
Abstract
Reactive oxygen species (ROS) serve as mediators of signal transduction. However, mechanisms of how ROS influence the target molecules to elicit signaling event have not been defined. Our laboratory recently accumulated evidence for the role of protein carbonylation in the mechanism of ROS signaling. This concept originated from experiments in which pulmonary artery smooth muscle cells were treated with endothelin-1 to understand the mechanism of cell growth. Endothelin-1 was found to promote protein carbonylation in an endothelin receptor- and Fenton reaction-dependent manner. Mass spectrometry identified proteins that are carbonylated in response to endothelin-1, including annexin A1. Our experiments generated a hypothesis that endothelin-1-mediated carbonylation and subsequent degradation of annexin A1 promote cell growth. This mechanism was found also to occur in response to other signaling activators such as serotonin and platelet-derived growth factor in smooth muscle cells of pulmonary circulation, systemic circulation, and the airway, as well as in cardiac muscle cells, suggesting the universal role of this pathway. We also discovered a process of decarbonylation that defines transient kinetics of carbonylation signals in certain conditions. We propose that protein carbonylation and decarbonylation serve as a mechanism of signal transduction. Antioxid. Redox Signal. 12, 393–404.
Reactive Oxygen Species and Cell Signaling
Traditionally, reactive oxygen species had been considered to nonspecifically and indiscriminately react with biological molecules, including lipids, DNA, proteins, and small molecules, and cause inhibition of biologic events (27). The concept of the role of reactive oxygen species in oxygen poisoning that might resemble radiation damage was developed by Gerschman, Gilbert, and co-workers in 1954 (21). Subsequently, the discovery of superoxide dismutase by McCord and Fridovich (47) was a pivotal point that confirmed the importance of reactive oxygen species in eliciting damage and that biological systems have antioxidant defense mechanisms.
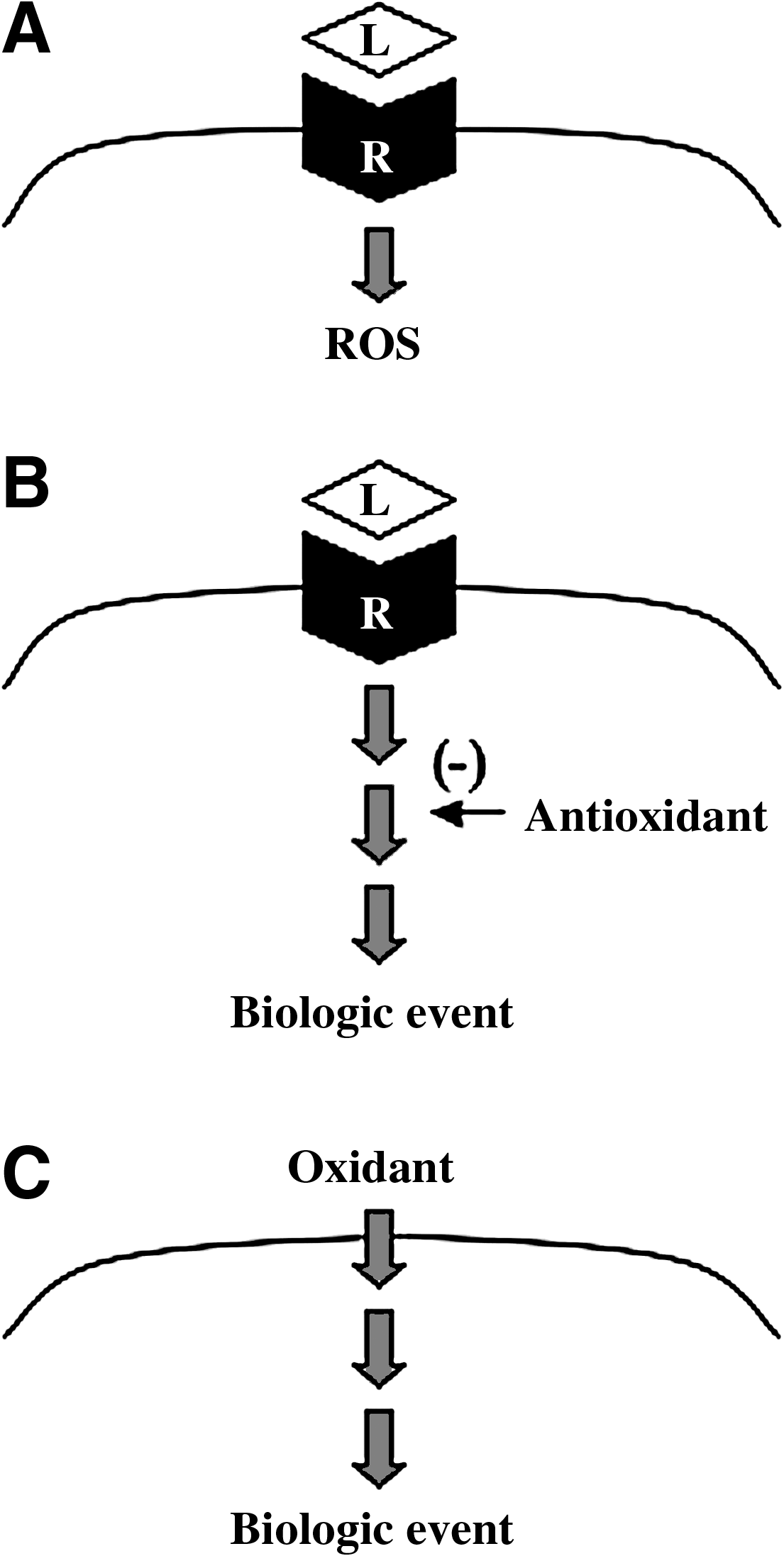
However, through observations that (a) ligand–receptor interactions produce reactive oxygen species, (b) antioxidants block signal transduction, and (c) reactive oxygen species can stimulate signaling events (Fig. 1), reactive oxygen species have been proposed to serve as second messengers in signal transduction processes (58, 67, 69). During the 1970's, the concept that H2O2 was the second messenger for insulin signaling emerged (12, 46). In 1991, H2O2 was proposed to be a second messenger for the activation of NF-κB transcription factor and human immunodeficiency virus (HIV) gene transcription in T cells (58). This proposal, which gained considerable attention, was derived from the authors' experimental observations that H2O2 activates NF-κB along with previously published data showing that an antioxidant N-acetylcysteine inhibits HIV activation (53) and NF-κB activation (63) induced by tumor necrosis factor-α or phorbol 12-myristate 13-acetate. In aortic smooth muscle cells, Rao and Berk (52) demonstrated that ROS promote cell growth. Subsequently, in these cells, ROS were reported to mediate signal transduction induced by angiotensin II (22) and platelet-derived growth factor (PDGF) (66), and nonphagocytic NAD(P)H oxidase was found to be a primary producer of ROS in cell signaling mechanisms (22). In bovine pulmonary artery smooth muscle cells, serotonin activates the production of superoxide (35, 36) and H2O2 (37). In these cells, it was proposed that NAD(P)H oxidase is the source of reactive oxygen species. In contrast, Lawrie et al. (33) reported that serotonin produces ROS by monoamine oxidase A in human pulmonary artery smooth muscle cells. Monoamine oxidase A catalyzes two-electron reduction of O2 to H2O2 and oxidative deamination of serotonin to 5-hydroxyindole-3-acetaldehyde. Endothelin-1 has been reported to produce reactive oxygen species via NAD(P)H oxidase, and antioxidants block endothelin-1-induced cell proliferation in fetal ovine pulmonary artery smooth muscle cells (74, 75). Thus, roles of reactive oxygen species in cell signaling have been supported by a large number of studies and are now widely accepted (69).
