxidation. The ability to switch substrate utilization by a number of regulatory mechanisms is a hallmark of a healthy heart (557).
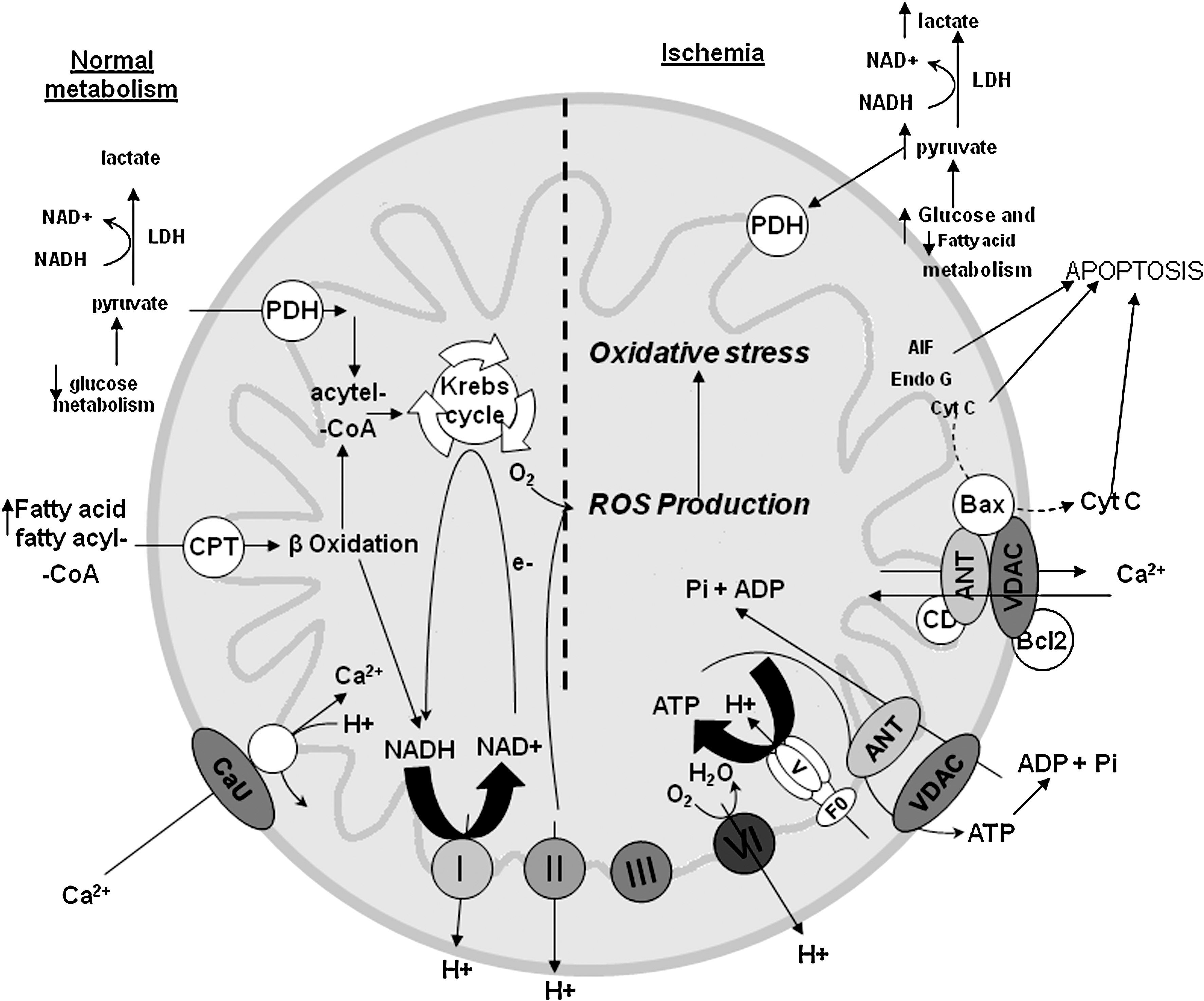
In contrast, remodeling in failing hearts results in impaired ability to oxidize both fatty acids and glucose (539). This is due to a downregulation of enzymes involved in β-oxidation (469, 493, 539), along with an impaired ability to utilize glucose due to suppression of glycolytic activity, and to decreased ability of the cardiomyocytes to take up glucose (559). In the extreme, the work capacity of the heart is limited not by the availability of metabolic substrates or O2, but by the impaired ability to consume the available substrates (267, 539). As Taegtmeyer noted it, “… the heart fails in the midst of plenty” (558). The acute and chronic maladaptation of certain metabolic enzymes in the disease state occurs on a background of normal physiological metabolic regulation.
The mitochondrial content and hence oxidative capacity of both skeletal and cardiac muscle cells are diminished in heart failure (203). This results in limited work in the failing heart due to limited free mitochondrial energy (214, 416). Indeed mitochondria isolated from explanted human hearts exhibiting severe cardiomyopathies show significant depression of state 3 respiration and lower respiratory control indices (517). Thus, the altered metabolic pattern observed in heart failure has a clear impact on the energetic state of the heart. The potential consequences of a diminished energetic state include an impaired ability of the heart to work and respond to acute and chronic stresses. Metabolic dysfunction can precede and may play a role in initiating structural remodeling and mechanical malfunction (191, 561). For example, evidence exists that metabolic remodeling precedes and contributes to inducing functional and structural remodeling of the heart in diabetes (560, 640). Thus, therapeutic strategies for metabolic modulation are potentially of great value.
An important treatment goal is attenuation or reversal of postinfarction remodeling and heart failure. Interestingly, drugs that inhibit NHE appear to provide benefit via a mitochondrial mechanism. Rats subjected to coronary artery ligation exhibited, 12 or 18 weeks later, cardiac hypertrophy, and increased mPTP opening and decreased state 2 and 3 respiration with complex I and II substrates in mitochondria isolated from these hearts (271). Daily postligation oral treatment with a NHE-1 inhibitor largely attenuated the decreases in respiration and reduced mPTP opening. The mechanism of mitochondrial protection by NHE-1 inhibitors in this model (272) appears due in part to attenuation of the downregulation (lower mRNA levels) of mitochondrial transcription factors that accompany hypertrophy and remodeling (i.e., nuclear respiratory factor 1 and 2, transcription factor A, mitochondrial encoded cytochrome c oxidase subunit 1, nuclear encoded cytochrome c oxidase subunit IV, and PGC-1α).
Although the idea of treating metabolic cardiac dysfunction is not new, the targeting of mitochondria in cardiomyocytes has gained an unprecedented degree of interest lately. A variety of metabolically targeted therapies have already been applied to improve cardiac metabolic function clinically. These include, but are not limited to, stimulation of pyruvate dehydrogenase (PDH) by inhibiting pyruvate kinase (54) with dichloroacetate (DCA). This approach has been shown to increase lactate uptake in the heart and to increase mechanical work and efficiency in heart failure patients (54). Whereas the use of DCA is very effective in stimulating pyruvate oxidation, its use is limited by its low potency (blood levels in mM range) and short half-life (538).
Several drugs that inhibit fatty acid oxidation have shown promise for a number of cardiac disorders. For example, etomoxir (inhibitor of the fatty acid transporter carnitine palmitoyl-transferase 1 (CPT 1) has been tried clinically to treat chronic heart failure (288, 623). However, long-term usage of this drug is associated with toxicity and heart failure (538). Other drugs, such as trimetazidine, commonly prescribed as an anti-angina drug (68, 171), and ranolazine (currently being tested for its effect on heart failure) directly inhibit β-oxidation enzymes and potentially other metabolic enzymes (68, 538). Irrespective of their individual mechanism of action, these drugs have two fundamental effects in common. They all induce a substrate switch from fatty acid to glucose and they improve the efficiency of ATP production (171).
In the larger context, metabolic dysfunction of mitochondria plays a role in the initiation and progression of a number of health concerns. Therefore, targeting metabolic dysfunction by enhancing glucose utilization and pyruvate oxidation at the expense of fatty acid oxidation, as in cardiac ischemia, appears to be a valid therapeutic approach (Section IX,C). Some of the therapeutic approaches may also be applicable in other organs or tissues. The intense research into developing more sophisticated techniques for assessing and treating mitochondrial disorders in a wide range of human diseases should be based on rational approaches in order to understand the underlying mechanisms of mitochondrial dysfunction by which to develop new pharmacological therapies.
C. Mitochondria and diabetes
Peripheral insulin resistance characterizes diabetes mellitus with a defect in insulin secretion by pancreatic β-cells. Alleviating the metabolic disorder can reduce or prevent the development and progression of diabetes (27). Intense studies are currently underway to identify novel therapies for a disease that is responsible for the deaths of millions worldwide and has caused crippling disabilities in millions more. The etiology of diabetes has both genetic and environmental components (27). Disruption of mitochondrial function is also implicated in the etiology of the disease. For example, mitochondria of type 2 diabetic patients have reduced ETC capacities (401). The ability of pancreatic β-cells to regulate blood glucose levels is dependent on mitochondrial ATP production. Indeed, the impairment in glucose homeostasis is associated with a severe ETC defect caused by a deficiency in the mtDNA-encoded cytochrome c oxidase. Therefore, defective mtDNA can result in mitochondrial dysfunction that could lead to the development of diabetes as a result of impaired insulin release. Patients with “mitochondrial diabetes” (352) and mitochondrial dysfunction have reduced glucose-stimulated insulin secretion, a finding that underscores the importance of normal mitochondrial function in β-cells (50,51).
Whether mitochondrial dysfunction is at the center in the etiology of insulin resistance in type 2 diabetes, or the underlying cause of impaired β-cells function in type 1 diabetes, remains unresolved. Nonetheless, insulin action results from a cascade of events from insulin-induced conformational change of insulin receptor subunits on the sarcolemma to autophosphorylation of tyrosine residues. The insulin receptor then acquires tyrosine kinase activity to phosphorylate the intracellular insulin receptor substrate (IRS) family of molecules (528). These events lead to activation of downstream PI3K and activation of Akt, which mediates most of the metabolic actions of insulin. Phosphorylation of the IRS by GSK3-β inhibits the tyrosine phosphorylation of IRS catalyzed by the insulin receptor.
Akt induces translocation of GLUT4 to the cell membrane resulting in increased glucose uptake. Mitochondrial dysfunction or reduced mitochondrial number could impair the insulin signaling cascade. For example, impaired mitochondrial function and subsequent impediment of fatty acid oxidation could lead to increased intracellular fatty acyl-coA and diaylglycerol content and consequently to activation of PKC. PKC is thought to trigger a serine kinase cascade that phosphorylates serine residues of the IRS, consequently blocking the insulin signaling pathway (528). Activation of insulin receptors also leads to translocation of activated Akt to mitochondria in cardiac muscle (635). Akt translocation to mitochondria is thought to represent a link between insulin receptor signaling and mitochondrial dysfunction in diabetic myocardium. Insulin action likely plays a major role in the regulation of myocardial OXPHOS because insulin receptor KO mice showed decreased OXPHOS and exacerbated ventricular dysfunction (511). Indeed, in a recent study, Yang et al. (635) reported that insulin modulates myocardial OXPHOS by the PI3K-Akt pathway in diabetic myocardium. Furthermore, insulin was shown to increase complex V activity in control mice; this was blunted in mice with a high fat/high fructose diet, but was increased in the streptocozin diabetic model (528).
Increased mitochondrial ROS production during hyperglycemia may be another way mitochondrial dysfunction contributes in the pathology of diabetes. Undeniably, mitochondrial ROS production and the concomitant oxidative damage may contribute to the onset and progression of both types 1 and 2 diabetes (280, 627). This is because elevated glucose and/or fatty acids may lead to greater O2 consumption, and coupled with the higher ΔΨm, to more ROS production (528). The mechanisms by which ROS may contribute to the pathophysiology of diabetes have been discussed extensively (265, 281, 405). Excess mitochondrial ROS in β-cells inhibits OXPHOS leading to a decrease in ATP for glucokinase (expressed in β-cells) and the low ATP/ADP ratio will result in inactivated KATP channels and impaired insulin secretion. Chronic oxidative stress on peripheral tissues ultimately leads to organ damage. Indeed, evidence for a connection between elevated blood glucose and oxidative stress has been obtained from experiments on endothelial cells, wherein increasing the glucose level was found to increase net cytosolic ROS levels (184, 651). The ROS generated in response to hyperglycemia may represent the proximal defect that eventually leads to other pathological consequences of the disease. This suggests that therapeutic strategies to limit mitochondrial ROS production or to increase the rate of ROS scavenging may be useful adjuvants to conventional therapies designed to normalize blood glucose (643).
When administered to mice, mitochondrial-targeted ROS scavengers, such as ubiquinone and vitamin E, have shown great efficacy against cell damage associated with high glucose (494). However, clinical trials using α-tocopherol (vitamin E), ascorbate (vitamin C), coenzyme Q, and α-lipoic acid have yielded ambiguous results. Artificial antioxidants such as SOD mimetics may be more potent than their natural counterparts, but their usefulness in clinical trials has not been determined (391, 498). In addition to detoxifying ROS with mitochondria-designed antioxidants, another possible approach is to target the ΔΨm. In this case, one could lower the state of depolarization (mild) by overexpressing UCPs or by titrating the dose of DNP, which has been shown to improve serum glucose, triglycerides and insulin levels in mice (88).
