
Abstract
Redox biochemistry is increasingly recognized as an integral component of cellular signal processing and cell fate decision making. Unfortunately, our capabilities to observe and measure clearly defined redox processes in the natural context of living cells, tissues, or organisms are woefully limited. The most advanced and promising tools for specific, quantitative, dynamic and compartment-specific observations are genetically encoded redox probes derived from green fluorescent protein (GFP). Within only few years from their initial introduction, redox-sensitive yellow FP (rxYFP), redox-sensitive GFPs (roGFPs), and HyPer have generated enormous interest in applying these novel tools to monitor dynamic redox changes in vivo. As genetically encoded probes, these biosensors can be specifically targeted to different subcellular locations. A critical advantage of roGFPs and HyPer is their ratiometric fluorogenic behavior. Moreover, the probe scaffold of redox-sensitive fluorescent proteins (rxYFP and roGFPs) is amenable to molecular engineering, offering fascinating prospects for further developments. In particular, the engineering of redox relays between roGFPs and redox enzymes allows control of probe specificity and enhancement of sensitivity. Genetically encoded redox probes enable the functional analysis of individual proteins in cellular redox homeostasis. In addition, redox biosensor transgenic model organisms offer extended opportunities for dynamic in vivo imaging of redox processes. Antioxid. Redox Signal. 13, 621–650.
I. Introduction: Redox Biosensing Between Aspiration and Reality
A. The need for novel redox probes
The study of redox processes in biological systems is a very broad and diverse field. It is always complex, frequently unclear, and sometimes downright confusing. One factor contributing to widespread confusion is the conceptual lumping together of diverse redox processes, as if they could be treated as one entity. Terms such as “cellular redox state” or “redox environment” are frequently used without further definition. Such terms seem to imply that a redox state is something like a global characteristic that does not require reference to any specific redox pair, or that the different redox pairs of the cell are in thermodynamic equilibrium, so that one redox pair can be used as a proxy for all the others. Such implicit assumptions are highly unrealistic. Cells harbor a multitude of redox pairs which are kinetically controlled and far from equilibrium with each other. Many kinds of redox reactions are thermodynamically favored, yet kinetically, sterically, or compartmentally separated. The enzymatic by-passing of kinetic inhibition is a powerful and fast-acting control principle in biology. The function of redox catalysts is to allow thermodynamic equilibration between two redox pairs in the right place and at the right time. For example, NADPH oxidase enzymes act as a highly regulated kinetic passageway for electron transfer between NADPH/NADP+ and O2/O2 •- (63). Because of barriers and their selective suspension by catalysts, the spatio-temporal pattern of intracellular redox changes is highly nonuniform between and within cellular compartments. Likewise, it is increasingly recognized that individual redox systems have distinct functions and are subject to independent regulation. Thus, a key task is to define the functional role of individual redox systems by looking at them specifically.
We are now at a stage where long-cherished views about “oxidants,” “oxidative stress,” and “antioxidants,” and their relationship with organismal health, are increasingly being questioned. Beneficial roles of oxidants (90, 94), and potentially harmful effects of antioxidants (99, 104) continue to be recognized. Obviously, broad concepts and umbrella terms (like “oxidative stress,” “ROS,” and “antioxidants”) lack the conceptual acuity that is needed for the understanding of the underlying causal relationships. After all, most cellular redox couples entertain electron transfer relationships in a dynamic and context-dependent manner. Obviously, a fine-grained view is needed, namely the quantitative measurement of clearly defined individual redox relationships in their specific cellular context, with high spatio-temporal resolution. Unfortunately, our ability to do this is very much limited.
Consequently, the redox probes we seek for the future must be highly specific for defined individual redox-active species or redox pairs. By responding sensitively, instantaneously, and reversibly, these probes have to allow compartment-specific real-time measurements in living cells, tissues, and whole organisms. They should not perturb cellular behavior and be easy to use. As if that were not demanding enough for a single probe, a whole collection of such redox probes will be needed, covering the spectrum of biologically relevant redox active molecules and redox pairs. High on the wish-list are probes for superoxide (O2 •-), H2O2, nitric oxide (NO•), glutathione (reduced form: GSH; oxidized form: GSSG), and ascorbate (reduced form: AA; oxidized form: dehydroascorbate, DHA). Not least, the redox status of proteins involved in the transmission of redox signals such as oxidoreductases (e.g., thioredoxin) and peroxidases (e.g., peroxiredoxins) is of fundamental interest as well.
B. The limits of conventional redox measurements
This review is about genetically encoded redox probes, because they promise to overcome many of the problems associated with conventional redox measurements. Briefly, these problems fall into two broad categories. The first major problem relates to the disruption of cells. Examples are measurements of glutathione, ascorbate, NADPH, and thioredoxin (Trx), by use of enzymatic assays, HPLC, or gel mobility. The major advantage is high specificity for the redox couple in question. Typically, both the oxidized and reduced form can be quantified and redox potentials estimated. The major disadvantage though is cell disruption, which creates oxidation artifacts, precludes dynamic measurements, and implies that calculated redox potentials are at best apparent redox potentials lacking resolution for specific subcellular compartments. For example, while several glutathione assays are reproducible and statistically robust, they all suffer from the inherent problem that tissue extraction and cellular disruption unavoidably destroys information about compartment-specific glutathione redox states.
The second major problem relates to the limitations of redox-active fluorogenic dyes when they are applied to living cells. Thiol-reactive dyes allow estimation of cellular glutathione content but cannot deliver information about the redox state of the glutathione couple (4, 81, 114, 134). Most oxidant-sensitive fluorescent dyes are characterized by at least partial nonspecific behavior, irreversibility, and lack of compartment specificity. The most widespread method to detect H2O2 in intact cells is the oxidation of 2′,7′-dihydrodichlorofluorescein (H2DCF) to the fluorescent compound 2′,7′-dichlorofluorescein (DCF). However, oxidation of H2DCF can occur in the absence of H2O2 and is stimulated by metals, peroxidases, and cytochrome c (102, 115). Like many other oxidant-sensitive dyes, H2DCF can only be considered a qualitative marker of undefined pro-oxidative processes. However, it should be noted that improved chemical probes are now being developed. For example, Christopher Chang and colleagues synthesized probes that are specifically triggered by H2O2 (16, 83, 85) and reversibly reacting probes that enable dynamic measurements (84, 86). Advanced chemical probes are thus prone to play important roles in the future and should be considered complementary to the fluorescent protein-based redox probes which are the subject of this review.
II. Redox-Sensitive Fluorescent Proteins
Although considerable progress is being made in the development of chemical probes (86), there are still major limitations regarding reversibility, quantitation, and subcellular targeting. Many of these limitations were overcome by the development of the first genetically encoded redox probes that allowed the quantitation of a dithiol–disulfide equilibrium within live cells. Green fluorescent protein (GFP) and its variants are resistant to common proteases and stable under physiological pH conditions, thus providing an excellent scaffold for the development of biosensors for physiologically important parameters. Therefore, it is not surprising that GFP attracted major interest for the development of redox-sensitive probes, which are now about to transform redox research towards more quantitative and dynamic approaches. The groups of Jakob Winther and James Remington independently developed GFP-based redox-sensitive probes by placing artificial disulfides onto fluorescent protein (FP) scaffolds. Redox-sensitive yellow fluorescent protein (rxYFP) and redox-sensitive GFP (roGFP) both allow noninvasive quantitative imaging of the associated dithiol–disulfide equilibrium (27, 92, 93). The set of redox-sensitive FPs was recently supplemented with a circularly-permuted YFP variant (cpYFP), which appears to be responsive to O2 •- (126). Here, we will discuss the properties and some applications for these probes.
A. Common properties of FPs
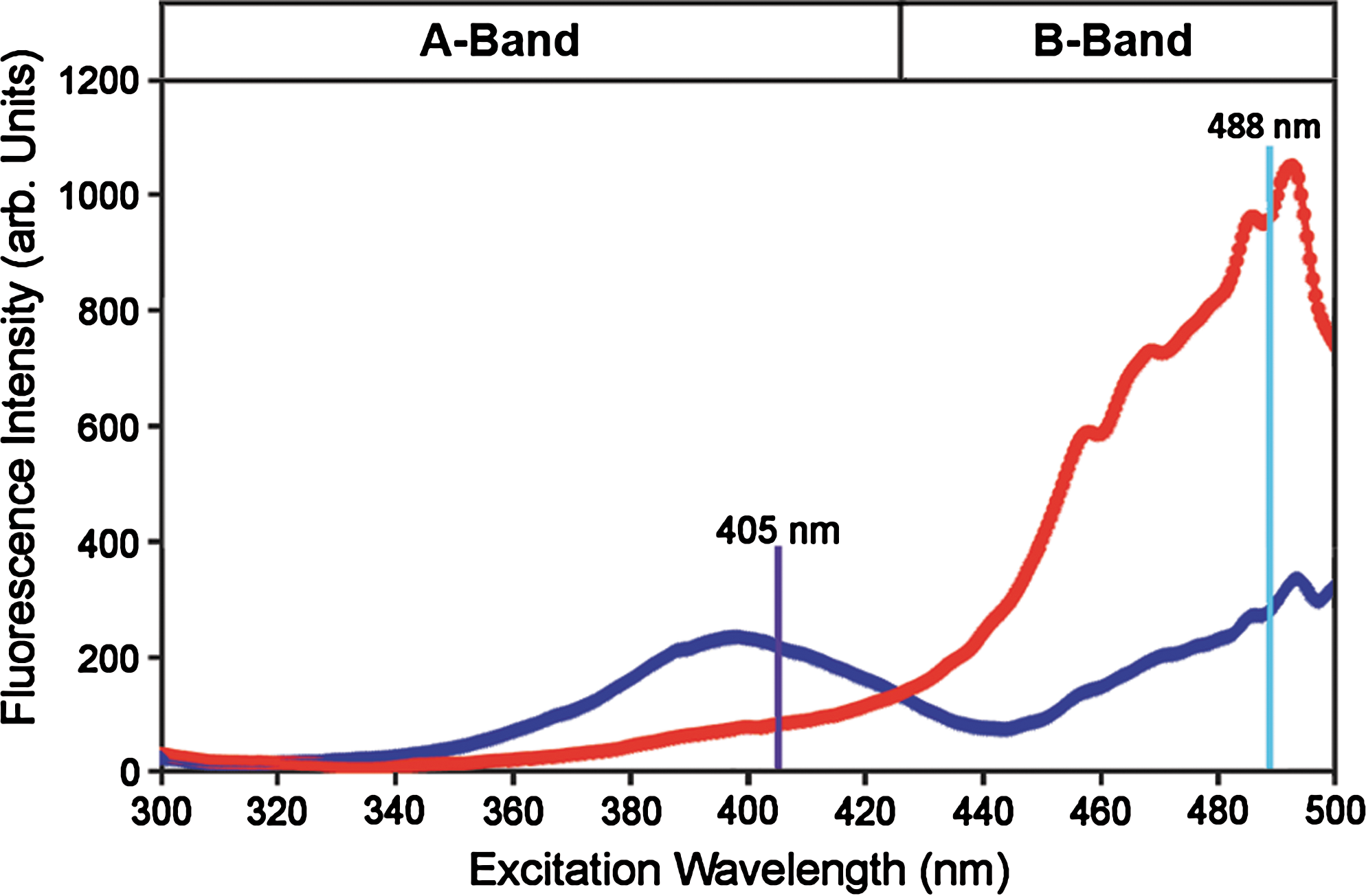
Wild-type GFP (wtGFP), originally isolated from the jellyfish Aequorea victoria by Osamu Shimomura (111), is a soluble protein of 27 kDa. The 238 amino acid long polypeptide forms an 11-stranded β-barrel shielding an internal α-helix, which runs up the axis of the β-barrel, against the surrounding medium (91, 133). The wild-type chromophore of GFP is formed through intramolecular cyclization of the three amino acids S65/Y66/G67 (Fig. 1A). Subsequent oxidation leads to formation of a conjugated system of π-electrons capable of absorbing and emitting visible light. Wild-type GFP has two excitation peaks which reflect different protonation states of the phenol group of Y66 within the chromophore (Fig. 1B), but only a single emission peak with a maximum at 509 nm. The first excitation peak with a maximum at 395 nm corresponds to the protonated neutral form of the chromophore (A-band) and the second peak with a maximum at 475 nm corresponds to the de-protonated, anionic form (B-band) (9, 30). Following excitation at the A-band (e.g., with the laser wavelength 405 nm) the excited neutral chromophore (A*) converts to the unrelaxed anionic form (I*) by transferring the phenolic proton from Y66 to the carboxylate of E222. This process is called excited state proton transfer (ESPT) (17) and involves a hydrogen bonding network surrounding the chromophore. The proton transfer between Y66 and E222 is mediated by a structural water molecule and the hydroxyl group of S205 (Fig. 1A). ESPT is the reason why excitation at both bands leads to emission of green light of similar wavelength (Fig. 1B) (78). Importantly, the dual excitation-single emission behavior of GFP can be exploited for ratiometric measurements.

In general, the degree of chromophore protonation is determined through interactions with the surrounding residues and thus can be affected by changes in protein composition and structure. The most relevant amino acids are those involved in the ESPT hydrogen bonding network (i.e., S205, E222, S65) and those in close proximity of Y66 (i.e., N146, H148, T203). For instance, chromophore deprotonation is significantly favored when S65 is replaced by threonine. The S65T mutation, leading to enhanced GFP (EGFP), thus shifts the major excitation peak to 488 nm (53). On the other hand, a way to manipulate FP emission is to alter chromophore stacking interactions. In YFP, the additional T203Y mutation leads to π-electron stacking between the chromophore and the aromatic side chain of Y203 (117, 124). Although generally considered to be a very rigid structure, the β-barrel has some flexibility and minor conformational changes can lead to alterations in chromophore protonation or π-electron stacking. Such changes have been exploited in roGFP and rxYFP.