Molecular Targets of Oxidant Signals
For the mechanism of oxidant signaling, the receptor activation has been shown to produce reactive oxygen species via NAD(P)H oxidase in various biological systems, and the mechanism of nonphagocytic NAD(P)H oxidase activation is well understood (22, 71). While a number of reports suggest that biologic events triggered by signaling stimuli involve reactive oxygen species, it is not clear exactly what the molecular targets of ROS in signal transduction mechanisms are. Reactive oxygen species targeting protein cysteine thiols has been a popular proposed mechanism (13, 49, 59, 72). However, the exact mechanism of reactive oxygen species targeting in cell signaling events has not been defined and is an important gap of knowledge in cell biology (Fig. 2A).
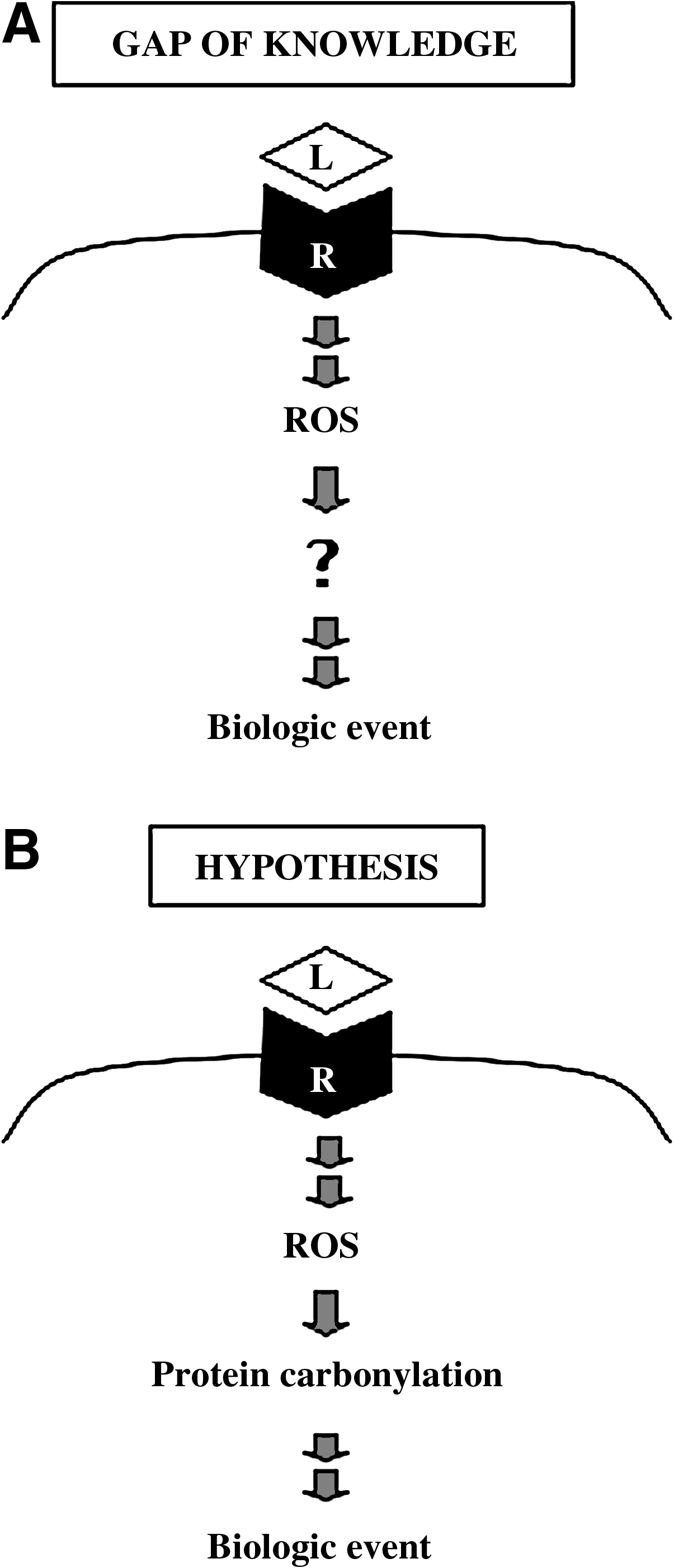
Our laboratory recently discovered that one mode of protein oxidation called carbonylation can be triggered in response to ligand–receptor interactions (78), revealing the possibility that protein carbonylation may play a role in reactive oxygen species signaling (Fig. 2B).
Protein Carbonylation
Reactive oxygen species can oxidize various biological molecules, including proteins, DNA, lipids, and small molecules. During various oxidative stress conditions, protein oxidation often results in the inactivation of protein functions. Carbonylation is one of protein oxidation processes that can occur directly on proteins, often in response to metal-catalyzed formation of hydroxyl radicals (39, 64). Oxidation of proline or arginine side chain forms glutamic semialdehyde, while lysine oxidation results in aminoadipic semialdehyde, introducing carbonyl groups (RR'C = O) in the protein structure. The hydroxyl group of threonine side chain can also be oxidized to form the carbonyl group. 2,4-Dinitrophenylhydrazine (DNPH) reacts with the carbonyl group of aldehyde or ketone and forms a hydrazone derivative (DNP) in the following reaction:
This hydrazone derivative can be detected spectrophotometrically. The availability of the antibody against dinitrophenylhydrazone-derivatized proteins has also allowed the use of immunological techniques such as Western blot analysis (39, 41, 42, 60). Measurements of carbonylated proteins have been well used as a protein oxidation marker in various oxidative stress and disease conditions in human and animal studies. Carbonylation of some proteins might result in a loss of protein functions. Further, carbonylated proteins have been shown to undergo proteasome-dependent degradation (25, 38 –40).
More recently, DNPH-derivatizable protein carbonylation formed by the addition of aldehydes generated from peroxidation of unsaturated lipids such as 4-hydroxynonenal to side chains of lysine, histidine, or cysteine residues has emerged to have important biologic functions (24, 57). In this article, we refer to this type of protein which is mediated through lipid peroxidation as “secondary protein carbonylation,” while metal-catalyzed oxidation directly of proteins as “primary protein carbonylation.”