Type 2 diabetes results in an elevation of plasma free fatty acid levels, an increase in β-oxidation in the heart, inhibition of PDH activity, and impaired glucose oxidation (353). The suggestion that diabetics would benefit from therapies that suppress fatty acid uptake and oxidation and increase pyruvate oxidation has been entertained in recent attempts to mitigate the disease. Indeed, PGC-1α, a member of a family of transcription co-activators that modulates cellular energy, mitochondrial biogenesis (344), and regulates ROS (242, 589), is intimately involved in disorders such as obesity, diabetes, and cardiomyopathy. In particular, its regulatory function in lipid metabolism makes it an inviting target for pharmacological intervention for treating obesity and type 2 diabetes. Overexpression of PGC-1α in mouse skeletal muscle increased glucose as well as the expression of proteins that are involved in fat oxidation and glucose transport (52). Thiazolidinediones (e.g., rosiglitazone) are a class of antidiabetic drugs that increase myocardial glucose utilization while lowering serum triglycerides (319). In skeletal muscle biopsy studies, it was shown that PGC-1α is reduced in patients with type 2 diabetes (446). Moreover, thiazolidinediones mediate their effect in part through the ability of PGC-1α to activate mitochondrial biogenesis and increase mitochondrial function (344). Consistent with this notion is the observation that mice deficient in PGC-1α were found to be defective in contractile protein function in skeletal and heart muscle. Further evidence show impaired ATP levels because the protein levels of ATP synthase and creatine kinase B were reduced in the diabetic patients (260). The PGC-1α signaling cascade may also alleviate diabetes in part by the induction of MnSOD (27).
D. Mitochondria and hypertension
The contribution of mitochondria in kidney disease and hypertension has gained attention recently. Loss of redox homeostasis and generation of ROS appear to play a critical role in the etiology of renal diseases and hypertension (406). Mitochondrial ROS may contribute to this pathogenesis and therefore mitochondria may be a target in the disease process. The kidney is intimately involved in the disease process of hypertension and the effects of ROS ultimately depend on the pro- and antioxidant pathways (406). In the kidney, the renin–angiotensin–aldosterone-system (RAAS) is key in the control of arterial blood pressure (ABP) and the pathogenesis of hypertension. Angiotensin II (AII), an oligopeptide, is a potent hypertensive hormone that causes peripheral vasoconstriction and also stimulates aldosterone release. Aldosterone, in turn, increases renal salt retention by acting on the distal tubule. All these actions can lead to increased ABP.
Recent landmark studies concluded that augmented O2 •− production underlies the pathogenesis of hypertension, and this was attributed primarily to AII (406, 410). For example, AII infusion in rats led to increased ABP and this effect was reversed by SOD treatment. In an in vivo model, Nozoe et al. (410) reported that mitochondria-derived ROS induced by AII mediated sympathoexcitation in the rostral ventrolateral medulla (RVLM), a brainstem site that maintains sympathetic vasomotor tone, resulted in a pressor response. Overexpression of MnSOD and administration of rotenone inhibited the AII-induced ROS production and attenuated the pressor response (410). In addition, the authors reported that depletion of extracellular Ca2+ with EGTA and blocking mCa2+ uptake with carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP), an uncoupler, also blocked AII-elicited mitochondrial ROS production (410). It was concluded that AII increases mCa2+ uptake, which leads to mitochondrial ROS production. The effect of rotenone to reduce ROS and to reduce ABP may be attributed to its effect on the ETC. This suggests that rotenone prevents forward electron transfer from complex I to complex III, a source of O2 •− generation. This is analogous to the effects of rotenone observed by others (18, 324) and to our recent study showing that amobarbital attenuated O2 •− generation during cardiac I/R injury (9).
Spontaneously hypertensive rats (SHR) exhibit ETC defects in complex I and II activities compared to normotensive Wistar–Kyoto rats (107). In a recent study, Kung et al. (313) demonstrated the importance of complex I in maintaining the high ABP in the SHR model. In their study, it was reported that in the SHR, microinjection of the adenovirus vector to overexpress eNOS in the RVLM reduced complex I activity and increased O2 •− and ONOO-, which were reversed with MnSOD transfection or decomposition of ONOO-. Co-transfection of MnSOD with eNOS prevented the rebound in ABP induced by eNOS overexpression in the SHR. Other studies reported an alteration in mCa2+ handling in brain mitochondria during hypertension (89). These studies demonstrate the contribution of RNS, ROS, and mCa2+ in regulating ABP through their actions on ETC complexes. A better understanding of the role of mitochondria in the etiology or progression of hypertension may lead to better design of drugs that target the root cause of the disease. One possible strategy would be the use of gene transfer (186) that would target mitochondria in the kidney, vascular endothelium, and in the sympathoexcitatory neurons of the RVLM. In support of this strategy, overexpression of SOD or catalase in the SHR by gene transfer reversed mitochondrial impairment, blunted ROS in RVLM, and mitigated sympathetic vasomotor tone (107).
AII also mediates cardiovascular dysfunction via ROS-induced ROS generation that culminates in cardiovascular pathology including hypertension. Ricci et al. (472) showed that increased AII initiates a signaling cascade involving PKC to generate NADPH oxidase (Nox) dependent ROS and RNS in the cytosol. Studies show that hypoxia-triggered mitochondrial ROS activate Nox-dependent ROS formation in pulmonary artery smooth muscle cells, resulting in more ROS/RNS production (467). Indeed cytosolic ROS/RNS do not directly mediate cell damage, rather they trigger mitochondrial ROS/RNS, which in turn leads to cell damage or death (174, 472). Apocynin and chelerythrine dramatically attenuate mitochondrial O2 •− generation in response to AII (174). Furthermore, AII stimulated Nox-dependent O2 •− acts on mitoKATP channels, or with NO• to form ONOO-, which damages ETC, leading to a feed-forward loop of more ROS/RNS generation and to further activation of Nox and more intracellular ROS production (174), thereby progressing to cardiovascular disease.
Other studies have reported similar crosstalk between mitochondria and Nox in AII-induced endothelial dysfunction and hypertension (174, 613). Blocking complex I, or preventing mPTP opening (613), or blocking the mitoKATP channel, could attenuate vascular dysfunction and ameliorate pathological conditions (174, 613). The actions of 5HD strongly support the role of mitoKATP channels in the dynamic duo between these two distinct sources of oxidative stress with distinct mechanisms (613). Interestingly, chronic nitroglycerin treatment results in the development of nitrate tolerance associated with vascular dysfunction; this might involve similar crosstalk, but in reverse order, between mitochondrial ROS and cytosolic Nox-dependent O2 •− generation. Specifically, mitochondrial ROS/RNS exit into the cytosol where they may activate vascular Nox in a PKC-dependent process (613).
Although it is generally understood that vascular Nox is a main source of ROS in cardiovascular diseases, the concept of mitochondrial-triggered activation of Nox is novel and is becoming attractive in other research areas. Mitochondrial control of Nox1 redox is implicated in breast and ovarian cancer (158). Therefore, strategies to target mitochondria as well as Nox-dependent oxidative stress or interfere with the crosstalk of ROS between mitochondria and Nox (613) represents a potential approach for use of targeted antioxidants in mitigating mitochondrial-related diseases that include hypertension.
E. Mitochondria and neurodegenerative diseases
The neurodegenerative diseases are a key health issue because they are profoundly debilitating (177). Mitochondrial dysfunction and the mechanisms that favor apoptosis and necrosis are believed to play a significant role in many neurodegenerative diseases (391). The central nervous system is particularly vulnerable to oxidative stress due to its high O2 demand and energy expenditure. Mitochondrial dysfunction leads to oxidative damage that is well documented in many of these diseases. Oxidative damage to mitochondrial membranes, enzymes, and the ETC components, culminate in impaired mitochondrial ATP production and facilitated mPTP opening (602). Recent studies have also implicated mitochondrial fission and fusion proteins (Section VIII.A) during the onset and progression of neurodegeneration. Specifically, Knott et al. (304) proposed that an imbalance in mitochondrial fission and fusion proteins might be the underlying common-thread that links most of these diseases to mitochondria. For example, hereditary mutations in the mitochondrial OPA-1 and mitofusin-2 proteins have been implicated in neurodegenerative diseases (304).
The neurodegenerative diseases include, but are by no means limited to, Alzheimer's disease (AD), Parkinson's disease (PD), Friedrich ataxia (FA), multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS), and the rare Huntington's disease (HD). Interested readers are referred to the following citations for more information (28, 398, 399). In addition, Table 2 summarizes some of the neurological diseases related to mitochondria disorders. Only a brief discussion of some selected diseases will be presented in this review. One salient observation in all these diseases is that even though the fundamental pathological mechanisms remain unclear, it is suggested from the evidence that mitochondrial dysfunction is a contributing factor at some level of the pathogenic process (177). These diseases are commonly associated with mutations in mtDNA, impaired bioenergetics, increased ROS production, and abnormal protein dynamics, including the mitochondrial accumulation of disease specific proteins (e.g., amyloid-β for AD, parkin in PD, mutant SOD1 for ALS, and frataxin for FD) (28, 133, 310, 362).
1. Alzheimer's disease
Alzheimer's disease (AD), the most common neurodegenerative disease, is characterized by accumulation of plaques in brain tissue and progressive cerebral neurodegeneration with advanced age (177). It is the most common form of dementia and is a complex neurological affliction that is characterized clinically by loss of memory and progressive deficits in cognitive ability (482). The underpinnings of the disease are mutations in the genes producing presenilin, the so-called amyloid precursor proteins (APP) (262, 296, 498). A mutation in the genes for the APPs is associated with expression of amyloid-β (A-β) (177). A-β is taken up into mitochondria with its mitochondrial targeting sequences via the TIM and TOM import machinery (247). After uptake, it becomes localized in the cristae of the matrix (247). Accumulation of A-β occurs in transgenic mice overexpressing mutant A-β peptide precursor protein, and is present in autopsied brains from AD patients (112). It is noteworthy that at an early stage of AD there is already a reduced number of mitochondria (247, 257), brain glucose metabolism is decreased (247, 287), and the activities of TCA cycle enzymes (79, 520) and cytochrome c oxidase are reduced (97, 247, 441) while there is enhanced cytochrome c release (177, 296), mPTP opening (384), and inhibition of OXPHOS (498).
AD and its association with mitochondria have been reported extensively in the literature (79, 112, 247, 257, 520). The mitochondrial dysfunction in AD is thought to be secondary to an increase in oxidative stress (498, 548) and or mCa2+ overload (384). The ROS generation increases the amyloidogenic process and sets up conditions conducive for further cell damage so that a destructive cycle of oxidative stress and mitochondrial damage ensues (177). Using brain mitochondria from 20-month-old diabetic Goto–Kakizaki rats, Moreira et al. (384) reported that treatment with CoQ10, a natural antioxidant and a highly mobile electron carrier between complexes I and III or complexes II and III, counteracts brain mitochondrial dysfunction induced by A-β neurotoxic amino acid sequences. Cyclosporine A or EGTA also reversed the adverse effects of A-β neurotoxicity on brain mitochondria (385). The use of chloroquinol, an antimalarial drug, has shown some effectiveness in ameliorating symptoms of AD in part by reducing Ca2+ accumulation and oxidative stress (177); mitochondrial A-β amyloidosis is also known to promote mitochondrial fission which may contribute to progression of the disease.
A detailed description of the etiology of AD and the controversy over the different proposals to reduce its incidence and severity is well discussed by Duchen (177). Our review addresses only the targeting of mitochondria as a possible therapeutic option in the treatment of AD. In AD as well as in PD the disease is propagated by its spreading to neighboring cells. It is therefore crucial that the root cause of the diseases be preemptively targeted if control of either of these diseases is to be achieved. A growing body of evidence suggests that a greater understanding of mitochondrial dynamics (fission/fusion), and the regulatory factors involved, may lead to novel therapeutic strategies for improving treatments and containment of the disease (155, 304).
2. Parkinson's disease
Parkinson's disease (PD) is the second most frequent neurodegenerative disorder after AD in the elderly (638). The etiology of the disease is not clearly defined. Present evidence suggests that PD is a multifactorial disorder probably caused by a combination of age, genetics, and environmental factors. The environmental link between mitochondria and PD was suggested nearly 2 decades ago when it was reported that a toxic byproduct of meperidine, 1-methyl-4-phenyl-1,2, 3,6-tetra-hydropyridine (MPTH), blocked mitochondrial ETC and produced symptoms similar to those seen in late stage PD (638). MPTH effects on mitochondria were energy depletion and increased ROS production (638). Chronic exposure to rotenone also reproduced a PD- like syndrome in rats that was associated with selective loss of dopaminergic neurons (254, 304). Studies have shown that mitochondrial genetic alteration, as in a missense mutation in the ND4 subunit of complex I, can lead to an atypical PD like disorder and loss of nigrostriatal neurons (254) as a consequence of oxidative stress.