B. Probes with disulfide bonds engineered into the GFP β-barrel
1. rxYFP
The introduction of the T203Y mutation converted EGFP to YFP, a red-shifted GFP variant with an excitation maximum at 513 nm and emission at 527 nm (124). Based on the assumption that dislocation of Y203 and accompanying changes in π-electron stacking would affect fluorescence significantly, Ostergaard and colleagues used YFP as a template for redox-sensitive variants (rxYFP) (92). Dislocation of Y203 was expected to occur when the β-strand 10 (aa 199–208) of the β-barrel of the YFP protein was forced into a slightly different position. To achieve such conformational change, pairs of cysteines were introduced in different positions on β-strands 7 and 10 such that the two residues are capable of forming an intramolecular disulfide bridge. Of four different combinations of cysteine pairs only the variant N149C/S202C gave a strong redox-dependent change in fluorescence (92). Formation of the disulfide bond between these two cysteines resulted in a 2.2-fold reduction of the emission peak without a significant shift in the excitation wavelength. YFP exhibits two absorption peaks for the neutral A-band (λmax = 392 nm) and the anionic B-band (λmax = 514 nm), with a clean isosbestic point at 432 nm (125). However, due to fluorescence quenching, the neutral form of YFP is nonfluorescent and thus recombinant rxYFP exhibits only a single excitation peak with a maximum at 512 nm similar to wtYFP (92). The midpoint redox potential E°′ of rxYFP was determined as −261 mV through redox titration of recombinant protein with 2GSH/GSSG buffers (92). Since the reactivity of rxYFP with glutathione is rather low, protein samples had to equilibrate for 21 hours and thus the adjusted 2GSH/GSSG mixture had to be carefully controlled for air oxidation of GSH.
Although fluorescence quenching of the neutral form of the chromophore in rxYFP would argue for exclusive usability of rxYFP as a single excitation wavelength probe only, Maulucci and colleagues report the use of rxYFP as a ratiometric probe (76). When expressed in human cells, the redox-dependent change in emission after excitation at 458 nm was only ∼50% of the change observed after excitation at 488 nm. This difference was used for ratiometric calculations (60, 76, 77). However, it is not entirely clear if this kind of ratiometry can separate variations in probe concentration from changes in redox state. The exact excitation spectra for reduced and oxidized rxYFP inside cells are not known. Because the reference wavelength (458 nm) is close to the isosbestic point of YFP, the fluorescence excited is very low and thus may be significantly obscured by background noise, which needs to be subtracted very carefully. This already implies that such analysis of rxYFP might be very sensitive to autofluorescence. Furthermore, the fact that the redox-dependent change in fluorescence points in the same direction for both excitation wavelengths inevitably lowers the dynamic range of the probe. Another issue that may be relevant for the interpretation of measurements is the sensitivity of YFP to changes in pH at near neutral conditions with a chromophore pK a of 7.0 (30) and the significant sensitivity of YFP to chloride (62,125).
2. roGFPs
The limitations due to lack of ratiometric properties were overcome with the development of redox-sensitive GFPs (roGFPs) (49). Starting from either wtGFP or EGFP, respectively, two cysteines were engineered into the positions S147 and Q204, which are located on β-strands 7 and 10 in close proximity to the positions mutated in rxYFP. The resulting redox probes were called roGFP1, which is based on wtGFP, and roGFP2, which is derived from EGFP. The two excitation maxima characteristic of GFP (∼400 nm for the A-band and 475–490 nm for the B-band) are maintained in the respective roGFP variants with band A being the main excitation peak in roGFP1 and band B dominating in roGFP2. Oxidation results in an increase in the A-band and a decrease in the B-band and an inverse behavior under reducing conditions, as shown exemplarily for roGFP2 in Figure 2. For both probes a clean isosbestic point at ∼425 nm was identified. With these spectral characteristics both roGFPs are bona fide ratiometric sensors.
RoGFP2 has been crystallized in both the reduced and oxidized states (PDB entries 1JC0 and 1JC1) (49). A comparison between the two structures highlights structural changes that likely explain the disulfide-induced shift in chromophore protonation. Formation of the disulfide bridge shifts one β-strand relative to the other and this causes several small structural rearrangements. Significant movements involve H148 (neighbor of C147 on strand 7) and S205 (neighbor of C204 on strand 10) (Fig. 3). H148 is located in H-bonding distance to the phenolic oxygen of Y66 and moves slightly away from the chromophore upon disulfide formation. The OH group of S205 (part of the ESPT hydrogen bonding network) moves relative to the water molecule and the E222 carboxylate. It appears likely that these changes influence the Y66 protonation state and thus explain the ratiometric shift in the excitation spectrum.

Midpoint redox potentials for roGFPs have been determined through titration with DTT, lipoic acid, or bis(2-mercaptoethyl)sulfone (BMES) (27, 49). Generally, the determined midpoint potentials of −288 to −294 mV for roGFP1 and −272 to −287 mV for roGFP2 are more negative than the midpoint potential of rxYFP. Although the use of different redox buffers for titration led to slightly different apparent midpoint potentials, the ranking of roGFP1 as more reducing than roGFP2 was found to be preserved (27). For future applications, Dooley and colleagues suggested consensus midpoint potentials of −291 mV for roGFP1 and −280 mV for roGFP2 (27). A more precise determination of E°′ for redox-sensitive FPs requires exact knowledge of the midpoint potential of the calibrating redox couple and avoidance of impurities in reduced and oxidized compounds.
In addition to the difference in midpoint potential, roGFP1 and roGFP2 also show some differences in their maximum achievable dynamic range for the change in fluorescence between fully reduced and oxidized probes (Table 1). If roGFPs are used as a single wavelength probe, they exhibit approximately a 3-fold change in fluorescence, which is only slightly better than the 2.2-fold change for rxYFP. However, because roGFPs are ratiometric, their dynamic range is significantly increased. With fully flexible filter-based detection systems such as epifluorescence microscopes or fluorescence plate readers, the probes can be excited exactly on the two excitation peaks, which would provide the largest achievable dynamic range. With excitation at 390 ± 5 nm and 480 ± 5 nm, roGFP2 has a dynamic range of about 12 (81). On appliances that use fixed wavelength lasers for fluorescence excitation such as confocal microscopes or fluorescence-activated cell sorting (FACS), the dynamic range is significantly lower due to off-peak excitation. The canonical laser wavelengths used for excitation of GFP are 405 and 488 nm. While the S65T mutation in roGFP2 shifts the excitation peak B for the anionic form of the chromophore to 490 nm, the anionic form of roGFP1 is excited with a maximum at 475 nm. When excited at 488 nm the dynamic range of roGFP1 is thus below 3, while roGFP2 would provide a theoretical dynamic range of ∼9 (107).
The table does not include all variants generated so far, but rather a selection considered most useful for biological applications.
dynamic range determined with the excitation wavelengths given in parenthesis.
Ostergaard et al. (92).
Hansen et al. (47).
Hanson et al. (49).
Dooley et al. (27).
Cannon and Remington (13).
Lohman and Remington (67).
Schwarzländer et al. (107).
N.D., Not determined.
3. Variants of rxYFP and roGFPs with altered properties
To match different needs and applications, it would be desirable to obtain a collection of probes with different midpoint potentials. For instance, to detect deflections from the steady state in both directions, probes with a midpoint potential matching the steady state redox potential of the environment under study are most appropriate. Along these lines, in addition to roGFP1 and roGFP2, four other variants, roGFP3 to roGFP6, have been engineered (Fig. 4; Table 1) (49).
RoGFP3 and roGFP4 differ from their siblings roGFP1 and roGFP2 in that they contain the disulfide bridge in position C149/C204. RoGFP5 and roGFP6 contain all four cysteines and thus can potentially form two internal disulfide bridges. However, none of these variants have been investigated in much detail so far and the presence of two disulfide bridges may render quantitative interpretation of the measured fluorescence ratio rather difficult. The variants roGFP3 and roGFP4, however, may have some potential for future applications. Although the observed dynamic range of theses variants is lower than that observed for roGFP1 and roGFP2, the midpoint potentials are slightly more negative. This is particularly true for roGFP3, which is derived from wtGFP, with the most negative midpoint potential of all roGFPs at −299 mV (49). Thus, in highly reducing compartments roGFP3 may have some potential for achieving more accurate measurements compared to other roGFPs.
The first generation of redox-sensitive FPs (rxYFP, roGFPs1-6) all had midpoint redox potentials of −260 mV or below, which made them most useful in reducing compartments, such as the cytosol, the mitochondrial matrix and chloroplast stroma (Table 1) (49, 92).
In the lumen of the endoplasmic reticulum (ER), rxYFP, roGFP1, and roGFP2 were fully oxidized, suggesting a redox potential higher than −240 mV (2, 49, 93). Thus, in oxidizing compartments the use of these probes is limited by their rather low midpoint potential. Recently, Lohman and Remington developed a new subfamily of roGFP variants derived from roGFP1, called roGFP1-iX, where ‘X’ denominates the insertion of different single amino acids into β-strand 7, adjacent to C147 (Fig. 4) (67). In conjunction with the exchange H148S the insertion resulted in an increased geometric strain in the disulfide, which renders the midpoint potential for the disulfide bond less negative. The least negative values were achieved with the insertion of leucine (−229 mV) and glutamate (−236 mV) (Table 1). Despite the very significant shift in E°′, it is not yet clear whether an E°′ of −230 mV is already sufficient for dynamic measurements in the ER.
To be reactive, thiols need to be de-protonated. The pK a values of the two cysteines in rxYFP and roGFPs are all located between 8.9 and 9.5 (47, 49), which results in mostly protonated thiols and hence low reactivity. Introduction of basic amino acids next to cysteines can lead to stabilization of the thiolate form and thus was expected to increase the reactivity of the thiols in a series of 2nd generation redox-sensitive FPs (Fig. 4). Insertion of two lysines close to the two cysteines plus an arginine in position 223 led to a 6-fold increased rate constant in roGFP1-R12 (13). The insertion of up to three positive charges near the disulfide in rxYFP (rxYFP200R/204R/227R) led to a 13-fold rate enhancement for the oxidation of rxYFP by GSSG (47). However, the achieved rate enhancements appear negligible for reactions in vivo, where the kinetic properties of redox-sensitive FPs are mainly determined through interaction with glutaredoxins (Grxs) (see Section III.A).
4. Interaction of rxYFP and roGFP with cellular redox pairs
When expressed in HeLa cells, the effective concentration for H2O2-induced oxidation of roGFPs was much lower than the concentration required in vitro (27). This observation suggested that oxidation of roGFPs inside cells is a catalyzed process. A link between roGFP oxidation and the cellular glutathione pool was indicated by a slight oxidation of roGFP in HeLa cells after a 2-hour incubation with 100 μM L-buthionine (S,R)-sulfoximine (BSO), a highly specific inhibitor of GSH biosynthesis. Furthermore, BSO-treatment also led to a highly increased sensitivity of roGFP to exogenously applied H2O2 (27).
At the same time, Jakob Winther and co-workers reported that rxYFP in yeast cells senses the glutathione redox potential (EGSH ) through interaction with Grxs (93). Thioredoxins, in contrast, were not capable of reducing or oxidizing rxYFP. A sensitive response of roGFP2 to depletion of cytosolic glutathione was shown in neuronal cells (121) and detailed studies in Arabidopsis thaliana confirmed that roGFP2, like rxYFP, is a quantitative biosensor for EGSH (81). Expression of roGFP2 in the partially GSH-deficient Arabidopsis mutant cad2, which is restricted in the activity of the first and rate-limiting GSH biosynthetic enzyme glutamate-cysteine ligase (GSH1) (21), resulted in an increased 405/488 nm fluorescence ratio reflecting a shift in EGSH towards less reducing values (Figs. 5B and 5C) (81). The detected shift in EGSH closely matched expectations based on only 30% remaining glutathione and the assumption of constant glutathione reductase (GR) activity. In a more severe GSH1 mutant, rml1, which contains less than 5% of wild-type GSH (12,120), roGFP2 was almost completely oxidized (Fig. 5D). Taken together, studies on rxYFP and roGFP strongly support the notion that within cells these proteins communicate with the glutathione redox couple through mediation by endogenous Grxs.

Several other redox-active compounds like NADPH and ascorbate, as well as oxidoreductases, such as Trxs and protein disulfide isomerases (PDIs), have been tested for interaction with roGFP in vitro, but none of these significantly affected the redox status of roGFP (40, 81) (Meyer et al., unpublished results). However, given the broad spectrum of oxidoreductases in the Trx superfamily and other thiol-reactive proteins in cells, it cannot be excluded that some of these proteins may be capable of interacting with roGFP. As long as the Grx-catalyzed equilibration of roGFP with 2GSH/GSSG is kinetically favored, all such theoretically possible side reactions of roGFP should be overruled and high specificity of roGFP for EGSH guaranteed. Translational fusion of an appropriate Grx to roGFP is a potent strategy to ensure constant kinetic coupling and fully reproducible probe specificity (see Section III.A).
5. The Nernst equation and its implications for using roGFPs
Based on the very well supported notion that roGFPs predominantly, if not exclusively, equilibrate with the 2GSH/GSSG redox couple, the redox behavior of roGFPs can be described and predicted by the Nernst equation, with various implications for the practical use of the probe. How fast the equilibrium is reached, or if it is reached at all, depends on the availability of Grx, which may differ between experimental systems. As discussed later on, the translational fusion of Grx to roGFP is a suitable strategy to ensure real-time equilibration (see Section III). The following calculations exemplarily apply to roGFP2.
a. Thermodynamics of roGFP2
Mathematically, the Grx-mediated exchange of electrons between glutathione and roGFP2 can be described by the Nernst equilibrium EGSH
= EroGFP2
, specifically
In this equation, R is the gas constant (8.315 J K−1 mol−1), T the absolute temperature (298.15 K), and F the Faraday constant (96,485 C mol−1).