Discovery of Activation of Protein Carbonylation by Ligand–Receptor Interactions in Mammalian Cells
It has been known that ROS can form carbonylated proteins and protein carbonyls have been well used as a protein oxidation marker. Recently, our laboratory provided a series of evidence that suggest the possible roles of protein carbonylation in signal transduction processes (44, 77, 78). The initial discovery was made in the study of the mechanism of endothelin-1 signaling in mammalian pulmonary artery smooth muscle cells for the purpose of understanding the pathogenesis of pulmonary hypertension.
Pulmonary hypertension can occur secondary to various lung and heart diseases such as acute lung injury, chronic obstructive pulmonary disease, sleep apnea syndrome, left ventricular failure, congenital heart defects, and post-thrombotic disease; and could worsen the prognosis of patients with various heart and lung diseases. Pulmonary hypertension can also occur in idiopathic and familial forms, which are rare, but often fatal. The incidence of pulmonary hypertension can also be increased by the use of some appetite suppressants. Other triggers of pulmonary hypertension include monocrotaline extracts, inhaled solvents, methamphetamine, cocaine, contaminated rapeseed oil, L-tryptophan, inflammatory disorders, sickle cell diseases, and infections such as HIV. Pathologic characteristics of pulmonary hypertension include vasoconstriction within the pulmonary vasculature, as well as histological abnormalities of the vascular wall with hypertrophy of smooth muscle, resulting in a mean pulmonary arterial pressure of >25 mm Hg at rest or >30 mm Hg during exercise. The increased pulmonary arterial blood pressure ultimately produces right ventricular heart failure and death. The median survival for patients with pulmonary arterial hypertension has been reported to be 2.8 years from the time of diagnosis (55).
The pathologic features of pulmonary hypertension include abnormalities of vascular structure and function in each compartment of the pulmonary circulation. The vascular lumen has a prothrombotic tendency, the endothelium produces excessive vasoconstrictors, and smooth muscle cells are overloaded with Ca2+. These events cause pulmonary vasoconstriction and vascular smooth muscle cell growth. By the time patients with pulmonary hypertension obtain clinical attention, pathologic lesions are developed, which include pulmonary artery medial hypertrophy, adventitial thickening, and neointimal lesions. Abnormal growth of smooth muscle cells is a main component of vascular remodeling, and is in part regulated by vasoactive factors such as endothelin-1 and serotonin.
Endothelin-1, a potent vasoconstrictor, is also a mitogen of pulmonary artery smooth muscle cells (28, 30). The activation of either ETA or ETB receptors can induce the proliferation of pulmonary artery smooth muscle cells (14) via generating reactive oxygen species (74, 75). Plasma endothelin-1 was found to be elevated in patients with pulmonary hypertension (65). Human studies have shown that an endothelin-1-receptor antagonist bosentan increased exercise capacity and improved hemodynamics in patients with pulmonary hypertension (10, 54). In animal models of pulmonary hypertension, increased endothelin-1 expression was observed (80) and endothelin-1 receptor antagonists blocked the progression of this disease (48).
The concept that protein carbonylation might play a role in cell signal transduction was initially developed through the experimental observations that endothelin-1 can increase protein carbonyls as early as 5–10 min in cultured bovine pulmonary artery smooth muscle cells (Fig. 3) (78). Endothelin-1 is a vasoactive peptide that causes smooth muscle contraction, as well as smooth muscle growth and survival (70). Our experiments were performed in order to understand the mechanism of endothelin-1 signaling in pulmonary artery smooth muscle cells that plays important roles in the development of pulmonary hypertension. Endothelin-1 has been shown to produce reactive oxygen species in pulmonary artery smooth muscle cells (74, 75), and these species have been proposed as signaling mediators for the growth of smooth muscle cells (52, 66). However, the exact molecular mechanism of reactive oxygen species signaling has not been defined. Cultured bovine pulmonary artery smooth muscle cells were treated with endothelin-1, cell lysates were prepared and derivatized with DNPH, and the samples were subjected to SDS-PAGE and immunoblotting with the antibody, which recognizes DNPH-derivatized proteins. As shown in Fig. 3, we observed that endothelin-1 increases carbonylation of various proteins with transient kinetics with a peak at ∼10 min, and carbonylation levels were subsequently decreased. Such kinetics is similar to protein phosphorylation such as ERK MAP kinase phosphorylation that can be triggered by various ligands (32, 70). The decrease in protein carbonylation (which we named ‘decarbonylation’) was surprising because protein carbonylation is thought to be quite stable and such rapid decrease in protein carbonylation was not known. We will discuss the protein decarbonylation mechanism more in a later section of this article. These experiments defined that endothelin-1 can promote protein carbonylation and decarbonylation.