Selective damage to mitochondrial complex I, with a concomitant increase in ROS within the dopaminergic neurons in the substantia nigra and an increase in cell death, is believed to be a major causative event in PD (254, 394, 497, 498). Some patients with PD have a high frequency of single nucleotide polymorphisms in the gene for MnSOD (216, 522). This alteration may contribute to an increase in ROS that can be reversed by treatment with tempol, a O2 •− scavenger that was reported to diminish the severity of PD syndrome (337). In patients with PD, administration of CoQ10 both enhanced the ETC function and reduced ROS. A large phase III clinical trial is underway to examine if high-dose oral CoQ10 will slow the progression of the disease (254).
Several recent lines of evidence have also implicated reactive nitrogen species (RNS) in neurodegeneration (638) leading to PD. There is growing evidence indicating that RNS are a major contributor to the pathogenesis and progression of PD. The association of NO• with PD is strengthened by studies that show induction of iNOS in glial cells contributes to the degeneration of dopaminergic neurons in a mice model of PD (638). NO• may contribute to the deterioration of dopaminergic neurons in part via ONOO-. ONOO- can block complex I function (Section IV,B), mainly by forming 3-nitrotyrosine and nitrosothiol (394, 638), and by increasing oxidation of cardiolipin (453). Other prevailing conditions in PD are the depletion of GSH (122, 453) and the susceptibility of complex IV to RNS and ROS (254, 453, 498). This may lead to a further increase in ROS levels and possibly to a vicious cycle of functional and anatomical deterioration.
The use of antioxidants to treat PD suggests that the primary mechanism of injury is via oxidative stress; metabolic therapies may not be effective since metabolic insufficiency appears to play no role in the disease (177). Recent data seem to support this contention. Trolox and GSH are both capable of slowing down the ONOO--mediated damage to these dopaminergic neurons (453). In this experimental setting, the NO• scavenger cPTIO only partially attenuated inhibition of complex I, whereas FeTPPS, a selective blocker of ONOO- and MnTBAP, completely blocked the inhibitory affects on complex I. Other approaches are to use molecules such as MitoQ and MitoVit E (ubiquinone and vitE homologues, respectively), which target and accumulate in the mitochondria and could both enhance ETC function and scavenging of ROS and prevent oxidation of cardiolipin, all of which may result in better functioning of the mitochondrial complexes (453, 498).
Recent evidence implicates a derangement of the mitochondrial-reticular network (Section VIII.A) in the etiology and progression of PD. Rotenone also initiates mitochondrial fission (304). Deng et al. (155) have proposed a direct genetic link of PD to mitochondrial dynamics. They showed that the pink1 (PTEN-induced kinase 1) gene of D. melanogaster interacts with the mitochondrial fusion/fission machinery. The PINK-1 protein is a serine-threonine kinase localized to the mitochondrial membrane via an 18-KDa amino-terminal in the mitochondrial targeting sequence (254). A pink1 deficiency in D. melanogaster resulted in a disorganized morphology similar to the parkin mutation and loss of dopaminergic neurons (254). The morphological changes could result from a dependency on OXPHOS for maintaining ΔΨm and an intact mitochondrial network (254). A knockdown of opa1, or overexpression of drp1, rescued the phenotype of muscle disintegration, mitochondrial abnormalities, and cell death in pink1 mutants. These studies demonstrate that pink1 promotes mitochondrial fission and/or inhibits fusion by negatively regulating opa1 function, and/or positively regulating drp1 (155). Rasagiline, an MAO inhibitor, has multiple effects on mitochondrial function, including stabilization of ΔΨm, which is necessary for maintaining the mitochondrial network, and has a PD-modifying effect (254). Thus, maintenance of the mitochondrial ΔΨm, a prerequisite for organizing the mitochondrial reticular network, is an essential feature in the strategy to alleviate PD.
3. Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS; Lou Gehrig's disease) is the most common adult-onset motor neuron disorder. It is characterized by selective neuronal (lower motor neurons in the spinal cord, brainstem, and motor cortex) degeneration (39, 586) and cell death. ALS can be fatal due to respiratory muscle weakness and complications of paralysis. The final stage of the disease ends with the patient's inability to initiate and control any voluntary movement. In the vast majority of affected people, the disease (90%) has no known cause and is termed “sporadic” ALS. In only a few victims is it inherited (familial ALS) (586, 634). The most common defects are in cytochrome c oxidase activity and in a mutation of sod1 (498, 634). Some investigators believe that mitochondrial dysfunction may be downstream of a primary disease process such as accumulation of a pathogenic protein. However, other recent studies have suggested that mtDNA damage plays a crucial role in the etiology of neurodegenerative diseases (634).
One potential consequence of mitochondrial dysfunction in ALS, as in other neurodegenerative diseases, is impaired energy metabolism and an increase in ROS emission. Indeed, impaired ETC function has been detected in muscle and spinal cord cells of ALS patients, and there is a significantly higher level of point mutations in spinal cord neuronal mtDNA. For example, there was observed an increase in 8-oxoguanine, a common marker of DNA lesion resulting from ROS, in the motor cortex, spinal cord, and plasma of these patients (222, 634). Of the small number of familial ALS patients, about 20% of the defects are mapped to the sod1 gene (586). The autosomal dominant nature of SOD1-associated ALS suggests a toxic gain of function for mutant SOD1 (586).
It is now firmly established that in the fraction of familial ALS linked to the sod1 gene, the expression of ALS-mutant SOD1 proteins is the ultimate cause of motor neuron death (586). It is reported that the mutant sod1 gene in mice leads to a high molecular weight, oligomerized species of SOD1 protein, which is toxic and accumulates in motor neurons (586) and could be involved in ROS formation (587). Studies showed that expression of mutant sod1 in cultured neuroblastoma cells resulted in increased levels of mitochondrial O2 •− production that was counteracted by MnSOD scavenging (189, 654). Other studies show a disturbance in mCa2+ homeostasis (99, 634). Overexpression of the mutant human sod1 gene in mice caused abnormal mitochondrial morphology [i.e., damage to IMM cristae in spinal cord motor neurons (302), disrupted cytochrome c, and decreased respiration (99)]. The altered SOD1 protein may interact with the pro-survival protein Bcl-2 which compromises its cell survival-promoting function. Indeed, increased levels of the pro-apoptotic proteins Bax and Bid, and decreased anti-apoptotic proteins Bcl-2 and Bcl-Xl, were found in spinal cord tissue of ALS patients (256, 442).
Other proposed mechanisms for oxidative damage in ALS include increased NO• production and decreased GSH levels. Increased NO• and O2 •− facilitates ONOO- production (Section IV,B), which can damage key mitochondrial enzymes (81, 99). Treating transgenic mice with L-NAME or preincubation with Mito-Q better preserved ETC function and improved respiration in transgenic, but not in nontransgenic mice. In addition, the antioxidant MnTE-2-Pyp (contains oxidized Mn3+) restored mitochondrial respiration, in part by the reduction of Mn3+ by complexes I and II to Mn2+ which degrades ONOO- (81, 99). Further studies are needed to understand why mutant expression of SOD1 and “sporadic” incidents cause motor neuron disease and until this mechanism is elucidated, the therapeutic approach targeting ALS via motor neuron mitochondria is very limited.
4. Friedreich's ataxia
Friedreich's ataxia (FA) is a clear example of a disease in which there are hallmarks of mitochondrial oxidative stress (362, 435). FA is an autosomal recessive disease that is the most common of the hereditary ataxias and is characterized by progressive damage with relatively early onset (435). The disease is characterized by high levels of iron (Fe) (362, 616) due to decrease frataxin, a mitochondrial Fe chaperone protein involved in regulating Fe within mitochondria (310, 362). Mutation of the genes encoding frataxin frequently leads to loss of activity by the protein (435), impairment of ETC complexes, and OXPHOS, and reduced levels of aconitase and other TCA cycle enzymes. These defects contribute to ROS production and a higher oxidative load in the cell (17, 484).
Therapeutic remedies for FA include idebenone, a CoQ10 analog and inhibitor of lipoperoxide, or the combination of idebenone and CoQ10, which improved neurological coordination (489, 490). The accumulation of Fe in the mitochondria suggests the potential for chelation therapy. Indeed, Fe chelators have been used to mitigate mitochondrial Fe load and to improve mitochondrial and cellular conditions. For example, recent studies show that Fe accumulates in the mitochondria of a mouse FA model, and that development and use of the mitochondrial permeable Fe chelator, desferrioxamine, reduced Fe load and prevents generation of OH• driven by the Fenton reaction or direct scavenging of O2 •−. In a D. melanogaster FA model, ectopic overexpression of scavenging enzymes restored the activity of aconitase (17); this is indicative and supportive of the theory that ROS are a salient feature in the pathogenesis of FA.
F. Neoplastic diseases
Mitochondrial involvement in the etiology of neoplastic diseases (ND) has been hypothesized since the 1930s, when the Nobel prize-winning German physician and scientist, Otto Heinrich Warburg, discovered that mitochondria in cancer cells do not efficiently generate energy (464). Unlike the neurodegenerative and ischemia-induced diseases discussed above, mitochondrial dysfunction in tumor cells tends to lead to cell survival and resistance to chemotherapy; hence, the term the “Warburg effect” (212, 485). In marked contrast to the therapeutic strategy for neurodegenerative and ischemic diseases, which is prevention of cell death, in ND the main goal of targeting the mitochondria is to kill malignant cells by inducing apoptosis (212). A growing body of evidence suggests that tumor cells exhibit increased intrinsic ROS stress due in part to oncogenic stimulation, increased metabolic activity, and mitochondrial dysfunction (56). Despite their variability, almost all neoplastic cells demonstrate enhanced uptake and utilization of glucose for glycolysis to generate ATP (212). The most glycolytic tumor cells were found to be most resistant to therapy and most aggressive in metastasis (212, 364).
A pivotal player in the switch from OXPHOS to glycolysis may be the HKII binding at the VDAC (Section II,A). As indicated previously, tumor cells adopt this phenotype for their survival. This includes most neoplastic cells that metastasize (364). HKII binding to VDAC prevents the pro-apoptotic proteins Bad and Bax from binding to the OMM, and so help immortalize these cells by gaining preferential access to newly synthesized ATP for phosphorylating glucose (136, 212, 364). Tumor cells are also better able to withstand an adverse microenvironment (hypoxia, acidosis, and shortage of growth factors) by virtue of their metabolic adaptation. Considering the key role of mitochondria in cell death, it appears that the relative mitochondrial “silencing” in neoplastic cells can, at least in part, explain resistance of most tumors to treatment.
Emerging insights from the Korsmeyer laboratory (101, 156) group identified a novel key intermediate phenotype that appears to be the lynchpin in the dynamic role of mitochondrial OMM permeability in apoptosis, and provided an important insight into the dysregulation of apoptosis in the neoplastic cell. Cancer cell (e.g., Hela and non-small cell lung) mitochondria are more resistant to OMM permeability because of overexpression of the Bcl protein family (195, 197, 386). Generation of sensitizer and activator pro-apoptotic peptides is robust in many neoplastic cells as disorganized metabolism and intracellular signaling generate robust stimuli for the activation of apoptosis. However, many of these cells also dramatically express Bcl-2 that in turn sequesters the pro-apoptotic peptides and blocks activation of apoptosis. Vigorous apoptosis ensues when Bcl-2 is inhibited by small molecule antagonists, such as ABT-737 (156, 212) or by short peptide antagonists (101), resulting in OMM permeabilization. This led Korsmeyer and Letai to propose the "primed to die" mitochondrial phenotype (333). The response of isolated mitochondria from neoplastic cells can functionally identify the anti-apoptotic peptide that is in fact preserving cell survival, based upon the small molecules or peptides that elicit cytochrome c release from their isolated mitochondria, so called “BH3 profiling” (156, 333). These findings have enhanced the understanding of the regulation of blockade of apoptosis, and have substantially increased an understanding beyond the relatively simple "dead or alive" Bax/Bcl-2 ratio concept that previously dominated the field. Identifying these tumor-specific changes responsible for the resistance to cell death is crucial for developing targeted chemotherapeutic agents (195, 197) based on a strategic approach to ensure normal cell survival.
The actual etiologies of ND are highly sought after. It is believed that apoptosis-regulatory genes (e.g., those encoding p53, PTEN and Bcl-2) are involved in the pathogenesis of many human cancers (136). PTEN is a phosphatase that inhibits Akt activation by reducing PIP3 levels (448). The mPTP whose constituents are different between normal and malignant cells play a role in this pathological process (136). Also it is postulated that this difference may explain resistance to apoptosis and the cancer-specific metabolic alterations (136) in these cells. For instance, HKII-VDAC interaction prevents binding of pro-apoptotic proteins binding to VDAC and thereby the induction of apoptosis (212,364). Consequently, a variety of compounds including avicins, which are pro-apoptotic, anti-inflammatory molecules with antioxidant properties, perturb mitochondrial function and initiate apoptosis in tumor cells (212, 364). Colon cancer cells HCT 116 express a small amount of Bak (383). Cisplatin, an anti-neoplastic drug kills colon cancer cells in part by activating Bak (276, 359, 563).