Here, it is important to realize the definition for OxDGSH
. The total concentration of glutathione (GSHtotal) refers to GSH equivalents (i.e., GSHtotal = [GSH] + 2[GSSG]). OxDGSH
is the fraction of GSHtotal that exists as [GSSG]. For example, OxDGSH
= 0.5 does not mean that GSSG and GSH are in a 1:1 molar ratio, it rather means that 50% of all GSH equivalents exist as GSSG (i.e., that GSSG and GSH are in a 1:2 molar ratio). As GSHtotal is an important variable in biological settings, it is useful to express the Nernst equilibrium relationship as a function of OxDGSH
, GSHtotal and OxDroGFP2
:
This relationship allows to make various predictions and estimations about the behavior of the roGFP2-based biosensor system (see following sections).
b. Measuring range and calibration of roGFP2
The relationship between OxDroGFP2
and the redox potential E can be derived from the right half of the preceding equation:
This relationship reveals that the response of the probe becomes increasingly nonlinear when approaching 5% and 95% oxidation, respectively, and thus renders the detected signal sensitive to background noise (Fig. 6). For practical purposes, changes below 5% and above 95% sensor oxidation cannot be sufficiently resolved in fluorescence measurements. At best, roGFP2 can be expected to measure changes in EGSH in the 80 mV-wide window between −320 and −240 mV (Fig. 6).
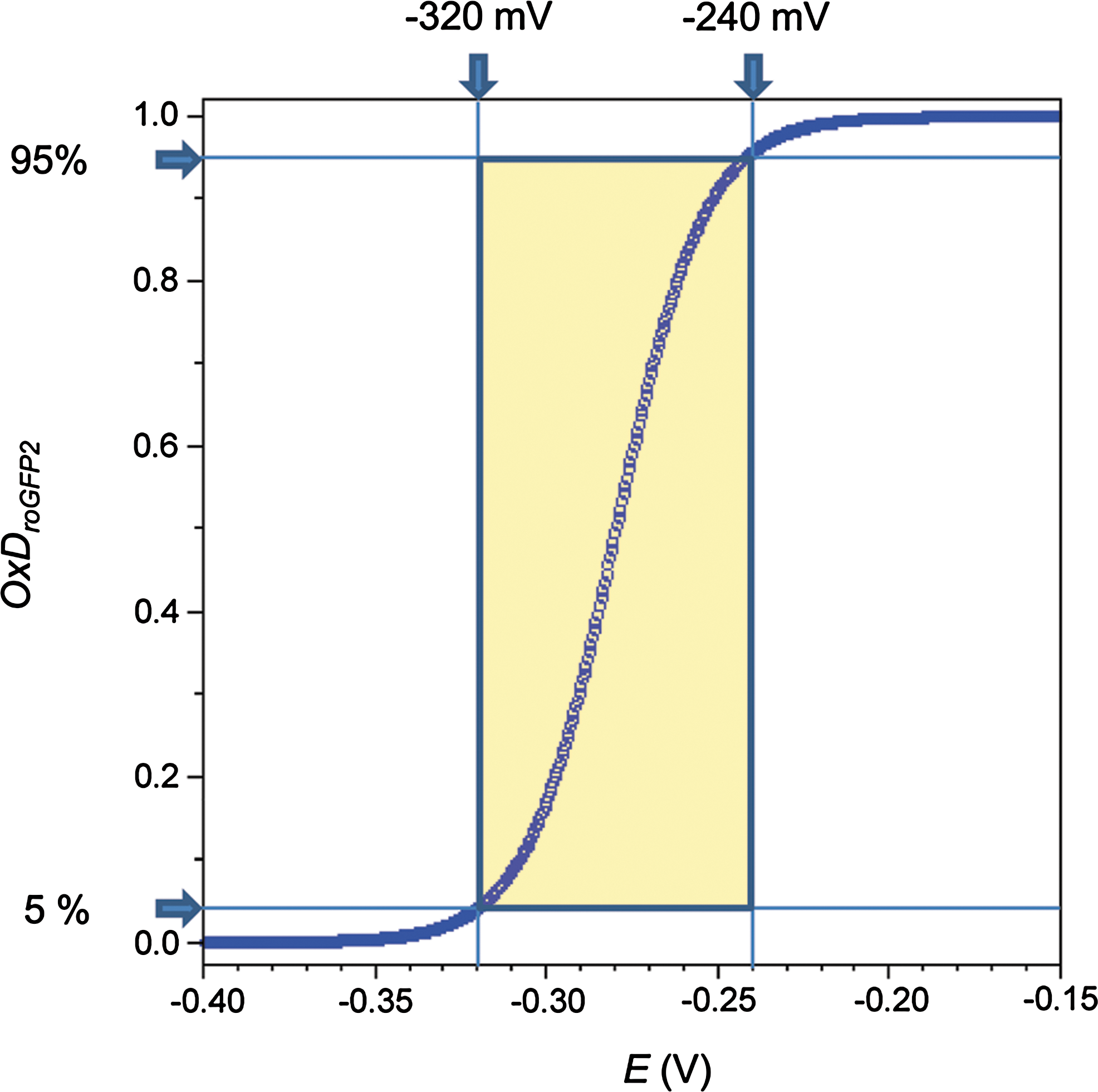
Suitably, the cytosolic EGSH of unstressed cells is typically found in the range between −320 to −300 mV, close to the lower end of the practical measuring range. The measuring range of roGFP2 is well suited for measuring EGSH in the cytosol and several other compartments, including the nucleus, mitochondria, and plastids. It is, however, not suitable for the ER, where it is oxidized at all times (81).
Given the available measuring range, is it feasible to derive absolute EGSH values from fluorescence data? Of course, it would be highly desirable to calibrate intracellular measurements of EGSH against independently confirmed standards. This, however, would require an independent compartment-specific approach to determine intracellular EGSH . Unfortunately, no such alternative method is available. Since absolute intracellular calibration is not feasible, a more indirect way of calibration has to be performed. In a practical approach, to estimate intracellular EGSH from the measured fluorescence ratio (R), externally applied cell-permeable reagents (typically dithiothreitol and diamide or H2O2) are used to deflect the probe in situ to the fully reduced and oxidized states. The resulting fluorescence ratios (Rred and Rox ) define the total range of the sensor inside intact cultured cells or transgenic tissues. Thus, the full set of measurements delivers six intensity values from which three ratios can be calculated (Table 2).
To establish a relationship between the macroscopic observables and OxDroGFP2 , several molecular quantities need to be defined: i405 red , i405 ox , i488 red , and i488 ox are the fluorescence intensities contributed by a single roGFP2 molecule at the indicated wavelength and redox state, Ntotal is the total number of roGFP2 molecules, Nred is the number of reduced roGFP2 molecules, and Nox is the number of oxidized roGFP2 molecules. Based on the assumption that the overall fluorescence intensity corresponds to the summation of fluorescence intensities from individual roGFP2 molecules, the relationships given in Table 3 apply.
By inserting the resulting equations (right column of Table 3) into OxDroGFP2
= Nox/Ntotal, the relationship between OxDroGFP2
and the macroscopic observables can be deduced. As a result, OxDroGFP2
can be calculated directly from the six measured intensity values:
Or, alternatively, after transforming the same equation, using ratios:
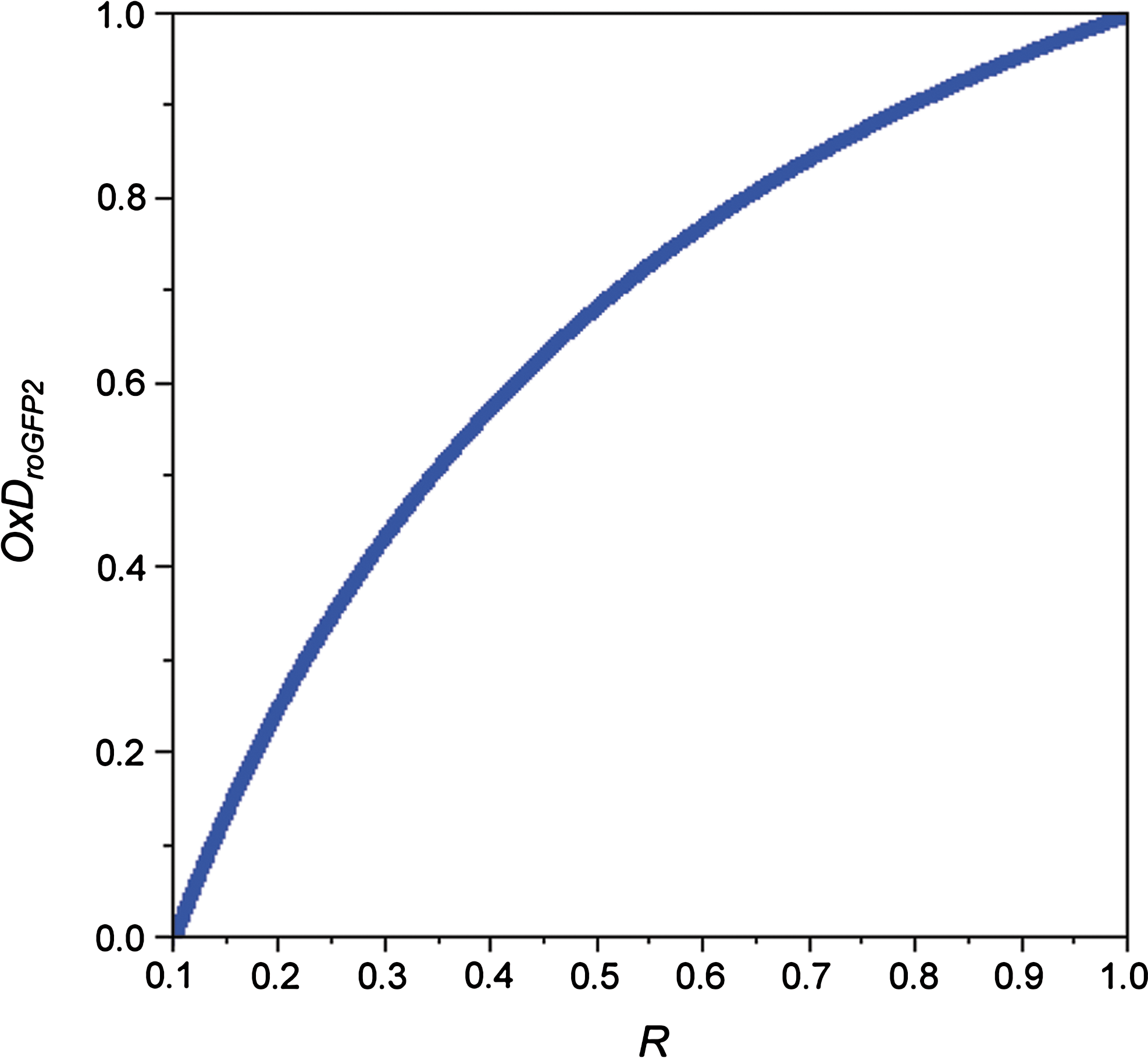
In this equation, the quotient (I488 ox /I488 red ) is most appropriately considered the ‘instrument factor’. As the intensity at both wavelengths is linearly dependent on OxDroGFP2 , the ratio R is not (Fig. 7). Only in the special case that the isosbestic point (425 nm in the case of roGFP2) is used as the reference wavelength for ratiometry (instead of 405 nm) the instrument factor is canceling out from the equation, which then describes a linear relationship.
Having obtained OxDroGFP2
, the corresponding intracellular sensor redox potential EroGFP2
can be calculated from the Nernst equation:
Finally, assuming complete equilibration, EGSH should equal EroGFP2 . Of course, the accuracy of the estimate is limited by measurement errors and differences in pH and temperature relative to the standard conditions. For a more detailed discussion of pH influence, see Section II.B.5.d below.
c. Sensitivity of roGFP2 towards changes in OxDGSH and [GSSG]
As the midpoint redox potential of roGFP2 (−280 mV) is much more negative than that of glutathione at physiological concentrations (ranging from −151 mV at 1 mM to −181 mV at 10 mM), even a small increase in the degree of glutathione oxidation will give rise to a large and disproportionate degree of sensor oxidation (Fig. 8). However, since EGSH depends on GSHtotal, so does the sensitivity of the probe towards changes in OxDGSH and [GSSG].