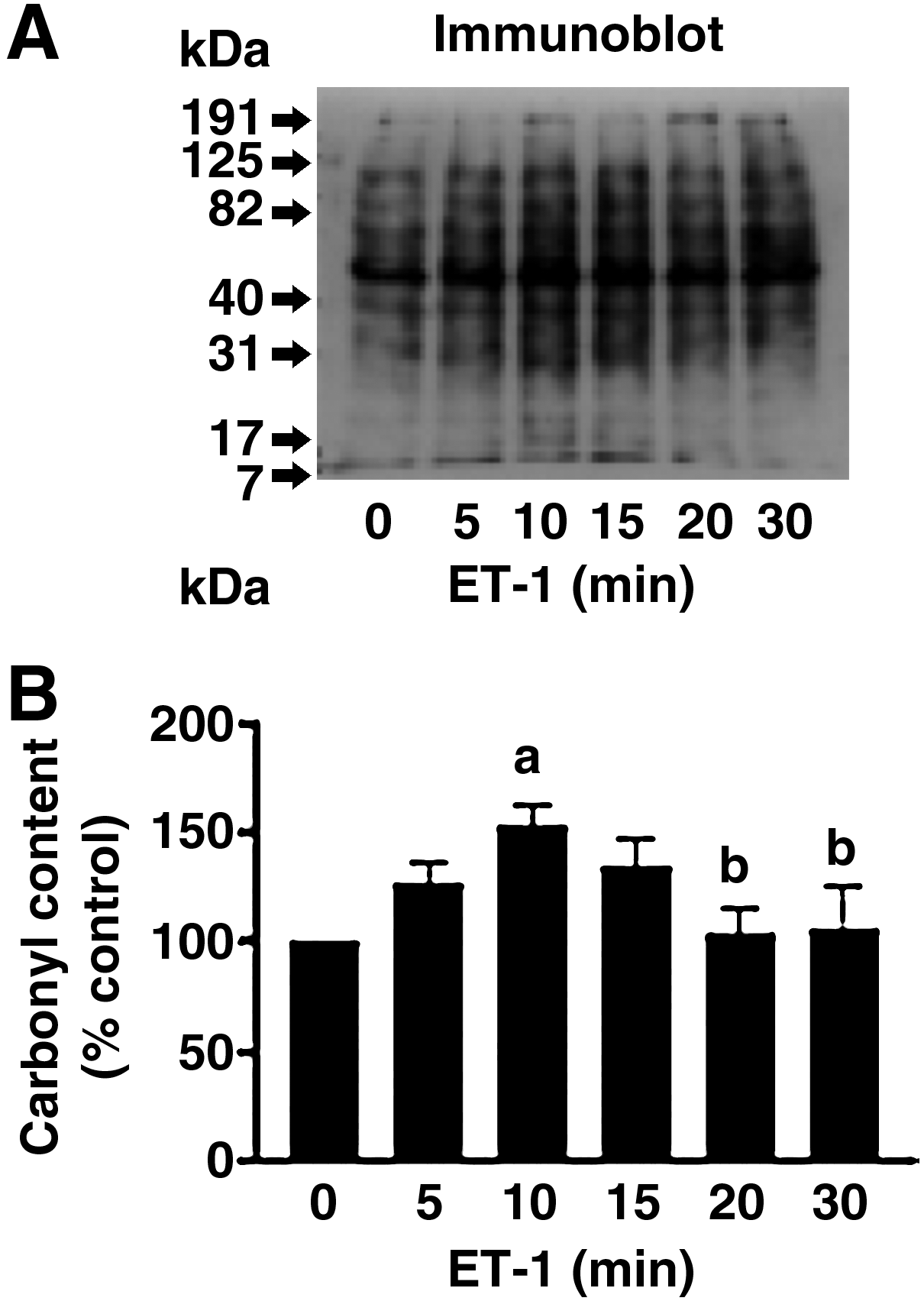
Soon after our discovery that endothelin-1 treatment of cells can promote protein carbonylation, we asked questions: (a) Is endothelin-1-mediated protein carbonylation receptor dependent? and (b) Is endothelin-1-mediated protein carbonylation metal-catalyzed oxidation (Fenton reaction) dependent? To answer these questions, cells were pre-treated with endothelin receptor antagonists (ETA and ETB receptor antagonist), hydrogen peroxide scavengers (ebselen and adenovirus expressing catalase), or an iron chelator (deferoxamine) before treatment with endothelin-1. Results showed that endothelin-1-mediated carbonylation of some of the proteins are indeed endothelin receptor- and metal-catalyzed oxidation-dependent (78). These are the first demonstration that metal-catalyzed protein carbonylation can be promoted in response to ligand–receptor interactions. The hypothesis that protein carbonylation promoted by reactive oxygen species may play roles in oxidant signaling mechanisms is supported by our observations that H2O2 at a concentration as low as 500 nM promoted protein carbonylation (78).
Subsequent studies have shown that, in addition to bovine pulmonary artery smooth muscle cells, protein carbonylation can occur in response to endothelin-1 in other cell types, including human pulmonary artery smooth muscle cells, bovine aortic smooth muscle cells, and human bronchial airway smooth muscle cells. Further, endothelin-1-mediated protein carbonylation was detected in tissue/organ models including cultured bovine pulmonary artery smooth muscle tissue and organ culture of rat pulmonary arteries (78). In addition to endothelin-1, other ligands which have been shown to produce reactive oxygen species, including serotonin (35, 36) and PDGF (66), were also found to promote protein carbonylation in cultured pulmonary artery smooth muscle cells (77). Our laboratory also found that serotonin promotes protein carbonylation in the right ventricular myocardium of the Langendorff perfused rat heart (44) as well as in the perfused rat pulmonary artery preparations. Furthermore, in vivo treatment of rats to chronic hypoxia, which promotes pulmonary hypertension and resultant right ventricular hypertrophy, promoted protein carbonylation in pulmonary artery (76, 79) and in the right ventricle (50).
Mechanisms of endothelin-1-mediated protein carbonylation appear to involve NAD(P)H oxidase, as the inhibitors of this enzyme suppressed carbonylation in bovine pulmonary artery smooth muscle cells. This is consistent with previous reports that endothelin-1 produces reactive oxygen species via NAD(P)H oxidase (74).
Mechanism of Serotonin-Mediated Protein Carbonylation
Serotonin plays an important role in the development of pulmonary hypertension and pulmonary artery smooth muscle cell growth (34). Serotonin is a potent vasoconstrictor and a mitogen of lung vascular smooth muscle. Evidence for the role of serotonin in the development of pulmonary hypertension was recognized in fawn-hooded rats, in which a genetic deficit in serotonin platelet storage and high plasma levels of serotonin are associated with susceptibility to developing pulmonary hypertension (56). Further studies showed that a continuous intravenous infusion of serotonin during exposure of rats to hypoxia potentiated the development of pulmonary hypertension (17). The role of serotonin in pulmonary hypertension is further supported by the results that serotonin transporter-deficient mice develop less hypoxic pulmonary hypertension (18). In mice, overexpression of serotonin transporter enhanced hypoxia-induced increase in pulmonary vascular remodeling, right ventricular pressure, and right ventricular hypertrophy (45). In patients with idiopathic pulmonary arterial hypertension, high levels of plasma serotonin were observed (29), and pulmonary artery smooth muscle cells grow faster than the cells from control subjects (19). The mechanism of pulmonary artery smooth muscle cell growth appears to involve serotonin transporter (34) as well as serotonin receptors (43). Serotonin transporter-mediated signaling events include the production of reactive oxygen species (35 –37) and also involve the MEK-ERK pathway (36) and Rho-associated kinase (43).
The mechanism of serotonin production of reactive oxygen species is controversial. In bovine pulmonary artery smooth muscle cells, Fanburg and co-workers proposed that serotonin produces ROS via NAD(P)H oxidase (35). In contrast, in human pulmonary artery smooth muscle cells, Rabinovitch and co-workers reported the role of monoamine oxidase A (33). In rat cardiac muscle cells, serotonin was reported to produce ROS via monoamine oxidase A (4, 5).