Another strategy used by neoplastic cells to confer protection is the overexpression of PBR (Section II), and Bcl-2 on the OMM (136, 194). Synthetic PBR ligands (e.g., melphalan) display increased cytotoxicity in a variety of rat and human brain tumor cell lines (194). Melphalan is considered a Smac mimetic and is used in the treatment of multiple myeloma (57, 110, 434); it induces apoptosis even in the presence of overexpressed Bcl-2 anti-apoptotic proteins in some of these tumor cells (205). Recent evidence indicates that the tumor suppressor protein p53 has extra-nuclear effects that contribute to its cell cycle-arresting and pro-apoptotic functions (195, 197). Smac/DIABLO can abrogate the protective function of IAPs (175, 597, 628), which confers chemoresistance in various cell types (109, 196, 231, 335). Cytoplasmic p53 can induce OMM permeabilization in part by direct interaction with the Bcl family proteins located on the OMM. Thus, since OMM permeabilization is impaired in tumor cells, its pharmacological induction constitutes a therapeutic strategy. In this case, the ultimate strategy would be to minimize the sequestration of large amounts of BH3-only proteins such as Bim in complexes with anti-apoptotic Bcl-2 proteins. In the absence of Bcl-2, Bim is not sequestered, apoptosis is triggered (315), and cell death is initiated.
Other strategies applied in mitochondria-directed therapies include increasing p53 and Bax, decreasing Bcl-Xl, and enhancing the activation of caspase 3. It was reported that the chemotherapeutic agent CD437 exerts its therapeutic effect by these mechanisms (136). It has been postulated that VDACs and ANT (ANT2 variant) are highly expressed in neoplastic cells when compared to normal cells because of the high glycolytic phenotype in tumor cells (525). The VDAC–ANT–HKII complex was shown to be a requirement for ATP transport in neoplastic cells (67). These studies support the notion that VDAC and ANT could be possible pharmacologic targets, or at least could be subjected to metabolic alteration in neutralizing neoplastic cells. Indeed, the upregulation of VDAC in tumor cells makes them susceptible to the anti-tumor effects of furanonapthoquinones, whose apoptotic activity is via NADH-dependent O2 •− generation (525). Another strategy involves the use of 3-BPR (Section II,A) that selectively enters and destroys tumor cells by targeting the HKII and the mitochondrial synthasome. This leads to rapid depletion of ATP and tumor cell destruction without harm to normal cells (364).
As an adaptation to an increase in size with limited nutrients and diminished O2, tumor cells are known to adapt to hypoxia by inducing the transcription of multiple genes via activation of the transcription factor hypoxia-inducible factor (HIF-1α). The proteins induced by HIF-1α are involved in regulation of glycolytic metabolism as well as tumor growth and angiogenesis. Therefore, preventing HIF activation may act to suppress tumor growth and cell proliferation. An O2 sensor may be central in this dynamic process that also clearly involves mitochondrial ROS (48). It is proposed that ROS, specifically H2O2, produced from complex III is a likely sensor for initiating and stabilization of HIF-1α during hypoxia (108). Therefore, a better understanding of how mitochondria function to initiate a hypoxic response will lead to development of therapies that target O2 consumption, ROS production, or alter key metabolite concentrations in mitochondria (48).
In recent studies, PGC-1α levels were downregulated in hepatic, breast, colon, and epithelial ovarian tumor cells (33, 647), while overexpression induced apoptosis occurred in epithelial ovarian tumor cells (Ho-8910), but not in CHO (Chinese Hamster Ovary) cells. It was suggested that the increased expression of PGC-1α results in upregulation of the pro-apoptotic gene Bax and downregulation of the anti-apoptotic gene Bcl-2 (33, 647). Moreover, PGC-1α-induced apoptosis was proposed to be mediated by a decreasing ratio of Bcl-2/Bax, which can lead to destabilization of mitochondria and release of cytochrome c (647). The finding that PGC-1α expression decreased in ovarian tumor cells and that increased expression promoted apoptosis suggests that PGC-1α might be involved in the pathogenesis of some cancers. The strategy of increased expression of the PGC-1α gene may be useful for cancer therapy; this again belies mitochondria as the epicenter of this strategy.
G. Other mitochondria-related diseases
1. Mitochondria and psychiatric disorders
Several studies have reported a role for mitochondria in the pathophysiology of bipolar disease (BD), major depressive disorders (MDD), including post-traumatic stress disorder (PTSD) and schizophrenia (SZ) (278, 480, 545). The features of mitochondrial abnormalities include deficiencies in OXPHOS and mtDNA deletion in the brain, and associations with mtDNA mutations/polymorphisms or nuclear-encoded mitochondrial genes (278, 545). It is suggested that the high rates of maternal offsprings with SZ and BD compared to paternal rates support the notion that increased risk for these diseases might be related to mitochondrial impairment (480). In SZ it is reported that there is an increased number of synonymous base substitution in mtDNA in the dorsolateral prefrontal cortex (DLPFC; Brodmann area 9/46) (480). In PTSD, decreased activity in the DLPFC, a brain area involved in the regulation of memory and preparation and selection of fear responses (545), was reported and it was proposed that mitochondrial dysfunction is a factor in the etiology of the disorder. Su et al. (545) studied dysregulation of mitochondrial genes in these brain areas (Brodmann) from postmortem patients with and without PTSD using human mitochondria-focused cDNA microarrays. The study revealed that a majority of the dysregulated genes were associated with mitochondrial dysfunction and neurological diseases. Some of the dysregulated genes appear confined in areas involved in neuronal function–survival and thus may be targets for neuropsychiatric drugs. The authors concluded that mitochondrial dysfunction is involved in PTSD “and provide the expression fingerprints that may ultimately serve as a biomarker for PTSD diagnosis and the drugs and molecular targets that may prove useful for the development of remedies for prevention and treatment of PTSD” (545).
Preliminary studies have shown that lower levels of brain energy metabolism are associated with MDD. Impaired energy metabolism in the brain detected by magnetic resonance spectroscopy suggests that mitochondrial dysfunction is an important component in MDD. A salient feature is that glucose is metabolized very slowly. It has also been proposed that altered mitochondrial OXPHOS malfunction is involved in psychiatric disorders and that brain mitochondria of SZ patients exhibit reduced complex IV activity in the frontal cortex; in other studies, low complex IV activity was highly associated with increased emotional and intellectual impairment, but not motor impairment. Other studies have reported reduced complexes II and III activities in frontal and temporal cortex of SZ patients (278, 480). A reduction in the activity of the complexes affects electron transfer and therefore may potentially interfere with mitochondrial metabolism and ATP production. In the brain this could lead to cell death as a consequence of disturbance in the regulation of intracellular homeostasis. Gerich et al. (207) showed a correlation between the spread of hypoxia-induced depression in rat CA1 region of hippocampal slices and ΔΨm depolarization. The author presented two basic questions to these findings: could it be mitochondrial depolarization that triggers the depression spread? If it is the mitochondria, how can the depolarization be transmitted to the cell membrane? A likely scenario according to the authors is that mitochondrial depolarization may reduce sequestration of Ca2+ by mitochondria and Ca2+ release from malfunctioning mitochondria; subsequent changes in intracellular ionic conditions and pH would be considered as putative signaling mechanisms.
However, a note of caution is that it remains to be resolved whether the occurrence of psychiatric symptoms in patients is just a coincidence or is more directly related to mitochondrial dysfunction, itself. The answer to this question requires further investigation. To date, dietary supplements, have been used to “fuel” mitochondrial function and in so doing mitigate the symptoms of psychiatric disorders. Some of these nutritional maneuvers are discussed below (Section XI,E).
2. Mitochondria and migraine headache
Initial studies have also raise the question that mitochondrial dysfunction may contribute to migraine headaches, at least in selected patients. Migraine headache is a neurological disorder believed to be a manifested in individuals with mtDNA sequence-related mitochondrial dysfunction (163). Indeed, epidemiologic evidence for frequent maternal transmission has implicated a role for a mitochondrial genetic background. The link of mitochondria and migraine headache is supported by the observation that it is frequently associated with a deficit in energy metabolism and in MELAS, the classic mitochondriopathies (163, 306). This is consistent with the observation that OXPHOS is impaired in migraineurs and increased numbers of sequence variants were detected in the noncoding control regions of mtDNA in migraineurs with occipital stroke (163). It is also conceivable that migraine headache may also be associated with mutation of nuclear genes that encode the ETC complex because it has been noted that other studies failed to show the connection between the headaches and mtDNA abnormalities (306). Regardless of the source of mitochondrial protein defect, the consequence of mitochondrial dysfunction when coupled with loss of intracellular Mg2+ in the brain, an occurrence in migraineurs, culminates in a metabolic shift that cause instability of neuronal function, which then enhances the development of a migraine attack (612). Disruption in mitochondrial metabolism may thus provide future pharmacologic targets for novel therapies against migraine headaches. Therapeutic strategies are briefly discussed in Section XI,E below.
X. Mitochondrial Pharmacology and Therapeutic Potential
The primary cause of most of the mitochondria-related diseases discussed in our review have multifaceted etiologies; hence efforts to develop effective drugs should be devoted to the design of individual new compounds that work in synergy to protect the mitochondrion. Current mitochondria-targeted drugs cover a wide range of pharmacological agents (556) (Table 3; Fig. 12). Some of these drugs target mitochondria directly, whereas others affect mitochondria as a secondary or side effect. Nonetheless, identification of the mitochondrion as a target of a drug may assist in better understanding of a drug's mechanisms of action and allow new perspectives for its application (556). Only selected aspects of targeting mitochondria for therapeutic benefit have been covered in all the sections discussed above. This is inevitable considering the broad nature of the subject and how mitochondrial function lies at the center of cell viability and cell death. This section will discuss briefly a) strategies for mitochondrial drug delivery, b) mitochondria-targeted drugs, c) maneuvers to protect against I/R injury, and d) other mitochondrial therapeutic approaches

A. Strategies for drug delivery to mitochondria
As a first step in designing a mitochondria-specific therapy, it is foremost important to develop a drug that can access the mitochondrial matrix. In recent years the search for new protective remedies against excess mitochondrial ROS has taken on a new sense of urgency. Some well-known scavengers like MnSOD have proved ineffective at preventing oxidative damage in animal disease models, presumably because they are unable to permeate the cell membranes. One general solution is to utilize the large ΔΨm and to attach a molecule, for example, one with antioxidant properties, onto a lipoid vehicle that can penetrate membranes (262). With ΔΨm approaching −180 mV in the nonphosphorylating state, for mitochondria-targeted drugs to work effectively consideration of the difference between the sarcolemmal and mitochondrial electrical gradients should be central to drug design. Specific delivery of mitochondria-targeted drugs directly into the mitochondrion appear pivotal for targeting mitochondria-related pathologies, including neurodegenerative diseases, cardiovascular diseases, and cancer (28).
To this end, numerous approaches have been attempted to increase the access of drugs into mitochondria. For example, rhodamine-123, a lipophilic cation, has the ability to penetrate the mitochondrion by using the negative potential gradient of the organelle as a driving force (Table 1) (28, 262). It has been used successfully to chaperone tethered compounds into mitochondria for cancer therapy, such as the anti-cancer drugs cisplatin and mastoparan. Some of these drugs display selectivity to mitochondria because of the high ΔΨm in cancer cells compared to normal cells (28, 262).
Other mitochondria-targeted drugs have utilized the lipophilic agent TPP+, which has been used by a majority of the nonpeptidic mitochondrial targeting agents. TPP+ has been used frequently to increase the incorporation of antioxidants into mitochondria. Examples include MitoQ and MitoVit E (Table 1) (28, 262). These compounds have been shown to effectively minimize oxidative damage in several experimental models (262). Indeed antioxidants that accumulate within the matrix provide better protection from oxidative injury than untargeted antioxidants (375). A mitochondria-targeted derivative of α-tocopherol (MitoVit E) and mitochondria-targeted ubiquinone selectively accumulate in the matrix when complexed with TPP+ (619) and this accumulation is associated with a more effective detoxification of ROS (375).
Tempol, a cell membrane amphilite, is broadly effective in dismutating O2 •− catalytically and it facilitates H2O2 metabolism by a catalase-like action to limit toxic OH• formation (619). The mitochondrial variant is termed mitotempol and it is the result of coupling tempol to TPP+. Mitotempol has been shown to be an effective scavenger of mitochondrial ROS; however, Wilcox et al. (619) have argued that it is no more effective than tempol itself in preventing O2 •− accumulation in mitochondria. A major fraction of cellular tempol is distributed to the mitochondria, which are the primary site for reducing tempol (619). Indeed, damage to mitochondrial respiration following incubation with 3-morpholinosydnonimine, which generates ONOO-, has been shown to be prevented by co-incubation with tempol (527). This raises questions concerning the rationale for using a mitochondria-targeting strategy for this particular drug. On the other hand, the cationic SOD2 mimetics MnTBAP and MnIIITE-2-PyP5+ accumulate in cardiac mitochondria after systemic injection (534). We reported that MnTBAP coupled with scavengers of H2O2 reduce mitochondrial ROS in a cardiac I/R model (90) and during hypothermia-induced ROS or RNS production (104).