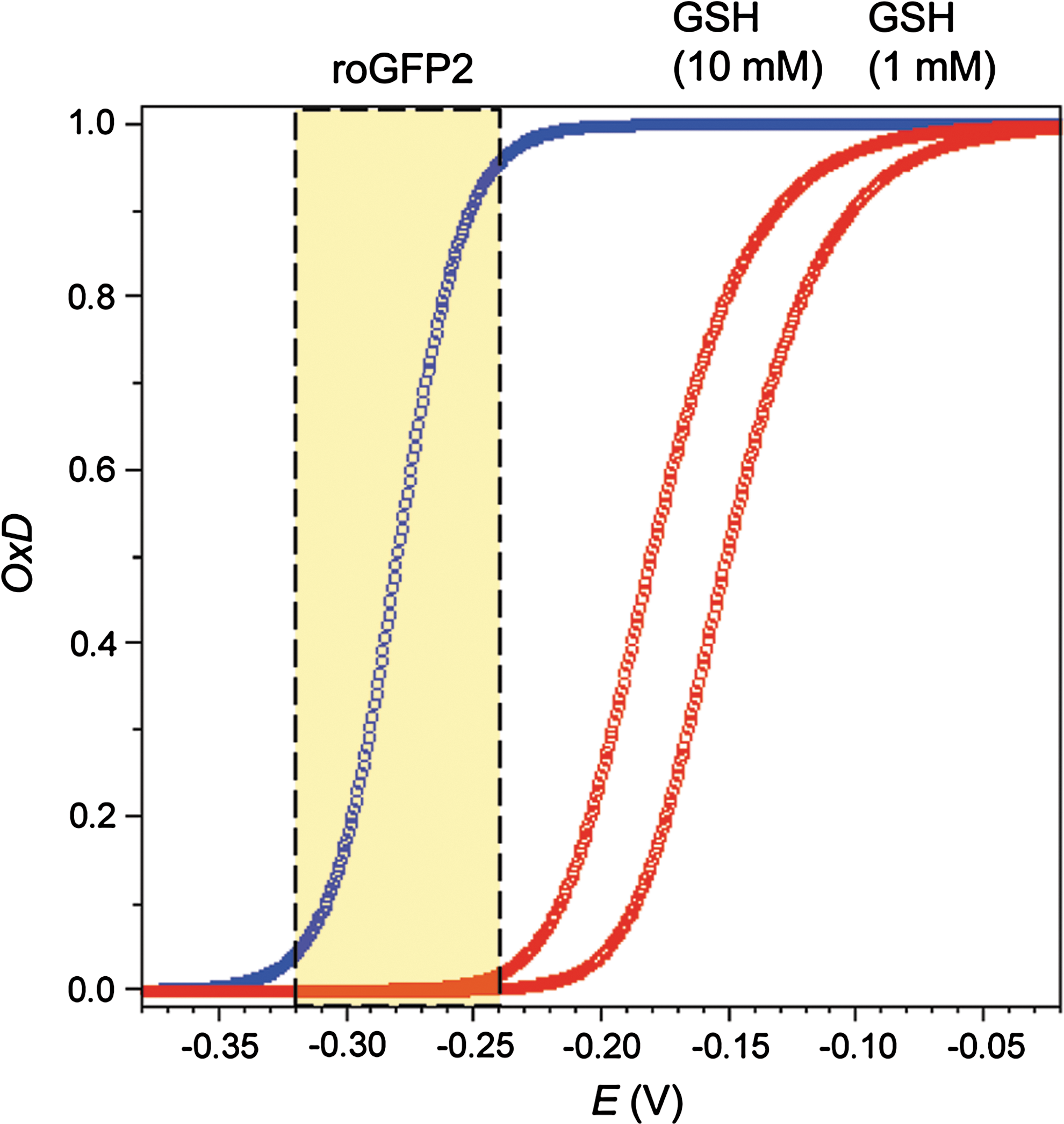
It follows that roGFP2 should be a more sensitive indicator of OxDGSH and [GSSG] in cells with low GSHtotal as compared to cells with high GSHtotal. Comparing overall cytosolic glutathione concentrations of 1 and 10 mM, broadly reflecting the variation between mammalian cell types, the difference in sensor oxidizablity is quite significant: At GSHtotal = 1 mM 50% sensor oxidation is achieved with a 10-fold lower OxDGSH (0.000088 vs. 0.00088) (Fig. 9A) and a 100-fold lower [GSSG] (44 nM vs. 4.4 μM) (Fig. 9B).

To quantify the ‘transmission’ between glutathione oxidation and sensor oxidation—as afforded by the difference in midpoint potential between the two redox pairs—the OxDroGFP2 /OxDGSH ratio at the sensor midpoint potential (−280 mV) can be considered. The ‘transmission ratio’ increases steeply with decreasing GSHtotal and is 10-fold higher at 1 mM than at 10 mM (Fig. 10).

Although the ‘transmission ratio’ becomes very high with decreasing GSH, at GSHtotal = 10 mM the ‘transmission’ is just sufficient to reliably observe changes in the lowest part of the physiological range between −320 and −300 mV. Specifically, at GSHtotal = 10 mM, the shift of EGSH from −320 to −300 mV corresponds to an absolute increase of [GSSG] from 200 to 930 nM, and a shift in OxDGSH from 0.00004 to 0.00018. As a consequence, the oxidation of roGFP2 increases from 4% to 17%. If, however, a redox-sensitive FP with a midpoint potential of −260 mV had been used instead of roGFP2, it could not reliably ‘perceive’ this change at GSHtotal = 10 mM, as its oxidation would just increase from 1 to 4%.
d. The influence of pH
How does pH influence the roGFP2 response? In principle, there are two ways by which pH can influence probes based on redox-sensitive FPs. Firstly, by influencing the fluorophore itself, and, secondly, by influencing the redox-sensing dithiol/disulfide pair. Direct influence on the fluorophore has been a concern, because the S65T mutation is expected to render roGFPs pH-sensitive. In fact, roGFP2 loses fluorescence at low pH, like other GFP variants with the S65T mutation (30). Fortunately, it turned out that pH-dependent fluorescence quenching affects both excitation wavelengths equally and that the calculation of excitation ratios effectively cancels out any pH effect in the range between 5.5 and 8.0 (107). The pH-insensitivity of the ratio was also confirmed for the fusion protein Grx1-roGFP2 (40), to be discussed later (see Section III.A.2).
What about the general influence of pH on thiol–disulfide equilibria? The reactivity of thiols and thus the redox potential is directly linked to the ambient pH because two protons are liberated during the formation of a disulfide bridge. As long as the pH does not approach the pK a of the thiols forming the disulfide bond, the redox potential will change by approximately 59 mV per pH unit (103, 127). Consequently, a change in pH leads to a simultaneous change in EGSH and EroGFP2 , into the same direction and by a very similar amount. Given a pK a of 8.92 for glutathione (103) and assuming a pK a of 9.0 for the roGFP2 cysteines (14), it can be expected that the difference in redox potential between the two redox pairs is not significantly influenced by pH changes in the physiological range (Fig. 11A). The predicted deviation from co-linearity (ΔΔE) amounts to just 2 mV at pH 8, but becomes more significant at higher pH (Fig. 11B).

This inference is supported by the in vitro observation that recombinant roGFP2 or Grx1-roGFP2 show the same response to GSH solutions adjusted to different pH in the range between 5.5 and 8.0 (Meyer et al., unpublished data; Gutscher et al., unpublished data). Thus, roGFP2 does not respond to pH-induced changes in EGSH and should be regarded an indicator for pH-independent changes of EGSH . It should be noted that pH nevertheless influences the kinetics of roGFP2-glutathione equilibration, which is faster at higher pH, attesting to the deprotonation of thiol groups involved in the exchange (Meyer et al., unpublished data; Gutscher et al., unpublished data). In conclusion, expressing roGFP2 in a compartment with different pH (e.g., mitochondria) should not influence its sensitivity to oxidation by GSSG (compare Section II.B.5.c). However, if absolute mV values are to be estimated from fluorescence data, a correction for the assumed pH difference is required.
6. Applications of rxYFP and roGFPs
The applicability of conventional redox-sensitive FPs is limited by the kinetics of equilibration with the glutathione system (which may differ between species and cell types). As complete intracellular probe equilibration can take tens of minutes, conventional redox-sensitive FPs are best suited to determine steady state redox conditions and to document redox changes that are sufficiently long-lived to allow the probe to equilibrate.
a. Measurement of steady state redox conditions
RxYFP and roGFPs were originally developed to visualize the formation of disulfide bonds in living cells and to conduct noninvasive quantitation of thiol–disulfide equilibria in distinct subcellular compartments (49, 92). Expression of these probes in the cytosol of Saccharomyces cerevisiae (92, 93), HeLa cells (40, 49), airway epithelial cells (19, 106), pulmonary artery smooth muscle cells (19), human lung fibroblasts (59), Arabidopsis thaliana, and tobacco (8, 57, 81) consistently showed that the probe redox potential in the cytosol is close to or even below −300 mV. From these measurements it can be concluded that cytosolic [GSSG] is in the low-micromolar to mid-nanomolar range and thus several orders of magnitude lower than assumed before on the basis of conventional measurements in tissue extracts (81, 93). Consistent with this view, rxYFP-expressing yeast and roGFP-expressing plant mutants lacking cytosolic GR display an increased fluorescence ratio and a shift of 30 to 40 mV towards less negative EGSH values (71, 93). Under severe oxidative stress the reducing function of GRs can be overruled. For example, treatment of Drosophila S2 cells with the redox cycling agent paraquat for 48–72 h led to severe oxidation of cytosolic roGFP (137).
The large overestimation of cytosolic [GSSG] by enzymatic or chromatographic analysis of cell lysates or tissue extracts likely results from the mixing of subcellular compartments and thus reflects the oxidized glutathione pool of the ER and, possibly, other organelles. In fact, all experiments in which redox-sensitive FPs have been targeted to the ER resulted in fully oxidized probes (49, 79, 81, 93). Oxidizing conditions in the ER lumen are generated though continuous formation of GSSG in oxidative protein folding (15). The ER membrane is considered to be impermeable to GSSG (64), thus keeping GSSG within the secretory pathway. In the absence of an ER-resident GR, it is thus not surprising that roGFP2 was found fully oxidized even in cell lines with defective oxidative protein folding (79). Of note, roGFP1 targeted to endosomes and lysosomes was also found to be oxidized, although these compartments have been expected to exhibit a reducing environment (2).
In mitochondria, chloroplasts and peroxisomes, roGFPs were found in the fully reduced state (49, 57, 106, 107). Despite the local production of ROS in these compartments, cells are apparently capable of maintaining a high degree of glutathione reduction. For plants, it has been shown that GRs are present in all three types of organelles (18, 31, 101). Taking into account that the plant mitochondrial matrix and the chloroplast stroma are potentially alkaline with pH values up to 8 (10, 66), the estimated glutathione redox potentials in these compartments may be as low as −360 mV (49, 57, 106, 107).
b. Measurement of induced redox changes
Beyond the ability to target roGFPs to different subcellular compartments for steady state redox measurements, the biggest advantage of roGFPs over conventional fluorescent dyes is the reversibility of the oxidation reaction and thus the potential for recording dynamic redox changes in living cells.
All studies published to date have confirmed the dynamic response of intracellular roGFPs to exogenously applied oxidants and reductants. Typically, the reversibility of the cytosolic or mitochondrial roGFP response was demonstrated by sequential incubation with H2O2 and DTT (49, 57, 81, 106, 107) or aldrithiol (2,2-dipyridyl disulfide) and DTT (27, 43). Similar experiments have measured the time course of probe reduction following an exogenous oxidative challenge. However, interpretation of kinetic probe behavior is difficult, because the isoforms and concentrations of Grxs may differ between cell types and compartments. For example, following probe oxidation with exogenously applied H2O2, roGFP2 in Arabidopsis mitochondria recovered very slowly (108). This response pattern has been discussed to indicate prolonged endogenous ROS production induced by exogenous oxidative stress. It cannot be excluded though that the slow reduction of roGFP2 in plant mitochondria is caused by lack of appropriate Grxs for catalyzing equilibration between roGFP2 and the matrix glutathione buffer. Out of a large number of 31 Grxs in Arabidopsis (80), only two have been experimentally confirmed to be present in mitochondria (18, 51, 54). It is not known whether these Grxs are capable of interacting with roGFPs.
Other experiments have looked at the response of ER-localized roGFPs following an exogenously applied reductive challenge. RoGFP1 targeted to the ER lumen in airway epithelial cells was almost fully oxidized but could be reduced with 500 μM DTT (106). After washout of the reductant, the fully oxidized state was re-established within 7 min. The roGFP re-oxidation process was also investigated in yeast cells (79). After initial reduction of ER-resident roGFP2 by DTT, it was possible to compare the kinetics of re-oxidation in wild-type and mutant cells. Severely delayed oxidation of roGFP2 in a knockdown of Ero1p supports the view that Ero1p is the major net generator of disulfides in the ER lumen (11, 34, 109, 118).
Another category of intracellular redox changes, closer to physiological relevance, are those caused by endogenously produced oxidants. In a recent study, roGFP was used to investigate the effects of complex II inhibitors on mitochondrial ROS production (42). Acute administration of thenoyl-trifluoroacetone (TTFA), an inhibitor of the ubiquinol binding site in complex II, resulted in a gradual increase in oxidation over a 30 min period. Oxidation induced by TTFA was significantly higher than that induced by inhibitors targeting the succinate binding site in complex II.