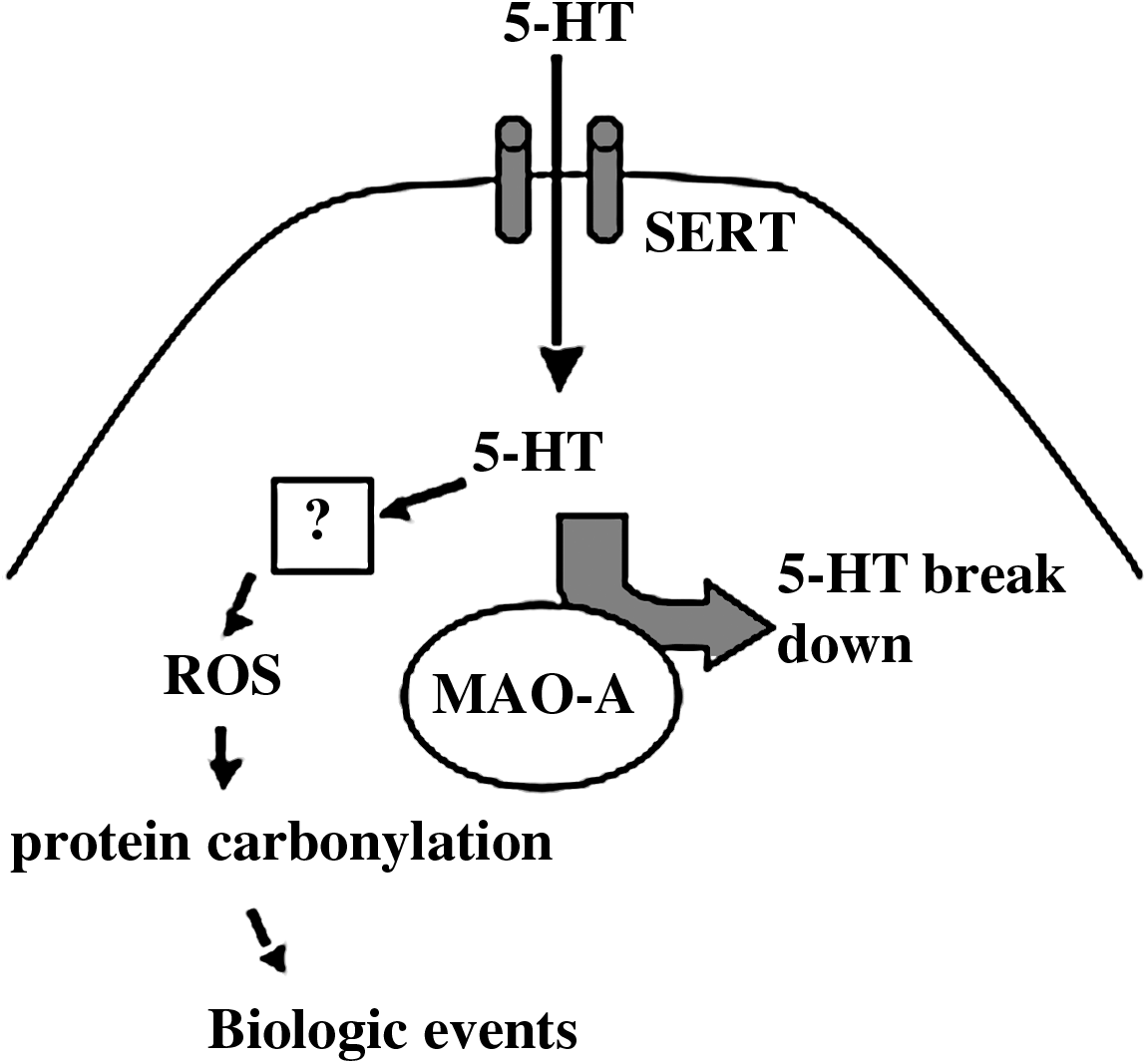
Our studies showed that monoamine oxidase inhibitors do not block serotonin-induced protein carbonylation, rather these inhibitors promoted carbonylation in the rat right ventricle; and that although the right ventricle has lower levels of monoamine oxidase A than the left ventricle, serotonin only causes protein carbonylation in the right ventricle (44). Thus, in the rat right ventricle, monoamine oxidase A is not the source of ROS, which promote protein carbonylation. This is similar to the previous finding by Fanburg and co-workers in bovine pulmonary artery smooth muscle cells that the monoamine oxidase inhibition enhanced serotonin-induced cell proliferation (34) and superoxide production (35). In bovine pulmonary artery smooth muscle cells, our laboratory has found that monoamine oxidase-A inhibition by clorgyline promoted protein carbonylation (unpublished results). From these observations, we proposed a mechanism in which monoamine oxidase A serves to regulate intracellular levels of serotonin which can target other mechanisms for the regulation of reactive oxygen species (Fig. 4). While further experiments are needed to determine the source of reactive oxygen species that are produced in response to serotonin, possible mechanisms include: (a) the activation of NAD(P)H oxidase to form superoxide, (b) serotonin radical formation and subsequent superoxide production, (c) promotion of nitric oxide synthase uncoupling to form superoxide, and (d) protein serotonylation which promotes reactive oxygen species production.
NAD(P)H oxidase has been shown to increase cytosolic ROS that in turn elicit signal transduction (8, 23). Results from Fanburg and co-workers suggest that although monoamine oxidase inhibition does not inhibit serotonin signaling, inhibitors of NAD(P)H oxidase effectively inhibit serotonin-induced elevation of superoxide and cell proliferation (35). Since serotonin effects are blocked by an inhibitor of serotonin transporter, the elevation of intracellular serotonin by monoamine oxidase A inhibition through a yet unknown mechanism may trigger NAD(P)H oxidase activation.
Perez-Reyes and Mason (51) described that serotonin can undergo oxidation to produce serotonin radical. This oxidation can be promoted by cytochrome c/Fe3+, ceruloplasmin/Cu2+, or superoxide. Interestingly, the reaction of serotonin with superoxide produces H2O2 and the products of reactions with metal ions are reduced iron and copper, generating substrates for Fenton reaction and hydroxyl radical formation. The serotonin radical can oxidize NAD(P)H, forming a NAD(P) radical, which can in turn reduces O2 to superoxide. Thus, reactions involving serotonin radicals can be sources of ROS as well as specific promoters of Fenton reaction.
Another cellular source of superoxide is nitric oxide synthase via uncoupling of this enzyme (20). While the roles of serotonin in nitric oxide synthase uncoupling and the formation of superoxide have not yet been demonstrated, it has been documented that serotonin transporter can interact with nitric oxide synthase and serotonin can modulate nitric oxide synthase activity (11).
Serotonylation of small GTPases on glutamine residues by transglutaminases was originally described by Walther et al. (73) and more recently shown to occur in smooth muscle cells (26). Thus, this mechanism may play important roles in the functions of intracellular serotonin, and possibly regulate the reactive oxygen species generation and protein carbonyl formation.
The hypothesis depicted in Fig. 4 suggests that, while monoamine oxidase A is also a source of H2O2, this enzyme largely serves to regulate intracellular serotonin, which through mechanisms described above, elicits the major ROS signaling pathways. These reactive oxygen species and H2O2 that is generated by monoamine oxidase A may function in concert to fine tune the specificity of target molecules to different reactive oxygen species for cell regulation.