Other strategies involve the use of specific precursor proteins that are synthesized in the cytosol; these often require sequence recognition by the import pathway to allow access into mitochondria (262). A novel class of cell-permeable antioxidant peptides that are selectively partitioned into the IMM independent of the ΔΨm has been reported recently (Table 1). These peptides, known as Szeto–Schiller (SS) peptides, have an aromatic-cationic motif that makes them cell permeable, so that they can selectively target the IMM in an energy-independent and nonsaturable manner. The best characterized member of the SS peptides is the SS-31, which can scavenge matrix H2O2 and ONOO-, due to its dimethyl-Tyr moiety, and it can also inhibit lipid peroxidation, reduce cytochrome c release, and reduce mitochondrial swelling. The SS-31 has demonstrated remarkable in vivo efficacy in reducing cardiac and brain I/R injury in animal models (123, 551, 553, 554, 574). Figure 13 shows that SS-31 peptide preserved cellular GSH levels and reduced infarction in mice subjected to 30 min middle cerebral artery occlusion (123). The variant SS-20, which inhibits mitochondrial ROS production, was effective against the MPTTH model of PD (553, 554). Their selectivity and specificity was designed to interact with mitochondria while minimizing undesirable side effects. It is noteworthy that both mitochondria-targeted catalase and SS-31 preserved insulin sensitivity by preventing mitochondrial oxidative stress induced by high fat diet in rodents (15).
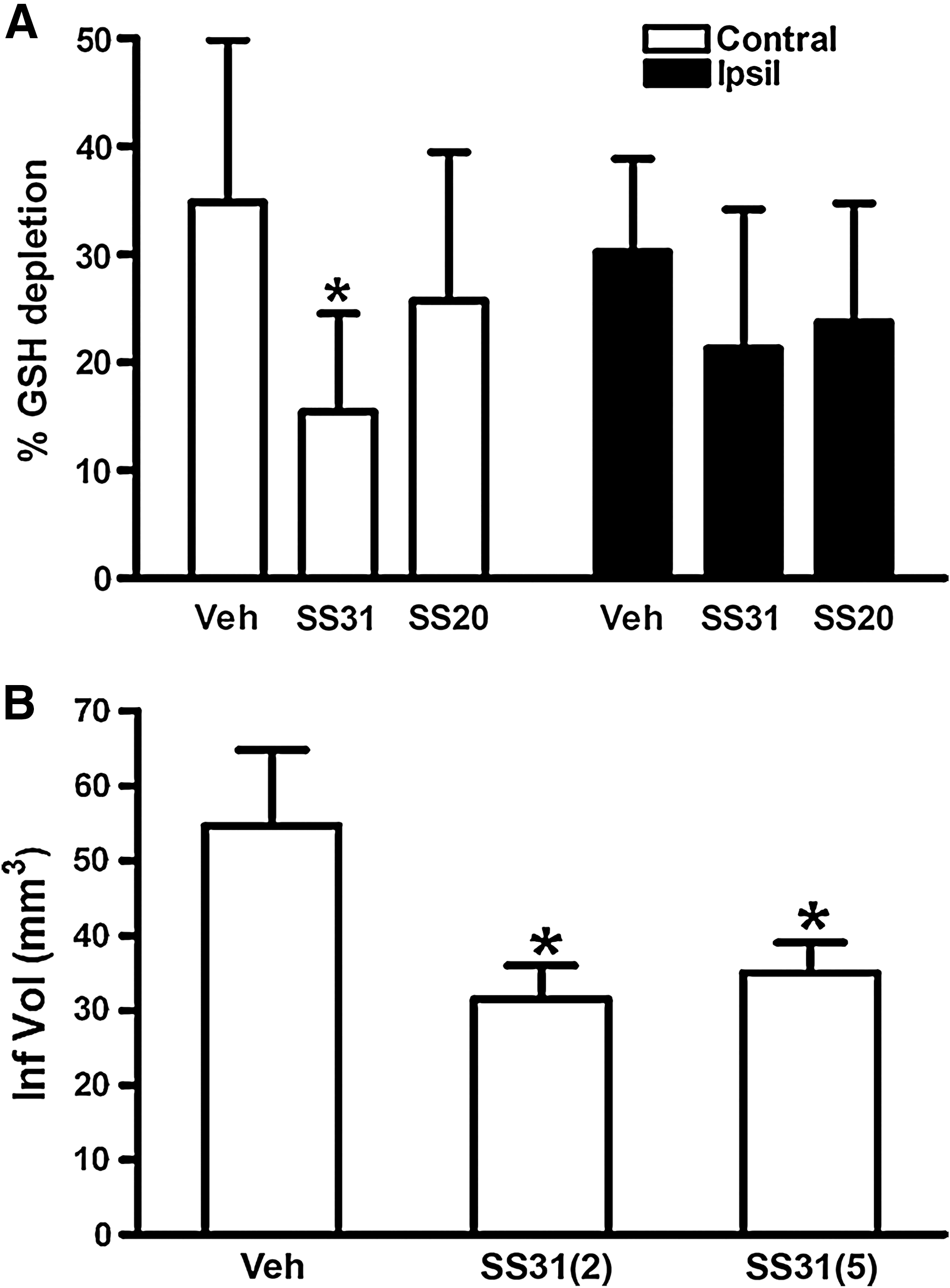
In a recent review, Armstrong (28) proposed the potential therapeutic application of mitochondrial targeting to include: a) delivery of antioxidants to prevent I/R injury, diabetes, and neurodegenerative diseases; b) delivery of apoptotic drugs that target Bcl-2 proteins or deliver toxic drugs to neutralize cancer cells; c) targeting of the mPTP in I/R and stroke; and d) use of uncoupling proteins or activation of endogenous uncoupling proteins in diabetes and obese patients. In addition to these approaches, other recent approaches include the use of techniques in molecular biology involving mitochondrial and nuclear genes, siRNA, and targeting of the mitochondrial reticular network and mitochondrial interactions with the nucleus and the ER. These new approaches include targeting the mitochondrial fusion and fission proteins, targeting the communication between ER and mitochondria via the IP3R response to cytochrome c release, and targeting oxidative stress and mitochondrial modulation of nuclear transcription factors (Section VIII). For interested readers, more information on strategies for delivering drugs to mitochondria can be gleaned from the literature, including the following references (28, 29, 262, 551 –554, 556, 574).
B. Mitochondria-targeted drugs
Mitochondria are ideal targets for therapeutic modification because they are key regulators of energy production, ROS production, and apoptosis. Mitochondria-targeted drugs are therapeutic agents that can directly target mitochondria to instigate apoptosis in cells (neoplastic cells) or protect against apoptosis (all other normal cells). For example, amiodarone, a class III anti-arrhythmic drug can be used to target mitochondria for reducing cardiac I/R injury. It can do this in part by preserving energy metabolism in the post-ischemic heart by inhibiting mitochondrial swelling induced by cytosolic Ca2+ overload (594, 595); however, other studies report that the drug worsens the damage to mitochondrial energy metabolism caused by I/R injury (594, 595). Amiodarone is also reported to exert inhibitory effects on complexes I and II and to reduce the activity of F1F0ATP synthase. It is thus important that these effects of amiodarone are tissue specific and concentration dependent (594, 595). These conditions complicate the targeting of mitochondria from one type of tissue to the next, since one type of tissue's mitochondria will likely respond differently to the drug (594, 595). This subject is discussed further in the Section XIII, Caveats and Limitations to Targeting Mitochondria.
Some antidiabetic drugs can accumulate in mitochondria as a result of the highly negative ΔΨm that favors preferential uptake into mitochondria. For example, metformin (N1,N1 -dimethylbiguanide), an antidiabetic drug, blocks complex I of the ETC because it accumulates in the matrix (427). These drugs are more effective in intact mitochondria than in submitochondrial particles. The positive charges on these drugs coupled with the ΔΨm in energized mitochondria makes it favorable for their accumulation in the matrix (427). Metformin has a broad spectrum of effects, which include fat breakdown, reduction of circulating free fatty acids with a modest inhibition of fatty acid oxidation, and an increase in glucose transport and glucose utilization (655). In other studies, metformin was shown to block mPTP opening as a possible mechanism of action to preserve cellular function. In the nondiabetic state, metformin alleviated some pathological conditions (e.g., inflammatory conditions in lung injury), in part by inhibiting complex I activity (77, 655). It is debatable if these actions of metformin also alter mitochondrial ATP production which would impair mitochondrial energy conservation as a mechanism of action. However, Zmijewski et al. (655) reported that the actions of metformin are not ATP dependent, whereas Brunmair et al. (77) reported that metformin not only blocks complex I, but also uncouples OXPHOS.
There are recent advances in understanding the complex interaction between mitochondria within a reticular network of mitochondria and mitochondrial interaction with other organelles (Section VIII.B C). This has opened a new potential therapeutic avenue for assessing and treating mitochondria-related disorders. For example, resveratrol, a polyphenolic compound abundant in grape skins, induces PGC-1α to express genes involved in mitochondrial biogenesis and OXPHOS (564). Increased expression of PGC-1α has also been shown to mitigate MPTH-induced cell injury in a PD model (254). How resveratrol treats PD is unclear. Resveratrol also reduces ischemic damage in brain (641) and cardiac cells (157). The effects observed experimentally suggest quite a promising therapeutic approach.
Other interventions to reduce cellular damage during a period of oxidative stress, as in post-ischemic injury, include enhancement of endogenous scavenging enzymes and nonenzymes. Pyruvate has been shown to be protective in various in vivo and in vitro models of oxidative stress (608). Wang et al. (608) showed that pyruvate can remarkably abrogate peroxide-induced toxicity in SK-N-SH neuroblastoma cells by a direct antioxidant protective effect on the mitochondrion, as evidenced by its dampening of mitochondrial O2 •− production and its preservation of ΔΨm. As a readily oxidizable substrate, pyruvate can bolster the cytosolic energy state and thereby provide the energy needed to maintain cellular function in the face of a metabolic challenge (608).
Permeabilization of the OMM and the IMM via mPTP opening (Section II) as a consequence of oxidative stress is an important underlying factor in most of the neurodegenerative diseases discussed above (Section IX,E). CsA treatment has been shown to prolong the survival of ALS transgenic mice compared to control (vehicle) treated mice (284). In PD models, daily doses of CsA showed partial preservation of striatal dopaminergic neurons and altered the progression of the disease in the ALS model (602). In the CNS, the effects of CsA is limited because it does not cross the blood brain barrier (284, 602). This restriction can be circumvented by various methods including direct administration of the drugs into the lateral ventricles of the CNS (284, 602).
Mitochondrial antioxidant systems are important in cancer chemotherapy. Tamoxifen, a synthetic nonsteroidal antiestrogen widely used to treat breast cancer, is known to have antioxidant and cardioprotective effects in part by induction of MnSOD (145). However, normal tissue injury is a major problem that limits the success of cancer therapy in protocols involving chemotherapeutic drugs (e.g., adriamycin). Generation of ROS is implicated in the toxicity of a large number of these agents and the injury is manifested at the mitochondrial level (145). These potential side effects will be discussed in Section XIV.
It is clear that the subject of mitochondrial pharmacology is vast and the above discussion only represents some examples. It is also clear that mitochondria are key factor in the etiology of numerous human mitochondria-related and nonrelated diseases, which can be mitigated by targeting the mitochondrion with specific drugs designed for uptake by the organelle. Interested readers are urged to examine the review literature for more information.
C. Approaches to improve mitochondrial function during ischemia and reperfusion
Mitochondria play a central role in I/R injury and hence are promising targets for novel anti-ischemic therapies. The importance of mitochondria as both targets and mediators of I/R is becoming increasingly recognized (113). Cardiac I/R result in mitochondrial dysfunction as shown by a decrease in oxidative capacity, loss of cytochrome c, and generation of ROS. Recent studies show that increased ROS is especially evident during late ischemia (9, 10, 90, 289, 475, 542, 591). Therefore, protection of mitochondrial respiration during ischemia could represent a new therapeutic approach in mitigating the deleterious effects of I/R injury.
A pioneering study by Ganote and colleagues (198) showed that inhibition of mitochondrial respiration could decrease contraction band formation and attenuate enzyme release during reoxygenation; they suggested that resumption of mitochondrial metabolism during reoxygenation can initially lead to deleterious consequences. We discussed earlier how a reperfusion-induced decline in cardiac function and accumulation of oxidatively damaged lipids were diminished when complex I was reversibly inhibited during early reperfusion with amobarbital (14), thus underlining the physiologic significance of mitochondrial ROS production to cardiac injury during reperfusion (113). We also discussed how amobarbital, when administered briefly before ischemia, preserved mitochondrial bioenergetics and improved cardiac function upon reperfusion. In addition, we noted suggestions on how rotenone preserved mitochondrial structural integrity and improved function (324).
Thus, there is support for the concept that mitochondrial function can be protected during global ischemia as well as on reperfusion as shown by reduced levels of mCa2+ uptake and O2 •− generation, and improved redox state after preconditioning (ischemic or pharmacological) (Section X,D–E) (476, 477), inhibition of NHE (10), hypothermia (475), and ROS scavengers (90). In each of these mitochondrial protective strategies, contractile function was improved and infarct size was reduced. In a recent study (12), we reported novel findings that hyperkalemic depolarizing cardioplegia was protective by means other than its effect on sparing high-energy phosphates; in the same article we also showed that lidocaine, a hyperpolarizing cardioplegic agent, also protected hearts in part by a direct action on the mitochondria, a concept supported by other studies (111, 566). Lipid-soluble local anesthetics could exert their cytoprotection in part by direct effects to attenuate complex I activity and to uncouple respiration (556). We found that lidocaine blocked complex I as suggested by an increased NADH without a change in FAD during lidocaine perfusion just before ischemia. This situation was similar to what we observed with the complex I inhibitor amobarbital (9).