Time-resolved roGFP imaging was used to analyze hypoxia-induced ROS formation in mouse pulmonary artery smooth muscle cells (25). Within minutes from the onset of hypoxia, OxDroGFP increased from ∼12% to ∼17%, a value that was maintained throughout the 30 min hypoxia treatment. After moving back to normoxic conditions, roGFP oxidation returned to base levels. The oxidation of roGFP during hypoxia was prevented by overexpression of catalase, implying a role for H2O2. A slight decrease in roGFP oxidation was observed during hypoxia in AML12 liver cells (43). It is, however, impossible to directly compare these observations because Desireddi et al. (25) present their data as % oxidation of roGFP, while Haga et al. (43) present the 400/480 nm fluorescence ratio. In addition, it is not clear whether the measurements are based on the same roGFP variant. As described in Section II.B, the fluorescence ratio and the achievable dynamic range depend on the roGFP variant and the excitation wavelengths employed. In contrast to the fast recovery of roGFP after return to normoxic conditions reported by Desireddi et al., Haga and colleagues found rapid oxidation immediately following re-oxygenation (43). Oxidation gradually increased over 60 min and was more pronounced after prolonged hypoxia. External application of N-acetylcysteine and catalase before onset of hypoxia prevented post-hypoxic roGFP oxidation. In the same study, adenovirally expressed roGFP was also used to monitor redox changes in the murine liver during ischemia and reperfusion. Similar to cell culture experiments, hepatic ischemia immediately led to increased roGFP reduction followed by a rapid partial oxidation within 5 min after ischemia ended (43).
c. roGFPs as topology reporters for membrane proteins
Current estimates indicate that 20%–30% of all predicted open reading frames in a typical genome code for transmembrane (TM) proteins (122), but the number, location and orientation of transmembrane domains (TMDs), as predicted by bioinformatics algorithms are often highly contradictory (29). Despite the general biological importance of TM proteins, tools for experimental analysis of the protein topology are limited. In yeast, the topology of TM proteins can be investigated in vivo using fusions with essential metabolic enzymes as topology reporters and expression of these fusions in a mutant background (58). This approach, however, is not feasible for most other eukaryotic cell types because suitable mutants are often lacking. An advanced method to analyze the topology of TM proteins in vivo is the redox-based topology analysis (ReTA) in which roGFP2 is exploited as a topology reporter (8) (Fig. 12). The only requirement for ReTA to be applicable is a distinct gradient in EGSH across the membrane the TM protein of interest is targeted to. Such a gradient exists across the ER membrane and thus all TM proteins passing through the ER are prime candidates for ReTA. The straightforward ratiometric analysis without the need of any calibration provides a binary readout for the orientation of the protein within the membrane (8). For further analysis of the entire protein topology, roGFP can be fused to truncated versions of the protein or the probe can be inserted into (putative) extra-membrane loops. The latter approach has been shown to be feasible by inserting conventional GFP into PIN5, an atypical member of the PIN auxin-efflux-carrier family in plants (88). Oxidized roGFP2 has a pK a of 6 (49) and thus could be quenched to a large extent in acidic compartments such as lysosomes, plant vacuoles, and the apoplast in plants. Loss of fluorescence on one side of the membrane would impede binary ratiometric readouts. Since most proteins in these membranes pass through the ER and their topology is assumed not to change during passage through the secretory pathway, it may still be possible to analyze their topology by ReTA within the ER (8). Another possibility to overcome fluorescence quenching in acidic compartments may be to use the pH-insensitive roGFP1, which is assumed to have a much lower pK a in the order of the apparent pK a near 4.5 determined for wtGFP (117).
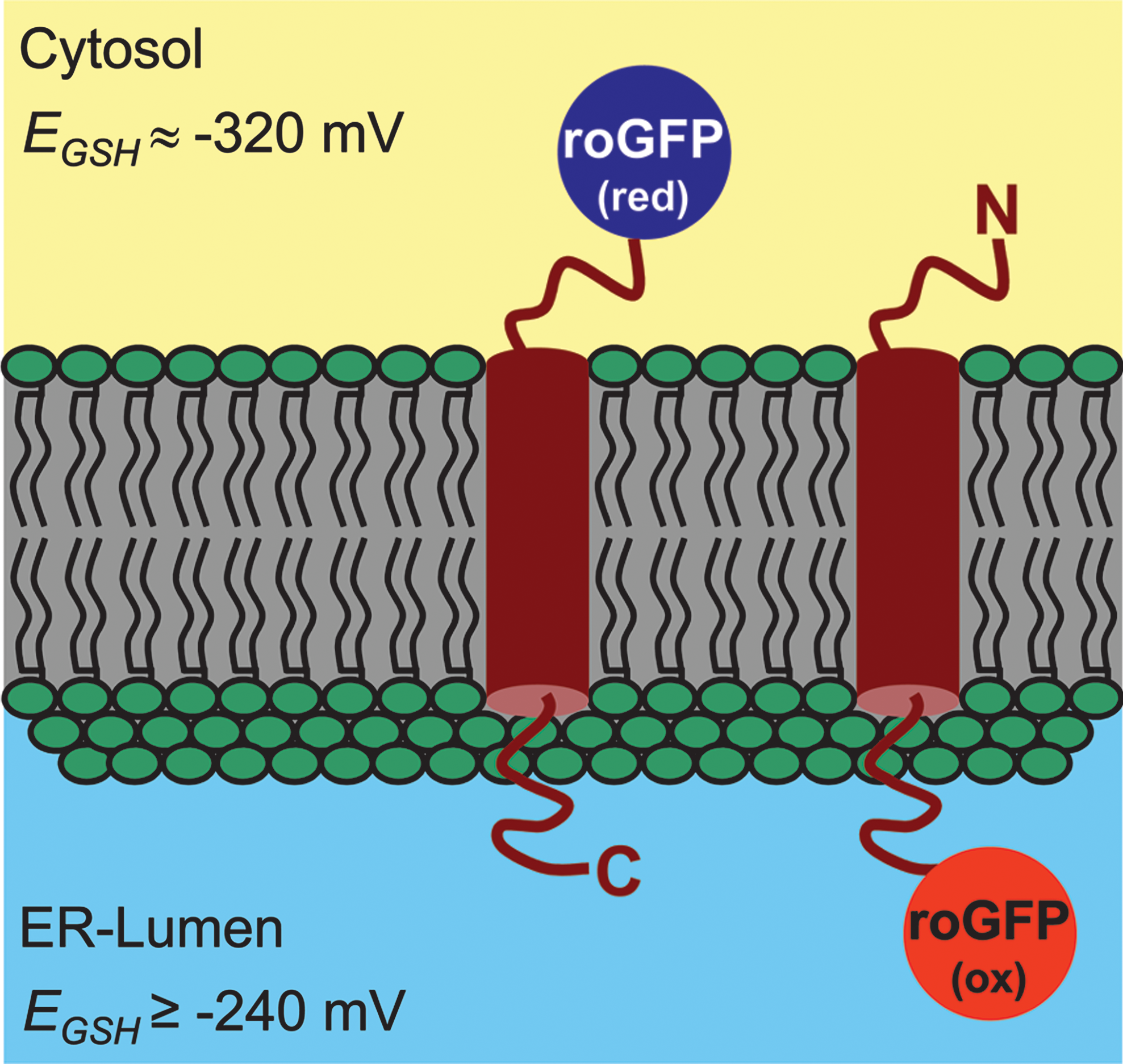
ReTA can be expanded to the analysis of protein orientation in nonsecretory membranes provided a sufficient EGSH gradient is present. For mitochondria it has been shown that rxYFP is partially oxidized in the intermembrane space (IMS), whereas the probe was considerably more reduced in the cytosol and the mitochondrial matrix (56). Based on this finding, ReTA should also be applicable to mitochondrial membrane proteins. Applicability of ReTA to TM proteins of the chloroplast envelope in plants would also depend on the ability to measure EGSH in the IMS of the chloroplast envelope. So far, however, no successful expression of any fluorescent protein has been reported for the plastidic IMS (e.g., (5)). Nevertheless, it can be envisaged that the topology of TM proteins in the inner membrane can be investigated in a mutant background specifically affected in plastidic GSH levels and thus plastidic EGSH . With these amendments ReTA is considered to be broadly applicable and the most simple-to-use method currently available for the analysis of TM protein topology in higher eukaryotic cells (8).
C. cpYFP as a putative probe for superoxide
O2
•- is the primary ROS generated by the photosynthetic and respiratory electron transport chains, as well as the Nox enzymes. Localized generation of O2
•- has been reported to accompany signaling and stress in both plants and animals (33, 63, 65). To understand the biological significance of such localized O2
•- production and the dynamics of associated signaling cascades, it would be highly informative to specifically record the formation of O2
•- as the first intermediate in such pathways. Rather unexpectedly, it was recently reported that cpYFP is capable of responding to O2
•- (126). In cpYFP, the amino and carboxyl portions of YFP have been interchanged with a short connecting linker between the original termini, while the axial α-helix containing the chromophore maintained its central position in the β-barrel. CpYFP was originally generated to develop so-called
When cpYFP was expressed in mitochondria, the probe apparently responded to O2 •- produced by the respiratory chain (126). In vitro, cpYFP was found to be five times brighter under strong oxidizing conditions imposed by incubation with aldrithiol as compared to ambient redox potentials between −7.5 mV and −319 mV set by incubation with a DTT redox buffer. Furthermore, cpYFP seemed to respond to enzymatically synthesized O2 •-, but not to H2O2 and ONOO-. Hydroxyl radicals and NO• caused a slight decrease of fluorescence rather than an increase (126). Similarly, NADH did not cause a significant change in cpYFP fluorescence. The authors interpreted fluctuations in cpYFP fluorescence as quantal bursts of O2 •- production (O2 •- flashes) within the matrix of individual mitochondria in quiescent cells under resting conditions.
The correct interpretation of changes in cpYFP fluorescence depends on the understanding of the underlying chemistry. Unfortunately, at this stage there is no good explanation for O2 •--induced changes of chromophore properties. In principle, O2 •- can act as an oxidant or reductant. O2 •- only rarely oxidizes biological compounds because the anionic charge limits reactivity with electron-rich centers (130). Nevertheless, Wang and colleagues considered the two residues C171 and C193 in the cpYFP molecule as potential targets of O2 •- (126). Indeed mutation of both cysteines to either alanine or methionine abrogates the sensitivity to aldrithiol, but it also largely reduces fluorescence of the entire molecule. C171 and C193, which are in close proximity to the chromophore on the central α-helix spanning the β-barrel, are equivalent to C48 and C70 in the parental GFP molecule. Loss of fluorescence was also reported earlier for the single mutation of C70 in GFP. While mutation of C48 does not affect the fluorescence of GFP, substitution of C70 proved to be deleterious to obtaining soluble, fluorescent protein (49). This deleterious effect makes the interpretation of the double cysteine mutant of cpYFP partly ambiguous. For further characterization of cpYFP, it would be necessary to study the properties of the respective single cysteine mutations.
While the interior of GFP is hardly accessible to small molecules, circular permutation of the protein results in obvious clefts in the β-barrel that may allow access for H+ and other small molecules to the chromophore. Indeed, cpGFP is more sensitive to acidic pH than EGFP and is acid-quenched below pH 6 (3). A change in protonation of the chromophore, however, should be detectable at both excitation wavelengths with opposite direction. In the case of cpYFP, such an effect of pH on the chromophore appears unlikly because a change in fluorescence was only observed with excitation at 488 nm, but not with excitation at 405 nm (126). In conclusion, more detailed studies are required before cpYFP can be adopted as a probe for superoxide.
III. Redox Relays Based on Redox-Sensitive FPs
Most recently, chimeric fusion proteins of redox-active enzymes and redox-sensitive FPs have been explored for their use as redox probes. A redox enzyme may be coupled directly to redox-sensitive FPs by electron transfer, thus forming a redox relay. The rationale behind this approach is the idea that redox enzymes will lend specificity and efficiency to a redox sensing process. The investigation of this principle was motivated by the fact that conventional roGFPs did not show responses to weak transient oxidative processes as they occur in a physiological setting (e.g., after growth factor stimulation) (27, 46). Since, on the other hand, roGFPs indicated pronounced redox differences between subcellular compartments (ER vs. cytosol), it was obvious that they nevertheless adjust to a steady state redox potential. Thus, it seemed that conventional roGFPs are limited by a slow response to oxidative changes (which may require tens of minutes) (13). As discussed, roGFPs are capable of equilibrating with 2GSH/GSSG, but in many situations this equilibration is too slow to allow the detection of transient and weak oxidative changes. Based on this consideration, it was hoped that the proximity of a covalently attached redox catalyst (present in a very high local concentration) in a 3rd generation of redox-sensitive FPs (Fig. 4) would facilitate rapid and complete equilibration with a defined cellular redox couple.
A. Coupling redox-sensitive FPs to glutaredoxins
1. rxYFP-Grx1p
Following the observation that rxYFP in yeast cells equilibrates with glutathione (93), the first investigation into the direction of fusion-based redox relays was done by the group of Jakob Winther. The fusion between rxYFP and yeast Grx1p (rxYFP-Grx1p) was primarily used as a biochemical tool to study the reaction mechanism of Grx. The fused Grx was found to operate through the monothiol mechanism. RxYFP oxidation was improved by a factor of 3300 and proceeded with almost absolute glutathione specificity (7). The crystal structure of rxYFP-Grx1p did not reveal direct contacts between the two protein domains, thus suggesting that it is the enhanced collision rate between the two domains that is responsible for the much improved thiol–disulfide exchange efficiency (44).
2. Grx1-roGFP2
As described in Section II.B.4, it had become clear that roGFPs, like rxYFP, interact with the glutathione system (81). This led to the question if Grx-based chimeric fusion proteins are suitable as redox probes for live imaging of redox changes in mammalian cells. To create a probe for EGSH , human Grx1 and roGFP2 were chosen as fusion partners (40). RoGFP2 was considered to be the most suitable redox-sensitive FP for the purpose of developing a biosensor. As discussed before, a general advantage of roGFPs over rxYFP is that they are ratiometric by excitation, thus making measurements insensitive to differences in sensor concentration and photobleaching (49). In addition, the midpoint potential of roGFP2 (−280 mV) is more negative than that of rxYFP (−261 mV), thus making the roGFP2 response more sensitive to oxidation by small increases in glutathione oxidation (compare Section II.B.5.c). roGFP2 was preferred over roGFP1 (49), mainly for three reasons: (i) roGFP2 is brighter, (ii) its two excitation maxima better match standard laser lines (405 and 488 nm), and (iii) the S65T mutation makes roGFP2 resistant to photoswitching, which may create artifacts by shifting fluorescence ratios (107). Due to their limited dynamic range, roGFP3 and roGFP4 (49) were not considered for the purpose of biosensor development.
a. The general concept behind Grx1-roGFP2
The intracellular response of nonfused roGFP2 is limited by the availability of endogenous Grx, which appears to be insufficient for rapid equilibration in many systems and situations. This also means that differences in Grx abundance and activity between species, cell types, subcellular compartments and developmental or environmental situations will influence the kinetics of equilibration between roGFP2 and glutathione, making observations much less comparable and reliable. To solve the problem of slow and variable equilibration, roGFP2-fused Grx is exploited to enforce continuous equilibration between the two redox pairs roGFP2red/roGFP2ox and 2GSH/GSSG. By covalently attaching the redox catalyst to roGFP2, its local concentration is dramatically increased. For approximation, assuming that Grx1 is restricted by the linker to an operating range of ∼75 Å, the local Grx1 concentration experienced by each roGFP2 molecule is ∼1000 times higher (∼1 mM) than the estimated absolute concentration of Grx1-roGFP2 in the cell (∼1 μM). The local Grx1 concentration experienced by roGFP2 is thus also at least three orders of magnitude higher than the concentration of endogenous Grx1 distributed in the mammalian cytosol (37). By making Grx an integral part of the probe, the biosensor becomes catalytically self-sufficient in securing rapid and efficient equilibration under all circumstances (Fig. 13).