Through two-dimensional gel electrophoresis of DNPH-derivatized bovine pulmonary artery smooth muscle cell lysate samples followed by Western blotting with the DNP antibody, our laboratory recently found that some of the proteins that are carbonylated in response to endothelin-1 and serotonin are different (unpublished results). These observations opened up the possibility that different reactive oxygen species might target different proteins in the cell. In the case of the difference between endothelin-1 and serotonin, different ROS generators may be involved. Thus, it is important to understand how serotonin might produce reactive oxygen species.
Identifications of Carbonylated Proteins
To identify proteins that are carbonylated in response to endothelin-1 in bovine pulmonary artery smooth muscle cells, we performed two-dimensional gel electrophoresis of the DNPH-derivatized cell lysate samples, followed by immunoblotting with the antibody that recognizes DNPH-derivatized proteins using the procedures previously described (3, 7). We defined the carbonylated protein spots of interest in two-dimensional gel electrophoresis gels by obtaining data from samples that are pre-incubated with endothelin receptor antagonists, ebselen (a glutathione peroxidase mimetic that eliminates hydrogen peroxide) and deferoxamine (an iron chelator) before the addition of endothelin-1 (78). We identified carbonylated protein spots that were carbonylated by endothelin-1 in an endothelin receptor- and Fenton reaction-dependent manner. Mass spectrometry identified some of these spots to be 8 proteins including annexin A1, annexin A2, phosphoglycerate dehydrogenase, phosphoglycerate mutase, heat-shock protein β1, peroxiredoxin 6, DJ-1, and cofilin-1 (Fig. 5) (78).

Functions of Carbonylated Proteins in Cell Signaling
Mass spectroscopy analyses revealed that annexin A1 is one protein that is carbonylated by endothelin-1 in bovine pulmonary artery smooth muscle cells (78). To confirm these results, we developed a technique to assess carbonylated proteins via immunoprecipitation and immunoblotting (78). Cell lysates were derivatized with DNPH, followed by immunoprecipitation with the antibody that recognizes DNPH-derivatized proteins. Precipitated proteins were then subjected to Western blotting with the antibodies against proteins of interest. Using this method, we confirmed that carbonylation of annexin A1, annexin A2, peroxiredoxin 6, and cofilin-1 were indeed promoted in response to endothelin-1 in bovine pulmonary artery smooth muscle cells (78). We decided to study further carbonylation of annexin A1 because annexin A1 has been shown to regulate cell growth and apoptosis (1, 9, 15, 31, 62) and these processes are important for the development of pulmonary hypertension. Using the immunoprecipitation/immunoblotting method we developed, we found that endothelin-1 can promote annexin A1 carbonylation in other cell types, including human pulmonary artery smooth muscle cells and human bronchial airway smooth muscle cells. Annexin A1 carbonylation was also found to be promoted in response to the treatment of cells with other growth factors such as serotonin and PDGF. Further, we found that in vivo treatment of rats with sustained hypoxia (10% O2) to promote pulmonary hypertension and right ventricular hypertrophy resulted in the promotion of annexin A1 carbonylation in the pulmonary artery and in the right heart ventricle (50, 79).
Annexin A1 functions to induce apoptosis and inhibit cell proliferation (1, 9, 15, 31, 62), thus a consequence of the suppression of the annexin A1 would be to promote cell growth and inhibit apoptosis, which would result in the characteristics of pulmonary vascular remodeling promoted by factors such as endothelin-1, serotonin, and PDGF. Our experiments, which examined the time course of annexin A1 protein expression in response to endothelin-1 in bovine pulmonary artery smooth muscle cells, revealed that the annexin A1 protein expression declines after annexin A1 gets carbonylated (78). This decrease in annexin A1 in response to endothelin-1 treatment can be blocked by pre-treating the cells with a proteasome inhibitor. Thus, we propose that endothelin-1 promotes carbonylation of annexin A1, which serves as a signal for the proteasome-dependent degradation. As noted above, degradation of annexin A1 would result in increased cell growth, establishing a mechanistic model for the promotion of