It has long been thought that most of the cellular injury occurred during reperfusion because mPTP opening, mCa2+ uptake, and O2 •− generation were more likely to occur after ischemia. However, recent studies have reported that ischemic injury can lead to a persistent defect in OXPHOS during early reperfusion (113, 115, 117). In an ex vivo reperfusion study of guinea pig hearts, SS-peptide (Szeto–Schiller) was reported to prevent myocardial stunning and to significantly improve contractility (551, 554). Restriction of oxidative metabolism during early reperfusion using a hypoxic reperfusate attenuated mitochondrial and cardiac damage (282, 513). Pharmacologic inhibition of mitochondrial respiration on reperfusion with amobarbital also decreased mitochondria-driven myocardial injury (14). Thus, there are a variety of strategies that can target mitochondria to interrupt the link between ischemic damage to mitochondria and mitochondria-mediated cellular damage during reperfusion.
D. Preconditioning
Preconditioning is a mechanism for reducing organ I/R injury on return of blood flow to the tissue. It is mediated after the removal of a protective stimulus (brief ischemia or a drug) some time before the onset of index (longer, damaging) ischemia. The stimulus does not directly induce cytoprotection but rather some downstream signaling factors are evoked to provide a lasting protection (memory effect). Ischemic preconditioning (IPC) and pharmacologic preconditioning (PPC) of the heart decrease mitochondrial damage from subsequent index ischemia (113, 350). IPC was identified as an endogenous cytoprotective phenomenon, whereas PPC has the advantage of not requiring brief episodes of ischemia to elicit cellular protection. The cellular and mitochondrial protection elicited from IPC or PPC involves a coordinated interplay of trigger and effector mechanisms (113). There is much circumstantial evidence that mitochondria-derived ROS play an important role to initiate IPC and PPC (126, 143), which are effected by intracellular protein kinase cascades, especially PKCɛ (150, 283) and PKG (135, 241). The key effector signaling pathways converge on mitochondria to modulate oxidative metabolism before prolonged ischemia (113) or to prevent mPTP opening on reperfusion (251, 261, 273). For example, IPC, pre-ischemic diazoxide treatment, and post ischemic CsA treatment were each found to reduce infarct size after I/R injury in rat isolated hearts and this protective effect was blocked by the mPTP opener atractyloside when given on reperfusion (319). However, protection by IPC against mPTP opening after reperfusion injury appears to be better achieved by blocking the factors that induce mPTP opening rather than by inhibiting mPTP directly with drugs such as CsA (273). In other examples, inhibition of apoptosis by PKCɛ involves the mPTP, which might be a principal target of this kinase (38, 150). The protective targets in mitochondria also involve the interaction of PKCɛ with complex IV of the ETC, resulting in increased activity and increased mitochondrial ETC efficiency, decreased loss of cytochrome c, preservation of ΔΨm, and inhibition of mPTP opening due to decreased ROS production (150). IPC also diminishes mitochondrial dysfunction after ischemia and confers protection in the brain (150).
The mechanisms of cytoprotection afforded by PPC, for example, anesthetic preconditioning (APC), has been intensely investigated and discussed (290, 408, 409, 428, 429, 476, 477) and among other signaling factors common to IPC, they also involve sarcolemmal and the putative mKATP channels (55, 585). IPC and PPC are proposed to activate mKATP channels, which may lead to mild uncoupling of mitochondrial respiration. The mild uncoupling is believed to result in partial depolarization of the mitochondria, either as a result of K+ entry or activation of KHE in response to K+ entry (Section II,B). It is suggested that this depolarization may reduce mCa2+ uptake, thereby reducing mCa2+ overload and minimizing cellular damage. A mild uncoupling effect without changing ΔΨm could also increase mitochondrial ROS production (253), which can then lead to activation of downstream end-effectors. The putative mKATP channel agonist diazoxide was proposed to mediate cytoprotection in part on the K+ channel as well as by exerting a direct effect on the mPTP (236). But diazoxide also attenuates complex II activity and TCA cycle supported respiration, which appears to result in O2 •− production and hence may contribute to the O2 •− generation during PPC (236, 243 –245). Interestingly, the mitochondrial O2 •− scavenger N-mercapto-propionyl-glycine was shown to block protective effects of diazoxide (432). This and other studies suggest that an indirect effect of diazoxide on complex II is to induce O2 •− generation (243 –245).
Recently, we reported that the putative mCa2+-dependent K+ channel (KCa) may also play a role in modulating mitochondrial bioenergetics and provide preconditioning cardioprotection against I/R injury. Mitochondrial KCa agonists, such as KATP agonists, appear to mediate their cytoprotection through ROS-dependent mechanisms as the mitochondrial ROS scavenger MnTBAP blocked the protection (542). In this study we reported that preconditioning with NS 1619, a well-known KCa channel agonist, protected the heart from ischemic injury (542). NS 1619 markedly decreased the deleterious increases in ROS and m[Ca2+] with I/R injury, better preserved redox state (NADH and FAD), and improved cardiac function. Paxilline, an NS 1619 antagonist, and MnTBAP, both abolished these effects (542).
It was also reported that preconditioning-mediated protection through mPTP opening with atractyloside abolished the beneficial effects of IPC or NS 1619 in isolated rat hearts and myocytes (95). Sedlic et al. (508) reported recently that isoflurane, like DNP, protects cardiomyocytes in part via mild decrease in ΔΨm, which attenuates ROS production under stress and leads to a delay in mPTP opening. Other preconditioning drugs have resulted in improved function along with improved tissue redox state (NADH and FAD), decreased cytosolic and m[Ca2+], and reduced O2 •− and ONOO- during I/R (106, 289, 409, 542). These studies support the notion that modulation of mitochondrial bioenergetics by preconditioning induces cytoprotective maneuvers that are mediated by mitochondria.
Hypothermic preconditioning (HPC) has been reported to exert an effect via mitochondria to protect against cardiac I/R injury (291, 404, 653). Ning et al. (404) showed that brief hypothermia before global cardiac ischemia improved mRNA levels encoding the mitochondrial proteins ANT1 and β-F1-ATPase in hypothermia-treated hearts. This preservation of mitochondrial proteins was associated with improved ATP levels and better contractility after I/R. Kahliulin et al. (291) reported that HPC preserved mitochondrial integrity by reducing mPTP opening. Susceptibility to mPTP opening was assessed by Ca2+-induced mitochondrial swelling, as determined by light scattering (291). They observed that HPC protection was equivalent, or for some variables more effective, than the classical IPC in protecting the heart. We also showed recently (unpublished data) that hypothermic exposure increased resistance to Ca2+-induced mPTP opening (250). To date, little is known about hypothermic postconditioning in preserving mitochondrial function in I/R injury. We are currently examining this phenomenon because of its enormous clinical utility.
E. Postconditioning
Preconditioning (ischemic or pharmacologic), as the name implies, must be applied before an ischemic event to be protective; this limits its usefulness clinically since these procedures are seldom instituted early enough to minimize infarction (585, 636). Reperfusion necessarily contributes to cellular injury as evidenced by the surge in ROS production, mCa2+ overload due to activation of NHE and NCE, and mPTP opening (600, 636). Postconditioning may protect against I/R injury, at least in part by maintaining an acidic pH during reperfusion, which may inhibit mPTP opening. Interventions aimed at modifying reperfusion, or postconditioning, ischemic or pharmacologic, limit cell injury. Postconditioning has the advantage that it can be applied after the ischemic insult has occurred (146, 236, 287, 300, 428, 547, 599, 600). Experimentally, ischemic postconditioning (IPoC) often involves several brief occlusions of the lateral anterior descending coronary artery in a regional cardiac ischemia model, or brief intermittent occlusions and reperfusion of the aorta in a global heart ischemic model. Pharmacological postconditioning (PPoC) is the intermittent administration and washout of drug for one or several cycles immediately on reperfusion to confer protection during the later reperfusion period.
Administration of a wide variety of drugs [e.g., G protein-linked receptor ligands, adenosine (355, 633), opioids (223, 428), insulin, and statins (428)] immediately on reperfusion has been shown to provide protection as powerful as IPC. Many of the signaling pathways invoked by IPC are also implicated in PPoC (428). Consequently, as in IPC, the end effector of PPoC may reside in the mitochondria (428). For example, the cardioprotective effect of volatile anesthetics given as a PPoC agent (anesthetic postconditioning (APoC)) appears to be mediated through the mPTP. Thus, cardioprotection by PPoC, which depends on recovery of mitochondrial function, might ultimately involve prevention of mPTP opening. Whether similar signaling pathways in IPC and PPC are involved in mediating IPoC and PPoC remains unsettled. It appears that there exist protein kinase cascade that are activated at the time of reperfusion by both IPC and PPoC (428). However, it is unclear if activation of the reperfusion injury signaling kinase (RISK) pathway and consequent interaction with the mPTP is the ultimate step in pre- and post-conditioning protection. Furthermore, Feng et al. (185) also showed recently that both IPC and PPoC might provide protection by reversing ischemia-induced ANT dephosphorylation and improving ATP production. However, in PPoC, it is unlikely that the pro-survival signaling pathways occur rapidly enough to avert injury from the initial injury during reperfusion. It is likely that other faster activated pro-survival factors may be elicited during the initial phase of reperfusion.
In a recent study, Pravdic et al. (456) showed that mitochondrial function is critical for the protection afforded by APoC. In their study it was shown that APoC with isoflurane better preserved a more acidic matrix pH during reperfusion (Fig. 14). Thus, exposure to volatile anesthetic during early reperfusion/reoxygenation may delay opening of mPTP and contribute to preservation of mitochondrial integrity. This novel finding implies a direct preconditioning effect of volatile anesthetics on mitochondrial bioenergetics independent of mKATP channels.
We also showed recently in a pilot study (593) that postconditioning with the KCa channel agonist, NS 1619, administered for 10 min during initial reperfusion after 30 min of global no flow ischemia reduced mitochondrial ROS, improved cardiac function, and reduced infarction compared to the untreated group. Interestingly, we also showed that paxilline, a putative NS 1619 blocker, given for 10 min on reperfusion increased mCa2+ overload and decreased mitochondrial redox state, when compared to untreated hearts (Figs. 15A and 15B) and worsened functional return (593). These results suggested that the putative mKCa channels are opened during early reperfusion after ischemia and intrinsically contribute to reducing mitochondrial damage and concomitant cellular injury. These kinds of studies could eventually provide a better understanding of cytoprotection after ischemia and lead to new therapeutic option to treat ischemic heart disease.

XI. Other Mitochondrial Therapeutic Approaches
A. Lipid replacement therapy
Many lipophilic agents penetrate the IMM freely, including fatty acids. Undissociated molecules of long-chain fatty acids can easily penetrate the membrane (556). The integral structure of the mitochondrial membrane is an essential aspect of this process. The role of lipids in regulating mitochondrial function in health and disease was discussed above in Section IX,B (Mitochondria and the failing heart). In this section the focus is on re-establishing the lipid structure, lipid replacement therapy (LRT), after damage due to oxidative stress. LRT is not just the dietary substitution of certain lipids with proposed health benefits, it is the actual replacement of damaged lipids of cellular and organelle membranes (401, 402). Because mitochondria are a major source of ROS and their membranes have a high susceptibility to oxidative damage, this makes replacement of mitochondrial membrane lipids a potential therapeutic strategy. Oxidative stress causes major changes in ETC function as a result of direct lipid oxidation (402), which induces important changes in membrane properties. It is believed that these deleterious changes in mitochondrial lipid composition are involved in some of the maladies associated with mitochondria and the toxic side effects of chemotherapeutic agents.
One example is doxorubicin treatment, which leads to increased ROS and mitochondrial dysfunction (35, 455). The common features include changes in mitochondrial membrane fluidity, permeability, and ETC function (400, 454). Such lipid derangements may necessitate treatments that could prevent the occurrence of the condition, for example with antioxidants, and once the lipid derangement has occurred, LRT could be initiated to repair the damage. It has been suggested that a combination treatment is more effective than either treatment alone in alleviating symptoms associated with the mitochondrial structural damage (402, 460). LRT has been used to mitigate damage to normal cells during chemotherapy without a reduction in therapeutic results (402).
Administration of NTFactor, a lipid oral replacement supplement, with vitamin supplements to scavenge ROS, has been reported to reduce unwanted effects associated with oxidative stress during chemotherapy (402). Earlier studies (460) also showed that use of sunflower or olive oil as lipid dietary sources in combination with α-tocopherol lowered mitochondrial hydroperoxide levels. These studies suggest that LRT plus scavenging could represent a useful adjuvant therapy in management of diseases where mitochondrial function is impaired as a result of excess ROS and alteration of lipid structure.