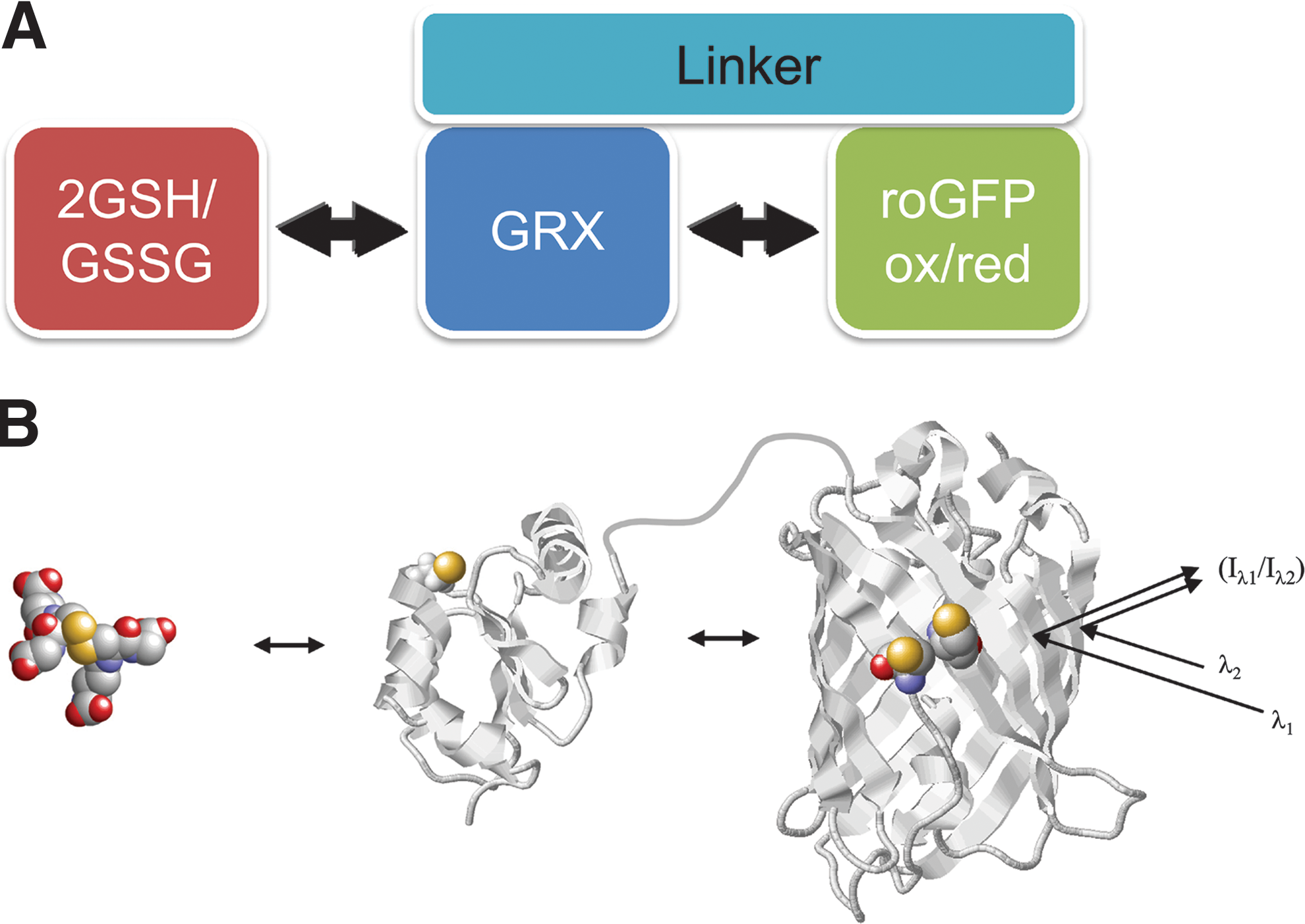
b. The molecular mechanism of Grx1-roGFP2
The response properties of Grx1-roGFP2 are based on the well established monothiol mechanism of Grxs (32, 98). This conclusion is supported by the finding that removal of the second active site thiol (C26) does not compromise the redox-sensing behavior of Grx1-roGFP2 in vitro or in live cell experiments (Gutscher et al., unpublished results). Likewise, the monothiol mechanism has also been established for rxYFP-Grx1p (7). In the oxidative response, the nucleophilic cysteine C23 of Grx1 specifically reacts with GSSG to form a mixed Grx1-glutathione disulfide intermediate. The latter reacts with one of the two thiols on roGFP2, which, as a consequence, becomes S-glutathionylated. The existence of S-glutathionylated intermediates has been confirmed by mass spectrometry (Meyer et al., unpublished data), but it remains unclear if one of the two roGFP2 cysteines is preferentially S-glutathionylated by Grx. In the last step, glutathionylated roGFP2 re-arranges to form the internal disulfide bridge (C147–C204). Importantly, the three consecutive steps of thiol–disulfide exchange are fully reversible. When an oxidative event fades away, [GSSG] and thus EGSH rapidly normalize, and the whole three-step thiol–disulfide cascade re-equilibrates (Fig. 14A). Grx1-roGFP2 is a truly dynamic probe because it is based on a completely symmetrical ping-pong mechanism (Fig. 14B).

c. Specificity of Grx1-roGFP2
Grx1-roGFP2 is expected to have a very high specificity for the 2GSH/GSSG redox couple as interactions of the roGFP2 domain with other redox couples are less likely to occur in the omnipresence of fused Grx. Grx by itself is highly specific for the interaction with glutathione and this specificity was also confirmed for the rxYFP-Grx1p and Grx1-roGFP2 fusion proteins (7, 40). Importantly, roGFP2 is not capable of interacting with the Trx system (see below), a fact that also contributes to overall probe specificity. When using Grx1-roGFP2 to investigate intracellular redox changes, it is important to realize that it is a probe for EGSH , no more and no less. As such, Grx1-roGFP2 will directly respond to changes in either OxDGSH or GSHtotal (with the exception of being mostly indifferent to pH-induced changes in OxDGSH , see Section II.D). Shifts in OxDGSH can be caused by several factors, including changes in [H2O2], but not restricted to it. It must be kept in mind that the response of intracellular Grx1-roGFP2 to H2O2 is indirect. Although direct oxidation of roGFP2 thiols by H2O2 is thermodynamically favored, it is kinetically very slow. The pK a of both thiols on roGFP2 is at least 9 (13) and without catalysis, thiols and H2O2 generally react very slowly (131). It is safe to assume that the response of Grx1-roGFP2 to physiological concentrations of H2O2 is always indirect in that H2O2 must first give rise to GSSG. Although it is expected that a rise in [H2O2] will typically entail an increase in OxDGSH , it should not be taken for granted to occur under all circumstances. For example, it is conceivable that under certain conditions H2O2 scavenging is dominated by peroxiredoxins and takes place without contribution by glutathione peroxidases. In this case, OxDGSH would not be significantly affected by H2O2.
d. Reliability of Grx1-roGFP2
Given the operating mode of Grx1-roGFP2, can we think of intracellular conditions that may compromise sensor function or even lead to false results? One possible concern is that the activity of the Grx1 domain could be subject to modulation by post-translational modification. Although there is no evidence for phosphorylation or other modifications, it should be noted that wild-type human Grx1 harbors three additional cysteines that are not essential for activity (C8, C79, and C83). It has been reported that oxidative modifications of these residues can negatively influence Grx activity in vitro (50), albeit the study used rather nonphysiological conditions and the in vivo relevance of this finding remains unclear. A roGFP2 fusion protein based on a Grx1 mutant lacking all three extra cysteines did not show a difference in redox behavior, neither in vitro nor inside cells (Gutscher et al., unpublished data). Thus, at present there are no indications that the Grx1-roGFP2 response is influenced by the Grx1 extra cysteines. However, to avoid uncertainties, it may be preferable to use modified versions of Grx1-roGFP2 lacking C8, C79 and C83.
Whenever a genetically encoded probe is (over)expressed in cells, there is a possibility of perturbation, especially if the probe is based on an enzyme. Is it conceivable that Grx1-roGFP2 disturbs intracellular EGSH , thus distorting its own measurements? This scenario appears unlikely. On the one hand, the reducing equivalents (thiols) introduced by the probe are negligible relative to the overall glutathione and protein thiol pools, both in the millimolar range (48). On the other hand, Grx1 is not known to create or destroy oxidizing equivalents and the redistribution of disulfide bonds catalyzed by Grx1 is not expected to alter EGSH . In fact, when roGFP2- and Grx1-roGFP2-expressing cells were compared, their steady state EGSH was not significantly different (Gutscher et al., unpublished data), strengthening the notion that the additional Grx1 brought into the cell by the fusion protein does not influence EGSH .
Another question though is if the additional Grx1 could influence other aspects of cellular behavior, for example, by impacting signaling. In mammalian cells, endogenous Grx1 has been found to be associated with ASK1 and procaspase-3 and may have a role in regulating these proteins (96, 112). Although it may be argued that fused Grx1 is poised to act on its fusion partner roGFP2, it cannot be excluded that roGFP2-linked Grx1 catalyzes glutathionylation/deglutathionylation of other proteins as well. Overexpression of Grx1-roGFP2 in HeLa cells did not influence proliferation rate or sensitivity to apoptosis relative to nontransfected cells (Gutscher et al., unpublished data). Nevertheless, as with other genetically encoded probes, the principal possibility of changing some aspects of cell physiology must be kept in mind. To minimize the potential risk of unwanted Grx interactions, it seems reasonable to use a distantly related Grx, for example, bacterial Grx for EGSH measurements in human cells. However, the performance of such alternative EGSH probes remains to be tested.
Finally, the structural integrity of GFP chimeric proteins is another general issue as proteolytic fragments may compromise the overall sensor response. In some cases, the linker must be optimized to minimize folding interference and proteolysis. When expressed in HeLa cells Grx1-roGFP2 did not show signs of degradation despite its rather long linker of 30 amino acids (Gutscher et al., unpublished data).
e. Sensitivity of Grx1-roGFP2
As discussed before, in addition to improved specificity, the major advantage of Grx1-roGFP2 is its rapid equilibration with the glutathione redox pair independently of endogenous Grx availability. Grx1-roGFP2 has been used to visualize changes in EGSH with minute resolution, as they occur during mitochondrial depolarization and receptor crosslinking (40). Rapid equilibration makes Grx1-roGFP2 responsive to oxidative events that are short-lived and weak, and it is these events that are easily missed by conventional roGFP2. As discussed before, no independent method for compartment-specific calibration is available. It is nevertheless clear that Grx1-roGFP2 detects nanomolar changes in [GSSG] against a backdrop of millimolar [GSH], in accordance with in vitro data and theoretical predictions. For example, the external application of a single H2O2 bolus in the low micromolar range (1–10 μM) leads to a reproducible intracellular probe response. Assuming that only a fraction of externally added H2O2 enters the cytosol (1) and that cytosolic H2O2 is not fully converted into GSSG (given peroxiredoxins and catalase), the sensor response almost certainly corresponds to the detection of nanomolar changes in [GSSG], possibly reaching down to the low nanomolar range.
f. Implications for redox signaling
Interestingly, the dynamic behavior of Grx1-roGFP2 inside living cells may have implications beyond its use as a biosensor. If it is assumed that natural Grx1 target proteins behave similarly to roGFP2, which seems reasonable, they should also be dynamically oxidized (S-glutathionylated) and reduced (de-glutathionylated) in response to physiological changes in EGSH (Fig. 15). Grx1-roGFP2 clearly demonstrates that glutathionylation can drive the formation of stable disulfide bonds, suggesting that Grx1 target proteins frequently convert S-glutathionylation into intra- or intermolecular disulfide bonds (if a second thiol is available nearby). Naturally, the extent of Grx1-catalyzed disulfide formation would depend on the respective dithiol/disulfide midpoint potential, which is certain to differ among target proteins. Although Grx1 is predominantly known for its reductive functions, protein S-glutathionylation is increasingly discussed as a possible pro-oxidative function of Grx1 (82). The dynamic behavior of Grx1-roGFP2 in living cells now clearly demonstrates that Grx1 catalyzes dynamically in both directions and that this happens readily within the physiological range of EGSH . Accordingly, a large set of thiol proteins may be redox-regulated by Grx1-mediated real-time equilibration with the 2GSH/GSSG redox couple. How-ever, Grx1-roGFP2 also demonstrates that co-localization between Grx1 and target protein is very important, and it may be that only those Grx1 target proteins in close proximity to endogenous Grx1 are efficiently coupled to EGSH . ASK1 and Procaspase-3 may be a case in point as these proteins seem to associate directly with Grx1 (96, 112).