pulmonary vascular thickening in which growth factors and mediators of pulmonary hypertension, such as endothelin-1, serotonin, and PDGF, cause annexin A1 carbonylation and degradation, resulting in the suppression of annexin A1 actions to promote apoptosis and inhibit cell proliferation, thereby promoting pulmonary vascular smooth muscle cell growth (Fig. 6). Our observations that annexin A1 can be carbonylated and degraded in bronchial airway smooth muscle cells also opened up an idea that mediators of asthma such as PDGF and endothelin-1 might promote airway wall thickening as well as possibly airway inflammation, since annexin A1 (also known as lipocortin, lipomodulin or macrocortin) was originally discovered as a glucocorticoid responsive anti-inflammatory molecule (6).
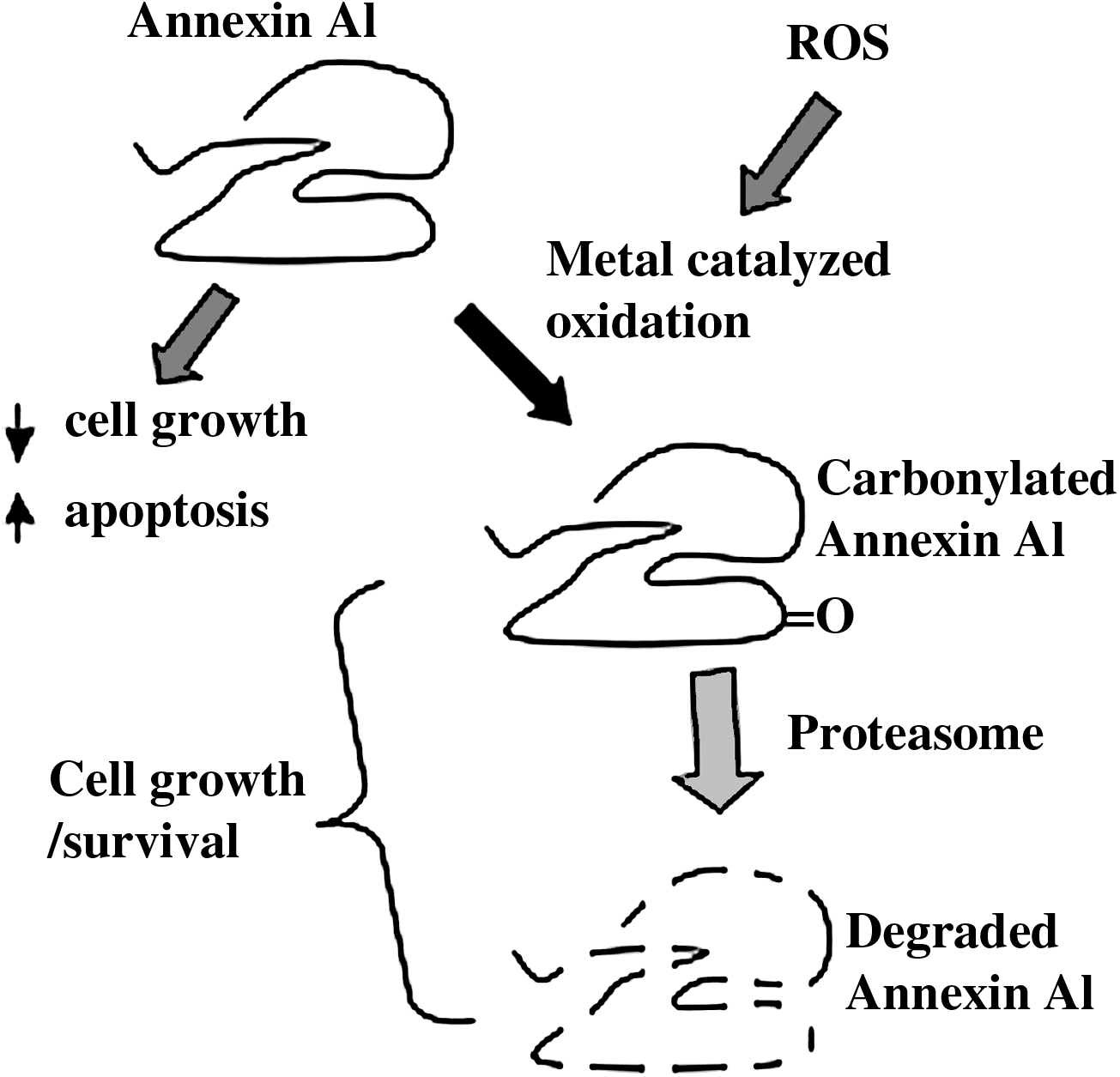
It should be noted that not all the carbonylated proteins get degraded by the proteasome. While annexin A1 gets carbonylated and degraded, we found that peroxiredoxin 6, another protein that was identified to be carbonylated by endothelin-1 (Fig. 5), does not get degraded. Thus, we propose that the carbonylation-degradation pathway occurs specifically to certain proteins, thereby conferring the specificity that is needed for cell signal transduction processes.
These results also opened up a question on what is the degree of carbonylation that is required to serve as cell signaling mechanisms. Large changes in specific carbonyl content are required for eliciting biological responses, or one amino acid modification may be sufficient to transduce signals? Answers to these intriguing questions may be revealed in the near future.
Protein Decarbonylation
The initial experiments we performed with endothelin-1 to treat bovine pulmonary artery smooth muscle cells revealed an interesting event in which carbonylated proteins can be decreased subsequent to the peak of carbonylation (Fig. 7A) (78). We named this process “decarbonylation.” Interestingly, this decarbonylation process occurs in bovine pulmonary artery smooth muscle cells, but not in bovine aortic smooth muscle cells, possibly revealing the regulatory differences in redox signaling in smooth muscle cells of pulmonary and systemic circulations. We initially hypothesized that the decrease is due to the degradation of proteins, as carbonylated proteins are known to be subjected to proteasome-dependent degradation (25). However, we found that the inhibition of the proteasome did not suppress the decarbonylation process (78). We subsequently came across the finding that the inclusion of β-mercaptoethanol in cell lysates before the addition of DNPH dramatically reduced the carbonyl content. Thus, we hypothesized that the mechanism of decarbonylation might involve reduction of carbonylated moieties by biological thiols, and accumulated a series of evidence supporting this hypothesis including: (a) an inhibitor of thioredoxin reductase can promote protein carbonylation without the occurrence of decarbonylation; (b) an inhibitor of thioredoxin reductase can suppress decarbonylation phase in response to the endothelin-1 treatment; (c) thioredoxin protein levels get increased during the decarbonylation phase in response to endothelin-1 in bovine pulmonary artery smooth muscle cells, but not in bovine aortic smooth muscle cells which do not exhibit endothelin-1 mediated decarbonylation (Fig. 7); and (d) during the decarbonylation phase, thioredoxin–carbonylated protein interactions increase. Thus, we propose the thiol-dependent reduction hypothesis for the mechanism of decarbonylation (78). While it is not likely that thiols directly reduce aldehydes or ketones, thiols may support enzymatic processes that are capable of reducing these carbonyl moieties in the biologic system.

However, alternative mechanisms are also possible for the decarbonylation mechanism such as further oxidation of carbonylated aldehyde or ketone to carboxylic acid, which eliminates reactivity with DNPH. Amici et al. (2) reported that oxidation of proline as well as arginine results in the formation of glutamyl semialdehyde, which is DNPH reactive. Reduction of the aldehyde carbonyl group results in the formation of alcohol, while oxidation forms carboxylic acid, as possible products of decarbonylation. Thiols may reduce glutamyl semialdehyde or may promote the oxidation process via promoting Fenton reaction by reducing metal ions (27) to elicit decarbonylation (Fig. 8A).