It is interesting that tumor cells adopt this strategy of LRT to protect themselves from death (164, 360). Indeed phosphotidylcholine remodeling results in an altered lipid profile and it is a characteristic of colorectal cancer cells (360). It was shown in these cells that HK binding to VDAC facilitated uptake of cholesterol and the elevated cholesterol content amplified the binding of HK to VDAC. This adaptive response, along with the observation that increased mitochondrial cholesterol blunts the ability of Bax to initiate dysfunction in cancer cells, makes them refractory to injury (443). It is evident that the therapeutic strategies adopted by neoplastic cells for survival involve mitochondria and this may pose yet another dilemma in the selective treatment of a specific disease by exclusively targeting cell specific mitochondria.
B. Transactivator of transcription proteins and mitochondrial therapy
The mitochondrial protein import pathways, specifically the mitochondria signal peptide-tagged cargo, have been used to deliver DNA molecules to the matrix. Thus, the targeting of covalently linked genetic information to mitochondrial peptide import machinery could potentially be used to rectify a mutant mitochondrial genome similar to classical gene replacement therapies that attempt to replace a corrected copy of defective nuclear genome (28). However, it is sometimes technically challenging to insert small DNAs to introduce small peptides in cells. The transactivator of transcription (TAT) protein-penetrating transduction system is being used to introduce small peptides into living cells (86, 507), as these peptides are often unstable and susceptible to cellular degradation. TAT-mediated protein transduction occurs in a rapid fashion that is independent of receptors and transporters (86, 238, 465, 507). Disorders of some mitochondrial proteins can be corrected by this system of uptake. The technique has been utilized in the isolated cell (86, 465, 507) and in the whole organ model where it has been shown to facilitate peptide uptake in the isolated heart (238).
TAT proteins with their “cargo” rapidly cross the cell membrane and enter mitochondria where they can replace mutated endogenous peptides (465). Rapoport et al. (465) showed recently that the TAT-mediated replacement approach could be used to correct for a mutated component of a protein complex, in this case the E3 subunit shared by the α-ketoacid dehydrogenases like pyruvate dehydrogenase (PDH). That is, this approach was used to increase the activity of the PDH complex, which before treatment showed low activity due to mutations in E3. TAT approaches could provide a new treatment for enzyme deficiencies as well as for other mitochondrial and metabolic disorders (465).
TAT-mediated transduction of the mutant form of Bnip3 (TAT-Bnip3ΔTM), a pro-apoptotic member of the Bcl family of mitochondrial proteins expressed in the adult myocardium, was also reported to reduce I/R-induced injury. The wild-type Bnip3 induced apoptosis in part by increasing the fragmentation of mitochondrial connectivity and causing mitochondrial dysfunction (i.e., loss of ΔΨm, increased cytochrome c and AIF release, and disruption of energy production) (238). An increase in our understanding of the role of these proteins and the strategies to manipulate them in mitochondria could be important in the development of effective therapies.
C. Molecular genetic approaches
Genetic maneuvers of various types have been attempted to reverse mtDNA-related diseases. These include suppression of mutant mtDNA expansion and manipulation of mtDNA replication by import into the mitochondrion of endonucleases that might selectively destroy a specific mutant sequence. In a recent study, Alexander et al. (13) showed that in cells heteroplasmic for the T8993G mutation that causes NARP (Section VIII,B), infection with an adenovirus, which encodes the mitochondria-targeted restriction endonuclease, led to selective destruction of mutant mtDNA. This led to a significantly increased rate of O2 consumption and ATP production and concurrently, decreased rate of lactic acid production in these cells, which is a marker of mitochondrial dysfunction. The specificity of this mitochondrial approach was demonstrated by the absence of nDNA damage (13).
Other potential therapeutic approaches involve overexpression of targeted proteins, for example CypD in cancer therapy, or its selective inhibition using a siRNA approach during I/R-related cellular injury (28). Other approaches are designed to impede the action of Bcl-2 by use of antisense technology to inhibit Bcl-Xl expression, or alternatively, to target pro-apoptotic Bcl-2 peptides to mitochondria (28). Other investigators have used similar approaches. For example, gene therapy using adenoviral Bax-delivering vectors has been successful in activating apoptosis (631) and a similar approach has been used to induce apoptosis using stable generated BH3 peptidomimetics designed to block Bcl-2 and to activate Bax and Bak (603). In another study, Li et al. (339) used a similar approach to develop Smac/DIABLO mimetics to inhibit the action of IAPs.
The inter- and intra-organelle communication between mitochondria as a group and mitochondria with ER and the nucleus are important factors in maintaining normal cellular function; disturbances in any of these complex systems or their coordinated activity will generate disease. The Bcl family of proteins is also believed to influence mitochondrial fission/fusion. For example, Bax and Bak promote fragmentation of the mitochondrial network, possibly by activating fission machinery; on the other hand, targeting these fission proteins through expression of Bcl-Xl prevented cytochrome c release (518). These findings indicate that Bcl family proteins can influence mitochondrial fission and fusion. However, another study reported that Bak blocks mitochondrial fusion to induce fragmentation. By this function, Bak may collaborate with Bax to permeabilize the OMM leading to apoptosis (72, 73). Thus these Bcl family proteins appear to be attractive therapeutic targets in fighting neoplastic cells (4, 131).
The disruption of the mitochondria to ER communication, required for normal cellular function, could be targeted by molecular approaches that would up- or downregulate the proteins involved in the link between these organelles. In the mitochondria–nuclear communication, overexpression of PGC1-α and or HSP70 and 60, which are involved in the importation of mitochondrial proteins, could also serve as targets for therapy to improve mitochondrial function. Other genetically targeted approaches have been used for IMM proteins and they have also furnished some effectiveness as a remedy against mitochondria-related pathologies (28). Yet, which, or if any of these mitochondrial genetic approaches will become clearly applicable in treating human maladies remains to be determined.
D. Mitochondria and caloric restriction
Morbid obesity is associated with numerous diseases from the metabolic syndrome to cancer. Indeed, obesity as a result of high-energy intake has been shown to increase the risk of age-related cognitive decline (622). Caloric restriction (CR; either a decrease in food intake or intermittent fasting) delays the rate of aging in many species and increases resistance to diseases (41, 129). In animal models of aging and neurodegenerative diseases, CR protected hippocampal, striatal, and cortical neurons, and ameliorated functional decline (622). CR has been shown to minimize age-related loss of mitochondrial function and biogenesis in several tissues, including heart, liver, skeletal muscle, and brain (190, 588) and to mitigate the severity of dysfunction. For example, studies have shown that the complete maintenance of skeletal aerobic contractile function with aging by CR is associated with improved mitochondrial function and a reduction of the age-related decline in mitochondrial capacity (255). The decline in mitochondrial biogenesis in obese animal has been associated with a decrease in PGC-1α (128). It was reported that the rate of decline of PGC-1α was lower in CR animals than in non-CR animals (255). This increase in mitochondrial biogenesis, coupled with increased oxidative capacity and lower ROS levels (62), might translate to less accumulation of oxidatively damaged mitochondria, thereby accounting for the protection of mitochondrial function with CR (255).
E. Mitochondria and dietary supplements
The potential contribution of impaired energy metabolism to behavioral disorders and migraine headache has been considered over the years. Initial studies correlated deficiencies in riboflavin or folate to the prevalence of depression. Higher dietary intake in folate and riboflavin in recent clinical trials in Japan was associated with lower prevalence of depressive symptoms in some patients (390). Riboflavin's (vitamin B2) effect is required for the formation of FAD+, which is critical for the TCA and as an electron donor to complex II. FAD+ is a key cofactor in folate-dependent methylation pathways that would lead to improved neuronal metabolism and improved function. Riboflavin in combination with CoQ10 has been shown to reduce neurological episodes associated with migraine headaches (505). The combined approach augments complex I activity and has been used in clinical trials to demonstrate their combined effectiveness for preventing migraine attacks (459). Niacin has also been used as a supplement to boost mitochondrial energy metabolism by increasing substrate availability to complex I; in this way it might act to deter/reduce migraine headache (459). Clearly, the efficacies of these approaches remain to be resolved, their side effects remain under scrutiny, and they are limited by the fact that some studies have failed to make any association of migraine headache or dietary supplements with specific mitochondria deficits.
XII. Mitochondria Age and Lifespan
A. Mitochondria and age-associated diseases
The term aging generally refers to organismal senescence, a process whereby cells lose their ability to divide after a number of cell divisions, and by a decline in the capacity of an organism to respond to stress. Although aging is not a disease, the frequency of many disease processes increases with age. Mitochondria are proposed to be critical components in the process of aging, in the regulation of the cell cycle, and in limiting cellular lifespan (401, 632). Mitochondria are in turn a primary target of the aging process, as evidenced by a decline in mitochondrial oxidative capacity in both skeletal and heart muscle with age (255, 292, 328). Aging has been associated with excessive oxidative stress and over production of RNS (401). Indeed, oxidative/nitrosative stress is thought to be an important contributor to the degeneration of long-lived postmitotic cells such as cardiomyocytes and neurons. This explains in part the relation between acquired cardiac and neurodegenerative diseases and aging (391).
In aged mitochondria, a defect in mtDNA is coincident with a decrease in complex IV activity and in some cases reduced activity of complex I (130). Although we were unable to show age-related changes in complex I activity in mitochondria, we did show a decrease in complex III activity, but only in interfibrillar mitochondria (328). These defects are conducive for increasing oxidative stress that further predisposes mitochondria to injury (Fig. 16). The exact mechanism for the relationship between impaired mitochondria and aging remains unclear. However, Melov et al. (371) recently reported that in older animals a characteristic mitochondrial “signature” of declining mitochondrial function could be reversed in part through exercise and suggested that a sufficient reserve capacity exists in the genome to reverse certain age-dependent expression profiles back to more youthful genotypes.
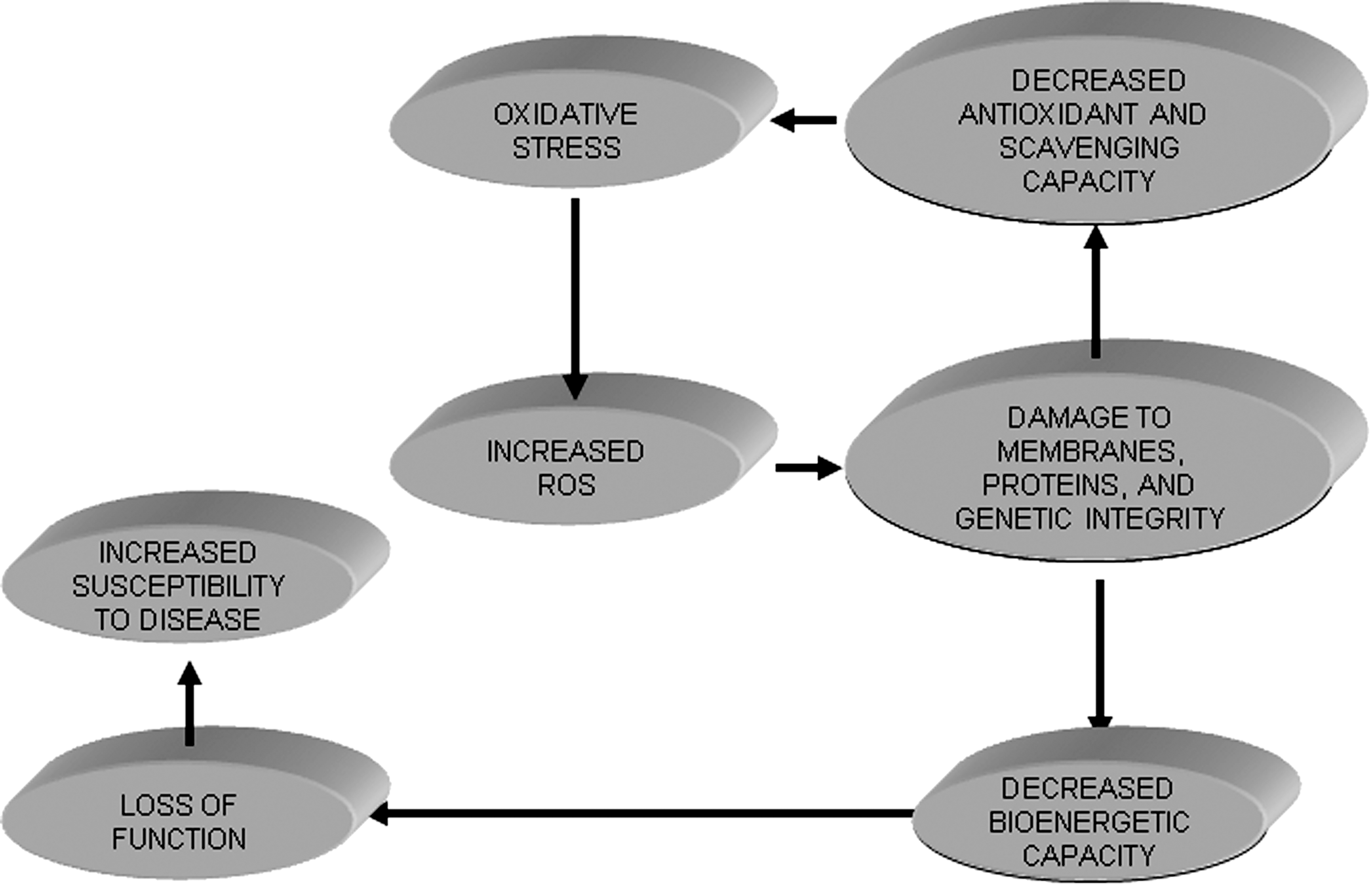
The current view is that mtDNA mutations accumulate at a rate that is several folds higher than nDNA mutations (292). This increase in mutation rate occurs primarily in postmitotic organs such as brain, skeletal muscle, and heart, compared to cells that undergo frequent mitosis (568). Thus the evidence, albeit tenuous, supports the notion that accumulation of ROS over time is wholly or partially responsible for aging. Certainly, increased oxidative stress has been associated with increased mtDNA strand breaks during aging and in the aged MnSOD-deficient mice (614). However, this belief has been questioned for vertebrate and invertebrate animal models of aging (172, 388). Furthermore, targeting of mitochondria to “reverse” the aging process and therefore mitigate age-related diseases has remained elusive. Indeed, overexpression of sod2 and catalase genes in the fruit fly D. melanogaster has yielded the opposite outcome, that is, lifespan was decreased instead of increased (16). In another study in the nematode C. elegans, Doonan et al. (172) showed that overexpression of sod genes did not alter lifespan, whereas other studies reported that ROS might in fact be responsible for extending the lifespan of C. elegans (590).