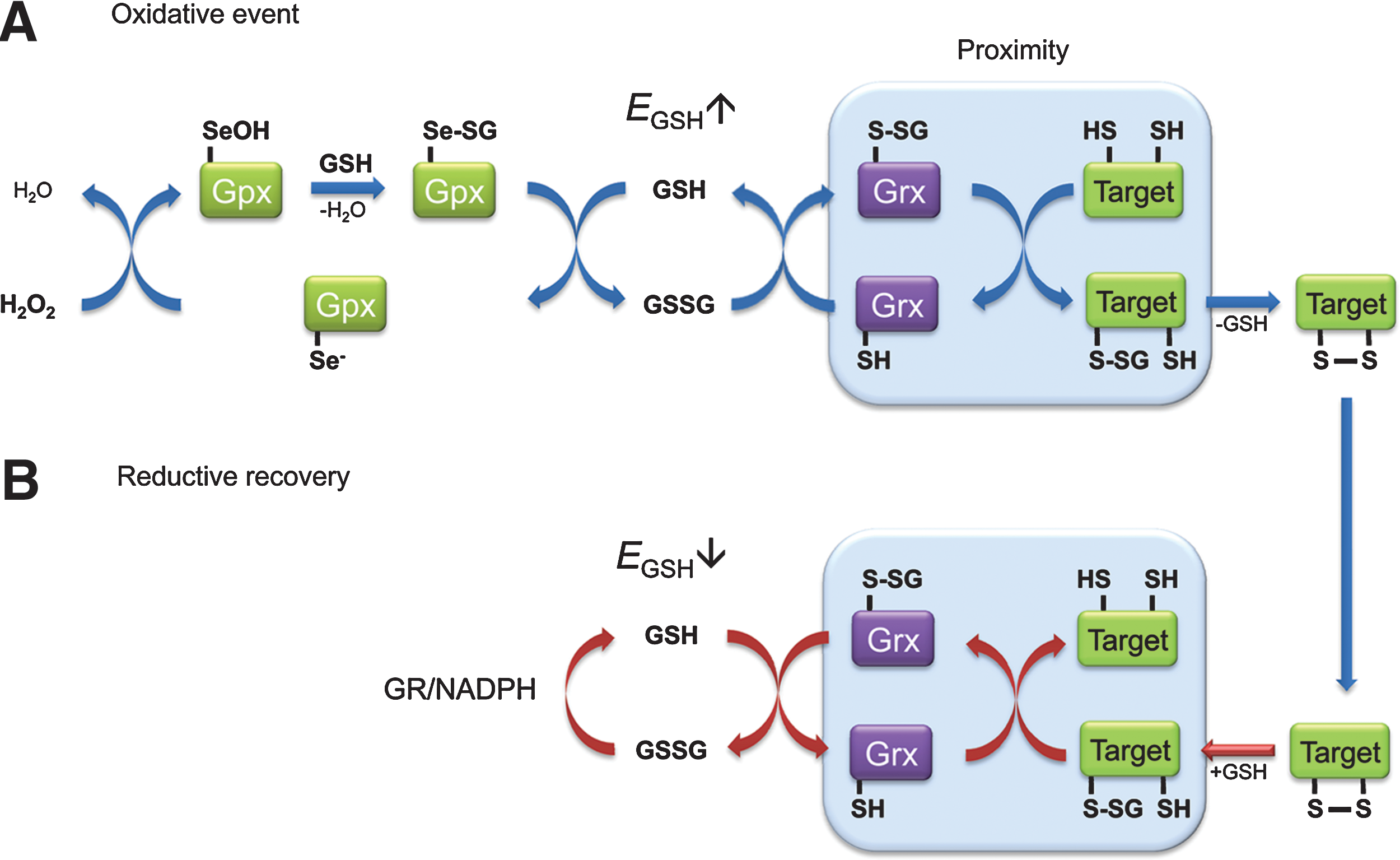
Grx-mediated protein oxidation may also help to understand the phenomenon of H2O2-based signaling. An important question is how H2O2 can oxidize redox-regulated proteins efficiently and specifically, despite competition by highly efficient and abundant scavenger systems. It has been suggested that regulatory protein thiol oxidation by H2O2 may sometimes, if not frequently, be an indirect process (130). The Grx1-roGFP2 probe potentially exemplifies such an indirect pathway (Fig. 15) in that H2O2 facilitates protein thiol oxidation through the GSSG intermediate. In other words, ultrasensitive H2O2-scavenging enzymes (e.g., glutathione peroxidases in mammalian cells or ascorbate peroxidase together with dehydroascorbate reductase in plant cells) may act as highly efficient initiators of oxidative signaling by generating transient bursts of GSSG. The resulting pro-oxidative shift in EGSH then triggers Grx-mediated protein oxidation in those oxidation-sensitive proteins that are in close physical association with appropriate Grxs, but not in others (Fig. 15A). Once EGSH normalizes by the action of GRs, Grx catalyzes the reduction of the same proteins (Fig. 15B). It may be speculated that Grx1-roGFP2 dynamically responds to endogenous H2O2 signals precisely because it mimics a natural pathway of regulatory protein thiol oxidation.
B. Coupling roGFPs to thioredoxins
1. The concept behind Trx1-roGFP2
The successful redox coupling of Grx1 and roGFP2 and the considerations regarding the increased local concentration and therefore probability of interaction (see Section III.A.2.a) suggested the possibility that a Trx-roGFP2 fusion protein may likewise serve as reversible probe for the Trx redox state. This possibility is conceivable for two reasons. First, Trx has been observed to become transiently oxidized during oxidative stress and redox signaling (45), thus suggesting that Trx is not kept in the reduced state at all times. Second, disulfide exchange between Trx and substrates is reversible in principle, and prokaryotic Trxs were previously shown to be able to act as oxidases (23, 113). Depending on the position of the equilibrium, Trx may thus oxidize roGFP by dithiol/disulfide exchange. In principle, this may allow visualization of transient Trx oxidation, potentially associated with oxidative signaling events. Assuming that specific and efficient equilibration between Trx1 and roGFP2 occurs in a fusion protein, the following Nernst relationship should apply:
The midpoint potential of the active site dithiol–disulfide pair of human Trx1 has been determined as −230 mV (128). The midpoint potential of roGFP2 is yet more negative (Fig. 16A), leading to the prediction that only a highly reduced Trx1 would keep roGFP2 in the reduced state. Any weak increase in Trx1 oxidation would translate into substantial roGFP2 oxidation. Specifically, a shift towards 2% Trx1 oxidation should entail 50% roGFP2 oxidation (Fig. 16B).
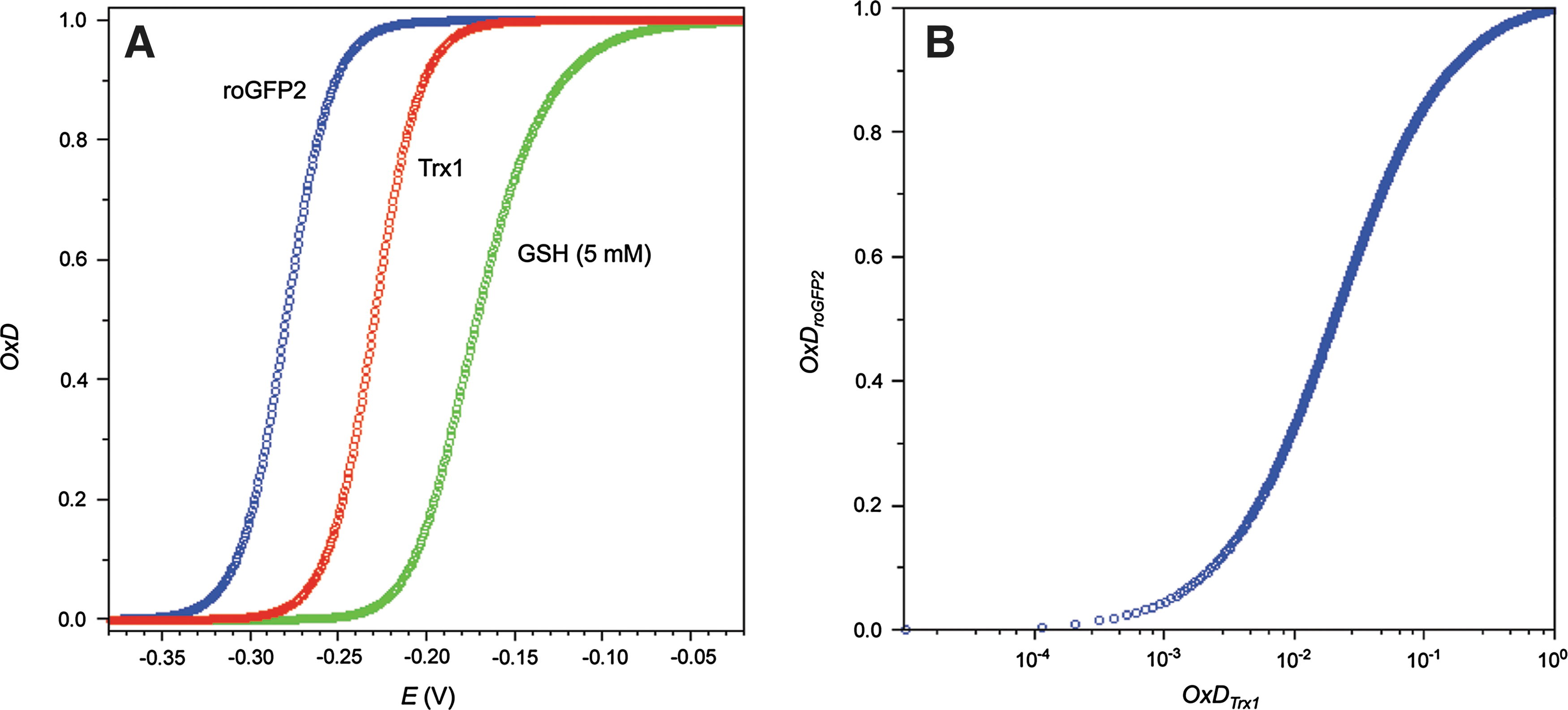
2. The geometrical limits of thiol–disulfide exchange
All these theoretical predictions assume effective equilibration by thiol–disulfide exchange. However, it turned out that human Trx1 does not interact with roGFP2. Neither does fully reduced Trx (coupled to the complete Trx system) target oxidized roGFP, nor does the reverse reaction take place (40). Likewise, the same observation applies to rxYFP and Trx (93). Why does roGFP2 readily interact with Grx, but not with Trx? It is not well understood by which criteria Trx1 targets particular disulfide bonds (while ignoring others, even if well exposed). The stereochemical conformation of the roGFP2 disulfide bridge is rather unusual. Bonds of this type are known as ‘right-handed staples’ or ‘cross-stranded’ disulfides (55, 105, 132). Interestingly, this atypical geometry is shared by several disulfides known to be readily reduced by Trx [e.g., a disulfide in the CD4 cell surface receptor (73) and another in BASI (barley α-amylase/subtilisin inhibitor)] (69). On these grounds, the disulfide on roGFP2 would appear to be a likely target for Trx. It is well exposed on the barrel surface and the ‘staple’ geometry is compatible with Trx-mediated reduction in other proteins. The most likely explanation for the resistance of the roGFP2 disulfide bond against Trx is a structural constraint imposed by the reaction mechanism. In general, thiol–disulfide exchange reactions proceed by bimolecular nucleophilic substitution (SN2). This implies that the three participating sulfur atoms (nucleophile, electrophile, and leaving group) have to be arranged in a straight line (Fig. 17). It is somewhat surprising that the implications of SN2 geometry for protein disulfide exchange interactions are rarely discussed in the literature, with only few exceptions (28, 52, 129).

The SN2 constraint likely explains why right-handed ‘staple’ disulfides are preferentially targeted by Trx in certain other proteins, but not in roGFP2 (or rxYFP). When a ‘staple’ disulfide bond is positioned on the edge of a β-sheet (as in BASI), the interface encountering the active site surface of Trx is relatively flat. The point in space that needs to be reached by the nucleophilic sulfur of C32 in order to mount an in-line attack is unobstructed by side-chains (Fig. 18). More specifically, it appears to be essential that the two residues flanking the target cysteine do not project their side chains into the direction of Trx. In fact, the β-sheet configuration effectively prevents steric interference of this kind. Any side chain (even alanine) projecting towards Trx would be predicted to get into steric conflict with W31 and P34 on Trx1, two highly conserved residues flanking the nucleophilic C32.

These considerations may also explain why the situation is fundamentally different in the case of the roGFP2-Trx interaction: The roGFP2 disulfide is not located at the edge of the β-sheet and the sulfur–sulfur axis of the roGFP disulfide bond lies flat and parallel to the barrel surface. Therefore, most likely, the nucleophilic sulfur of Trx, located in a narrow groove between W31, P34, and other residues, is sterically prevented from positioning itself for the SN2 reaction (Figs. 19 and 20A).