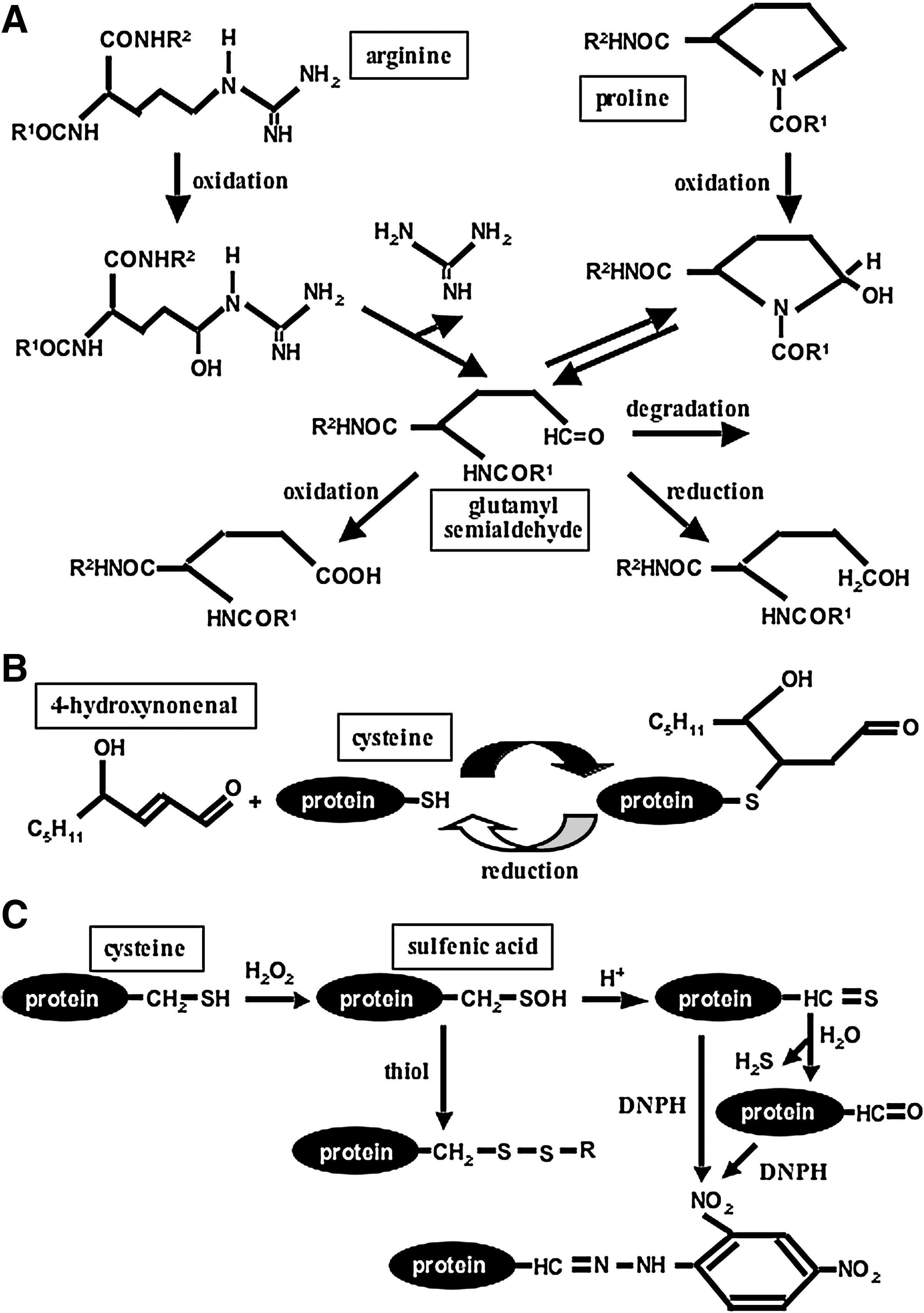
As noted above, the detected DNPH reactive component might be a result of secondary protein carbonylation through the addition of aldehyde from lipid peroxidation. This situation applies if the carbonylated residues are lysine, histidine, or cysteine (24, 57). The secondary protein carbonylation may be reversible, and the mechanism of decarbonylation may simply the deletion of added aldehyde from the amino acid side chain. The secondary protein carbonylation on cysteine residues may be particularly sensitive to reduction by thiols (61) and may be the mechanism for the observed decarbonylation process (Fig. 8B).
Recently, Dalle-Donne et al. (16) found that DNPH can also label cysteine sulfenic acids in their in vitro study using a cysteine-containing model peptide. This opens up a possibility that some of the DNPH signals we observed in immunoblotting experiments (particularly those which are not responsive to deferoxamine inhibition) are derived from cysteine sulfenic acids, rather than protein carbonylation and that apparent decarbonylation by thiols is due to the elimination of sulfenic acids (Fig. 8C). Using mass spectrometry, our laboratory, however, has determined that primary carbonylation does get promoted by ligand–receptor interactions (unpublished data) and is currently investigating if these primary carbonylation sites can be decarbonylated.
In summary, our initial studies have opened up the possibilities that protein carbonylation, as well as protein decarbonylation, might play important roles in the mechanism of redox signaling. Further investigations are needed to advance the knowledge of possible roles of protein carbonylation as well as decarbonylation in cell signaling. Molecular mechanisms of observed protein carbonylation and decarbony-lation during cell signaling also need to be determined. It should be noted that primary protein carbonylation/decarbonylation, secondary protein carbonylation/decarbo-nylation, and sulfenic acid formation and elimination may, in concert, play important roles in the regulation of cell signal transduction.
Clinical Implications
While our laboratory currently focuses on the studies of pulmonary hypertension and resultant right heart failure, possible roles of protein carbonylation in ROS signaling have implications to many other diseases.
Pulmonary hypertension is a devastating disease currently without known cure, with mean survival of 2–3 years after diagnosis. It is characterized by increased pulmonary blood pressure due to excessive vasoconstriction, as well as increased thickness of the pulmonary vascular wall. Increased pulmonary vascular resistance strains the right heart ventricle and leads ultimately to right heart failure. Enhanced growth of pulmonary vascular cells plays a role in the development of an irreversible condition that leads to increased pulmonary vascular resistance. Vasoactive agents such as endothelin-1 and serotonin are not only inducers of pulmonary vasoconstriction, but also are mitogens and anti-apoptotic factors that increase the smooth muscle cell number. Our studies have shown that mediators of pulmonary vascular smooth muscle cell growth including endothelin-1, serotonin, and PDGF that are known to trigger ROS signaling also promote protein carbonylation (77 –79). One protein that was identified to be carbonylated is annexin A1. Carbonylation of annexin A1 consequently regulates pulmonary artery smooth muscle cell growth through signaling to proteasomal degradation, thereby ceasing its antiproliferative and apoptotic activities. We propose a mechanism of ROS signaling that can be triggered by mediators of pulmonary hypertension in which protein carbonylation and subsequent degradation of annexin A1 leads to cell proliferation and survival. Consistent with these cell culture studies, we demonstrated that annexin A1 carbonylation and subsequent degradation occur in response to in vivo treatment of rats to chronic hypoxia to promote pulmonary hypertension. These results have opened up therapeutic possibilities for the use of inhibitors of metal-catalyzed oxidation and proteasome-dependent degradation for the treatment of pulmonary hypertension. Interestingly, this mechanism was also found to occur in the right heart ventricle as a signal to elicit right ventricular hypertrophy (50). Thus, agents that target this pathway may suppress both pulmonary vascular remodeling as well as right ventricular remodeling. In the right heart, we identified a transcription factor that can bind to annexin A1 and may regulate gene transcription of a major cardiac hypertrophic protein, GATA4 (50). Our laboratory continues to study the role of annexin A1 in cell growth signaling mechanisms in pulmonary vascular smooth muscle and in right ventricular cardiac myocytes.
In addition to pulmonary vascular smooth muscle cells, we found that protein carbonylation can occur in smooth muscle cells of systemic circulation, as well as in airway smooth muscle cells, suggesting the possible roles of protein carbonylation signaling in systemic hypertension, arteriosclerosis, asthma, and chronic obstructive pulmonary disease. Annexin A1 regulation in airway smooth muscle cells may particularly be important, as a major biologic action of annexin A1 is to elicit anti-inflammatory activities, which has clinical relevance to asthma.
As ROS signaling has been implicated in many other diseases, including cancer, diabetes, and neurological disorders, whether protein carbonylation occurs in response to appropriate signaling stimuli under these conditions should also be explored. It is likely that research on the mechanisms and functional roles of protein carbonylation will be an important area of biology in the next decade.
Footnotes
Acknowledgments
This work was supported in part by National Institutes of Health Grant R01 HL72844 and American Heart Association Grant-in-Aid (to YJS).