Some studies have consistently reported a role for mitochondrial ROS in aging-related maladies. This includes some of the neurodegenerative diseases discussed above (Section IX,E); others include presbyacusis, the hearing loss associated with aging as a consequence of the progressive deterioration of the cochlear mtDNA (391, 451). Antioxidants targeting mitochondria were reported to reduce this age-related hearing loss (127). Toxic aldehydes, including HNE, an end product of lipid peroxides, are known to accumulate in the brain in neurodegenerative disease. Mitochondrial aldehyde dehydrogenase 2 (ALDH2) detoxifies HNE by oxidizing the aldehyde group. ALDH2-deficient neuronal cells show increased vulnerability to HNE. The increased oxidative stress may in part explain some age-dependent neurodegeneration, such as loss of pyramidal cells and marked deficits in cognition. These neurological deficits could be mitigated by expression of ALDH2 (419). The implication is that patients deficient in the enzyme may benefit if treated with activators.
In the aged heart, ischemic damage to mitochondria is superimposed upon aging-induced defects in mitochondrial oxidative metabolism (327, 328, 330). The aged heart generally sustains increased damage during I/R (30, 192, 326). Isolated, buffer-perfused hearts from 24 mo-old Fischer 344 rats sustained greater myocardial injury after I/R than hearts from 6 mo-old adult controls (326, 349). Unfortunately, IPC and APC were found to provide minimal cardioprotection in the aged rat (1, 565) and in human hearts (376). Older patients with ischemic heart disease generally have impaired recovery of myocardial function after cardiac surgery or other cardiac interventions when compared to younger patients (471). There is evidence also that older patients are more sensitive to ischemic damage and have a poorer prognosis and higher mortality due to acute myocardial infarction (361). Thus, the potentially useful approach of conditioning to protect aged myocardium appears not to be very effective. This highlights the importance of considering other approaches to modulate mitochondrial oxidative metabolism to protect the aged heart.
In the aging heart, enhancing basal glucose uptake and lowering fatty acid oxidation by overexpression of GLUT1 in the heart protects against I/R injury (351). Also the observed decrease in OXPHOS in the older heart may be attributed to an aging-induced defect of cytochrome c oxidase (113). Studies have shown that acetylcarnitine reversed an aging-induced decrease in cytochrome c oxidase activity, as well as enhanced the maximal rate of OXPHOS in mitochondria to rates observed in the younger hearts (87, 329). Acetylcarnitine had no effect on the extent of myocardial or contractile recovery in the adult heart. In an earlier study, Hagen et al. (234) demonstrated that acetyl-L-carnitine restored mitochondrial cardiolipin levels in hearts of old animals compared to hearts of young animals and improved OXPHOS. As in the adult heart, the ETC contributes to mitochondrial damage during ischemia in the aged heart (329). Amobarbital treatment immediately before ischemia protected OXPHOS in the aged heart on reperfusion (Tanaka–Esposito et al. unpublished observations). These observations provide strong experimental support for the association of aging-related defects in mitochondrial metabolism to the enhanced cardiac damage in older hearts observed following I/R.
The increased susceptibility in the aging heart to I/R injury could also be attributed in part to an increased sensitivity to cell stress, possibly due to a reduction of antioxidants and chaperone proteins, altered mitochondrial respiration, and a shift from the survival pathway to the cell death signaling pathway. Thus, a better understanding of molecular and cellular mechanisms of aging could improve not only medical care of the elderly, but also may offer some hope in finding feasible solutions to slow down the aging process as well.
B. Mitochondrial p66shc and lifespan
The 66 KDa isoform of the growth factor adapter Shc (p66shc) is located in IMS and may play a role as a redox enzyme involved in ROS generation via electron transfer from reduced cytochrome c (92, 211). The protein is a signaling link between cellular stress and mitochondrial pro-apoptotic activity. Cellular oxidative stress led to phosphorylation of the protein and translocation into the mitochondrion led to activation of the apoptotic program (452). p66shc is implicated in lifespan determination and is in crosstalk with other lifespan proteins, p53, a tumor suppressor protein, and MnSOD to regulate oxidative stress in the organelle (436). As discussed above, accumulation of oxidative stress with age is considered a mediator of age-associated diseases (92). Mitochondrial p66shc is reported to contribute to tissue injury during I/R and it has been shown to contribute to aging of vessels and concomitant functional impairment (132). Redox-defective mutants of p66shc were unable to induce ROS production and swelling or to mediate apoptosis in vivo (132). Indeed mice deficient in p66shc (p66shc-/-) gene showed less ROS production and extended lifespan. The protein has also been implicated in the signal transduction pathway relevant to hyperglycemia-induced vascular damage, and hence, represents a novel therapeutic strategy in diabetic vascular complication (92), and could be a potential target of pharmacological approaches to slow aging (452).
XIII. Caveats and Potential Limitations in Mitochondrial Drug Targeting
Targeting the mitochondria for therapeutic purposes poses a dilemma of how to protect, while at the same time, preserve the normal aspects of cellular functions that maintain viability. A vivid example is the role of ROS as a physiological modulator of cellular function as well as a mediator of cell death (543). Given the multiple pathways potentially affected by a change in mitochondrial function, development of drugs targeting mitochondria requires judicious safety assessment and risk management (239). Muravchick (391) reinforced this in a recent article by stating that “therapeutic strategies that suppress or block the effects of putative pro-apoptotic agents may produce unintended interruptions of other cell functions and actually compromise viability”.
An emerging therapeutic dilemma is the systemic manipulation of the pro-apoptotic proteome of the OMM. In cancer therapy, an attempt to promote pro-apoptotic mechanisms in tumor cells may unwittingly worsen the pathological state of normal differentiated cells. As discussed above, the goal in cancer therapy, especially therapy against lymphomas, is to overcome inhibition of cell apoptosis in cells immortalized due to overexpression of Bcl-2. In contrast, the same inhibition of Bcl-2 in tissues with normal expression of Bcl-2, especially relatively postmitotic tissues such as the heart, may predispose to unanticipated deleterious cell death. There is an emerging concern that Bcl-2 blockade utilized in cancer chemotherapy may predispose to cardiomyopathy, especially if combined with other therapies including doxorubicin that can also result in cardiomyopathy.
The use of mitochondria as a therapeutic target can also be limited by the changes the organelle undergoes during the different phases of development. For example, the younger heart shows greater sensitivity to anesthetic preconditioning (APC) than the older heart (376), which may correspond to the period during which bioenergetic function may begin to decline. Ontogenetic defects in mitochondrial function that lead to depressed mitochondrial bioenergetics due to inherited mitochondrial cytopathies could result in altered responses to pharmacological interventions. For example, an anesthetic given in the APC paradigm could unintentionally lead to pathologic levels of ROS that cause cell damage (391) instead of the small amount of ROS needed to mediate cellular protection.
The desired effect of any targeted drug or gene delivery can be achieved only if the bioactive molecule is delivered to the destined organ and/or cell type, and also to desired subcellular location. To achieve this specificity of target, a more efficient and selective delivery vehicle should be constructed to enable transport of the bioactive drug to the desired mitochondrial population and site of mitochondria. As presented above, attempts to engineer such mitochondria homing devices is being actively sought. Although some of these agents, the SS-peptides, or use of TPP+ moieties, are currently being considered, whether these targeted entities can be delivered to the desired mitochondria in vivo is highly sought. This raises a critical concern about the specificity of their biodistribution, penetration, and their bioactivity and pharmacodynamics. These concerns remain to be resolved. Some of the examples given below illustrate the complications associated with mitochondria-targeted approaches.
The risk associated with using uncouplers to mitigate mitochondrial-related pathologies has been recognized. DNP, which wastes energy and produces heat by OXPHOS uncoupling, was once used as a weight-loss drug but was abruptly discontinued due to undesirable side effects and even death (570). Aside from their very limited window of action, another major problem with uncouplers is that they must be designed for tissue specificity to maximize their therapeutic potential and minimize side effects. A broad usage of uncouplers could lead to reduction in ATP production in untargeted mitochondria (218). Systemic induction of UCP2 in pancreatic β-cells could lead to decrease in ΔΨm, which in turn could make insulin release by the cells less responsive to changes in glucose concentration (218).
In pancreatic β-cells, mKATP channels are pivotal in regulating plasma glucose levels. Use of KATP channel inhibitors for treatment of type-2 diabetes could have a serious down side because KATP channels are widely expressed in a variety of tissues, including cardiac, skeletal, and nervous system. Blockade of KATP channels in non-β pancreatic cells during type 2 diabetes treatment may engender severe side effects. This could limit the potential beneficial effects of IPC or PPoC, which appear to use the KATP channel for protection (378). Therefore, potential undesirable effects related to the use of sulfonylurea drugs are cardiovascular complications (303). Thus, the challenge is to design uncoupling agents and other drugs that are mild and also tissue selective/specific to decrease ROS production without significantly affecting ATP production in other tissues (218, 378).
Another pharmacologic delivery problem is how to target brain mitochondria due to the relative impermeability of the blood–brain barrier (BBB). For example, MnTBAP is effective in ameliorating numerous pathological cardiovascular abnormalities and in extending the lifespan of sod2-deficient mice (369, 445). However, MnTBAP does not cross the BBB and consequently, ROS can accumulate in the brain of these animals and cause abnormalities such as ALS, tremor, and other movement disorders (233, 370).
It is evident that an overarching concern in development of mitochondria-targeted drugs is to make the distinctions between nonadverse drug effects that are physiologic, pharmacologic, or adaptive, and the adverse effects that lead to unacceptable deleterious consequences. Despite these limitations and challenges, the approaches outlined in this review have features that suggest a potential utility of targeting mitochondria for therapeutic purposes.
XIV. Conclusion and Perspectives
This review has focused on the growing body of evidence that mitochondria, although the major source of ATP, are also intimately involved in the etiology of numerous human pathologies. Different cells and tissues have distinct sensitivities and responses to mitochondrial dysfunction. These differences are probably due to the cell-type specializations that rely on particular functions of specific mitochondria (105). Appreciation of these differences will be important when considering mitochondrial therapeutic strategies to combat diverse groups of maladies such as coronary heart disease, heart failure, hypertension, diabetes, cancer, and neurodegenerative diseases. Some of these diseases have a potential for considerable “crosstalk” (254) as a result of mitochondrial oxidative stress, and similar pathways in the disease process may be involved or overlap. Therefore, the goal in development of effective treatments for each of these diseases becomes a more pressing issue. It is evident that much future work is required to develop novel and more tissue specific mitochondria-targeted approaches or interventions that will furnish a greater efficacy and selectivity for the disease of interest, but leading to fewer undesirable effects.
Footnotes
Acknowledgments
The authors wish to thank Mohammed Aldakkak, M.D. for his superb skills in generating the quality figures shown in this review article, and for his thorough critique of the manuscript. We would also like to extend our gratitudes to Johan Haumann, M.D., and Makut Sharma, Ph.D. for their careful review and critique of this manuscript, and to Daniel A. Beard, Ph.D. for his contribution in some aspects of this manuscript and for his financial support (HL094317). This work was supported in part from the National Institutes of Health (HL089514 to DFS, and HL073246 to AKSC, 2PO1AG15885 to EJL), VA Research Merit Award 8204-05P (to DFS), a Grant-in Aid from the American Heart Association (0855940G to DFS), and by the office of Research Development, Medical Research Service, Department of Veteran Affairs (McGuire VAMC, Richmond, VA to EJL and Zablocki VAMC, Milwaukee WI to DFS).
Abbreviations Used
Reviewing Editors: Enrique Cadenas, Andreas Daiber, Cherubino Di Lorenzo, Sanjeev Gupta, Sabzali Javadov, Jiri Neuzil, and Michael Roth