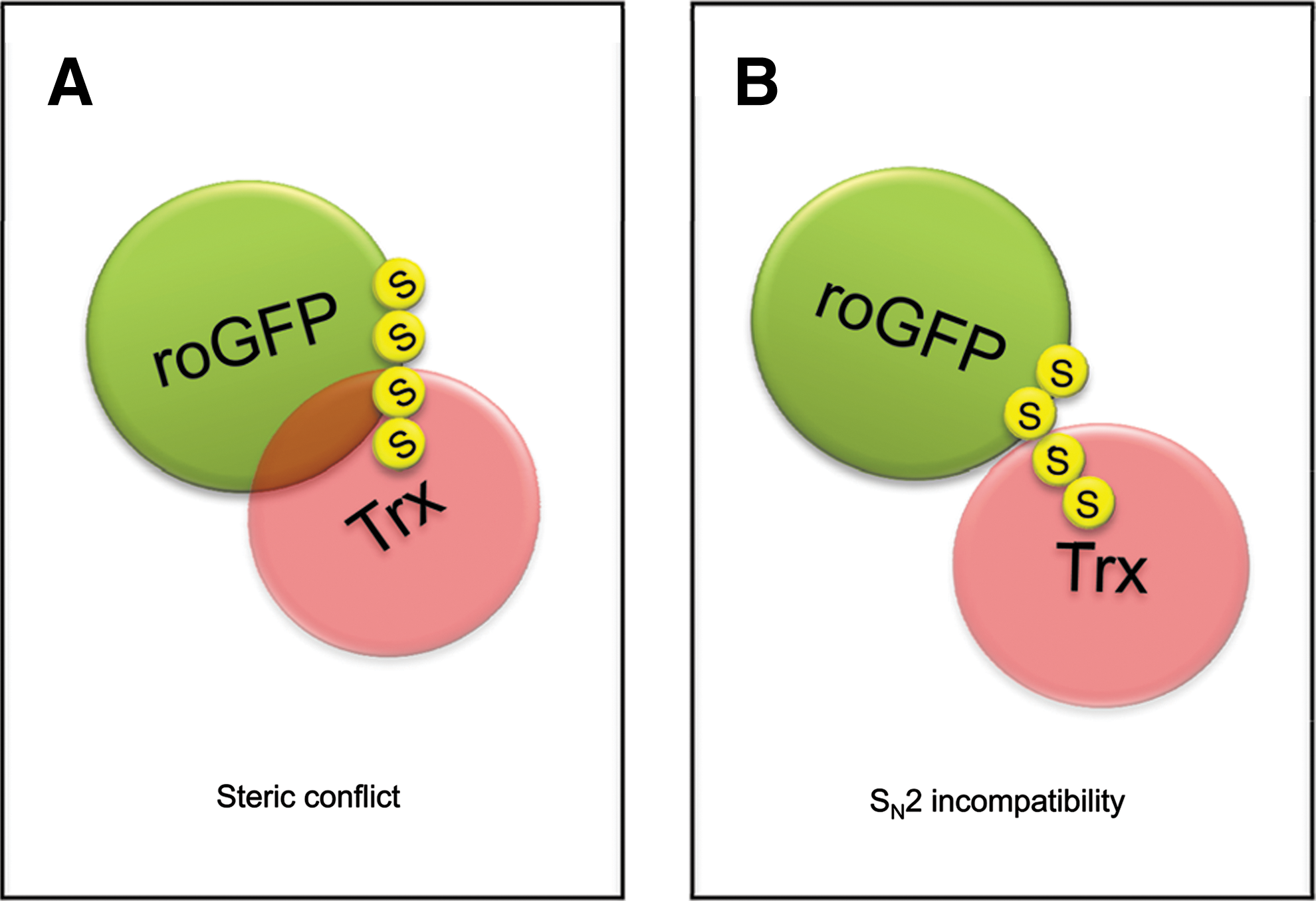
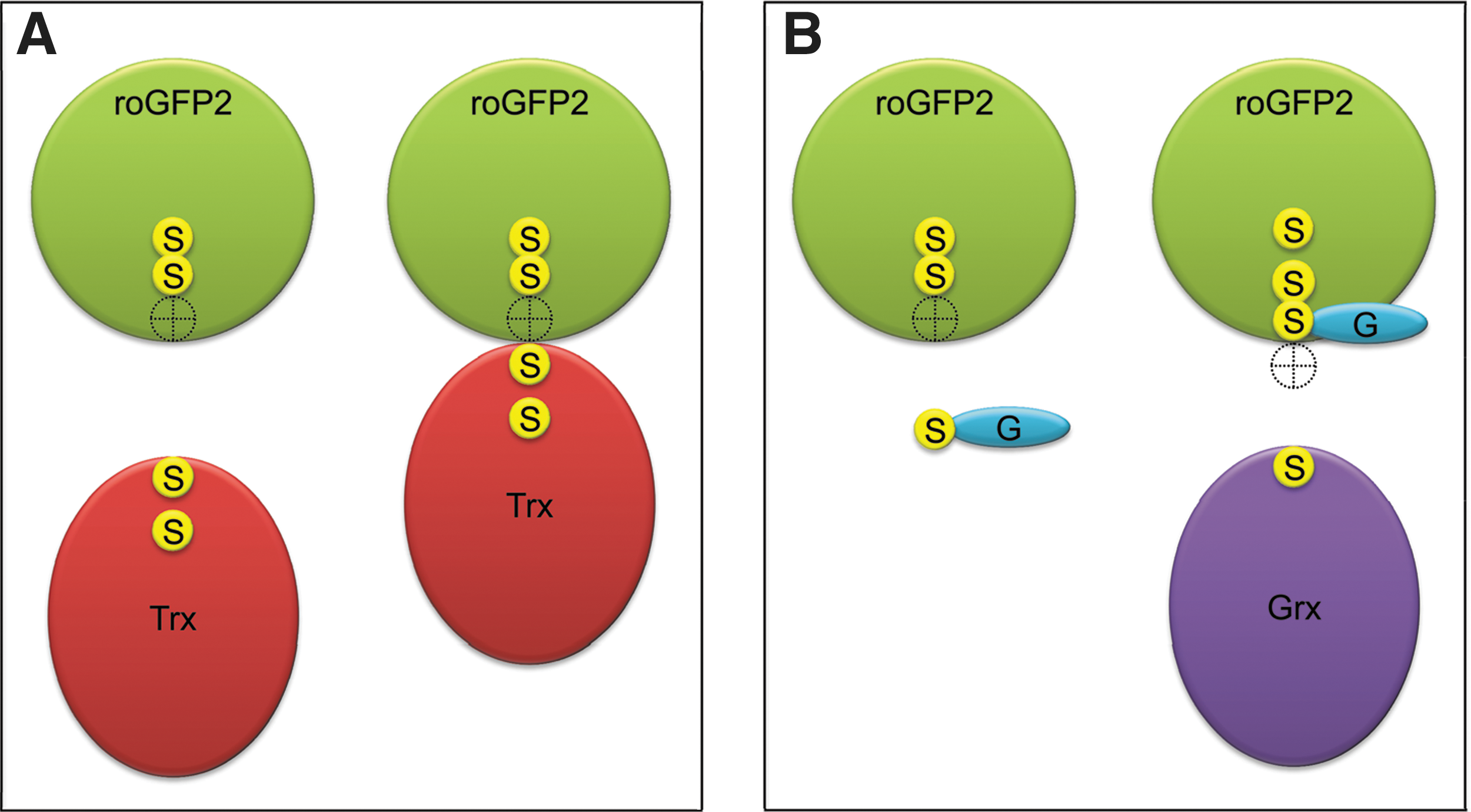
The same considerations may also explain why the roGFP2 disulfide bond is a very good substrate for the Grx/GSH system (Fig. 20B). Most likely, the small glutathione molecule can easily reach the position required for in line attack on the roGFP2 disulfide bond. The resulting roGFP2–S-SG mixed disulfide bond is much more accessible, because it is not as close to the barrel surface. In addition, the sulfur-containing bonds of roGFP2–S-SG are rotatable, thus offering many alternative positions for subsequent nucleophilic attack. This may explain why there is no steric problem for Grx to attack roGFP2–S-SG. It may be asked if Trx would also be able to attack and reduce the roGFP2-S-SG conjugate with its more accessible disulfide bond. In vitro experiments indicate that this is not the case (Gutscher et al., unpublished data), suggesting that either roGFP–S-SG is still not accessible enough for Trx, or that Trx somehow discriminates against protein–S-SG mixed disulfides as substrates.
In conclusion, it seems unlikely that a working Trx-roGFP redox relay can be created. On the positive side, the fact that Trx refuses to interact with the roGFP2 intramolecular disulfide bond (and also with the roGFP2-S-SG disulfide bond) strengthens the concept that roGFP2 (and especially Grx1-roGFP2) really are highly specific probes for 2GSH/GSSG. Of course, it remains possible that certain Trxs from other organisms or organelles, possibly those with a shallower active site groove (i.e., with a more exposed nucleophile), can engage in thiol–disulfide exchange with roGFPs. However, even if a disulfide-exchanging Trx/roGFP combination can be found, the usefulness of the corresponding fusion protein to visualize the native endogenous Trx redox state would be quite uncertain. The bulky roGFP domain may sterically hinder the interaction of Trx with its native redox partners TrxR and the peroxiredoxins. In that case the probe may not properly reflect the native redox state of Trx. Moreover, the interpretation of the probe response may be further complicated as slow interactions with the endogenous GSH/Grx system may take place in parallel. It seems that fresh ideas are needed to finally develop a genetically encoded redox probe specific to the Trx redox state (see also Section V.A.2).
C. Coupling roGFPs to peroxidases
1. The concept behind roGFP2-Orp1
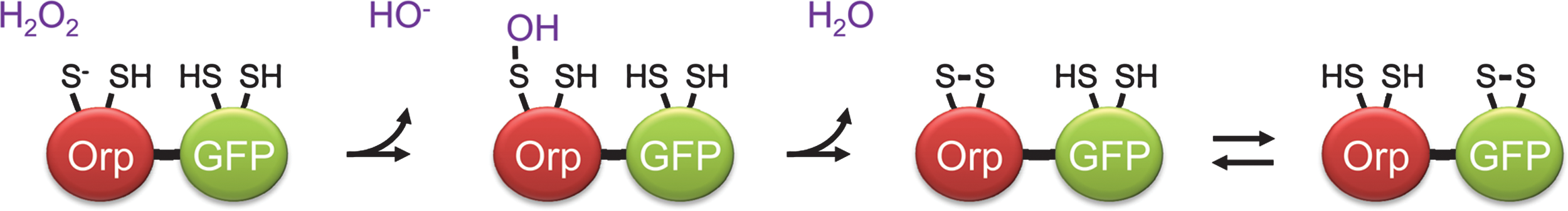
Is it possible to convert roGFPs into specific probes for H2O2 or other peroxides? Peroxiredoxins and glutathione peroxidases are highly efficient and selective H2O2 receptors. Intriguingly, some peroxidases have been found to transmit oxidation to other proteins. The best studied example is the regulatory oxidation of the transcription factor Yap1 by the peroxidase Orp1 (Gpx3) in yeast (24). This finding led to the question if Orp1 would accept roGFP in place of its natural target protein, Yap1. To test the idea, roGFP2 was fused to Orp1 to create a redox relay in which roGFP2 replaces Yap1 and the linker enforces proximity (41) (Fig. 21). In fact, Orp1 was found to form a highly efficient redox relay with roGFP2. In vitro, Orp1 mediated near-stoichiometric oxidation of roGFP2 by H2O2, converting almost every H2O2 molecule into a roGFP2 disulfide bridge. Moreover, the Orp1-roGFP2 redox couple effectively converts physiological H2O2 signals into measurable fluorescent signals in living cells (41).

The intracellular mechanism of the roGFP2-Orp1 redox relay was found to be based on dithiol-disulfide exchange between the two domains (Fig. 22). Oxidation of Orp1 by H2O2 first generates an intramolecular disulfide bond between C36 and C82. In a second step, the disulfide bond is transferred to roGFP2 by thiol–disulfide exchange (41). The Orp1 C36/C82 dithiol/disulfide midpoint potential was measured as −255 (± 8) mV at pH 6.0 (72), suggesting a value of −314 mV at pH 7.0 by extrapolation (based on the −59 mV per pH unit dependence). Considering thiol–disulfide exchange between Orp1 and roGFP2, Orp1 should be rather inefficient in oxidizing roGFP2 (50% sensor oxidation would require approx. 90% Orp1 oxidation). However, in situations where Orp1 encounters H2O2 frequently, it will be kept at 100% oxidation by H2O2 and thus transfer oxidative equivalents to roGFP2 very efficiently. Thus, Orp1 can be seen as a redox catalyst mediating electron flow between the H2O2/H2O pair (approx. + 1350 mV at pH 7) and the roGFP2ox/roGFP2red redox pair.
The roGFP2-Orp1 relay demonstrates dynamic behavior in living cells. Thus, it is important to ask how the probe is reduced inside cells. In principle, there are two possibilities. Similar to conventional roGFP2, the roGFP2 disulfide within the fusion protein may be reduced by GSH. This would depend on endogenous Grx and be rather slow. Based on the thermodynamic relationship between roGFP2 and Orp1, it appears more likely that roGFP2 passes the disulfide bond back to Orp1. This leads to the question of how Orp1 is reduced inside living cells. In yeast, Trx is the natural reductant of Orp1 and this may also be the case in mammalian cells. The −314 mV midpoint potential of Orp1 is compatible with its reduction by human Trx1
2. Implications for H2O2 signaling
Similar to the potential role of Grx1 in regulatory protein oxidation (Section III.A.2.f.), the fact that roGFP2-Orp1 converts physiological concentrations of H2O2 into protein disulfide bonds may as well have implications beyond its use as a biosensor, namely for the mechanism of H2O2 signaling. Apparently, Orp1 is not mechanistically restricted to use Yap1 as the recipient of oxidative equivalents and thus harbors a more general ability to act as a thiol oxidase for other proteins (41). In turn, this may suggest that certain other peroxidases, peroxiredoxins, and/or members of the glutathione peroxidase family have a similar intrinsic capacity to promote the oxidation of other proteins. In fact, the human selenocysteine-based glutathione peroxidase Gpx4 was already implicated in the disulfide cross-linking of protamines and is also capable of mediating the oxidation of roGFP2 in living cells (41). Close pre-positioning between peroxidases and target proteins likely is critical for the transfer of oxidizing equivalents. Only protein thiols close to the peroxidase active site are likely to intercept the peroxidatic cycle. If roGFP2-Orp1 is indicative of a more general principle of H2O2 signaling, a major task for the future is to identify and study natural peroxidase-target relationships in higher eukaryotic cells.
IV. Other Genetically Encoded Redox Probes
All genetically encoded redox probes discussed so far are based on redox-sensitive FPs. However, redox probes can also be developed from non-redox-sensitive FPs. In principle, any conformational change associated with protein redox state may be exploited to change the fluorescent properties of a FP which is not redox-active by itself. One possible approach is the insertion of cpYFP into a redox-active protein domain, in a way comparable to previously established non-redox probes (e.g.,
A. HyPer: A cpYFP-based redox probe
1. The concept behind HyPer
HyPer has been designed to serve as a specific indicator for intracellular H2O2 (6). It is built on the H2O2-sensing regulatory domain (RD) of the prokaryotic transcription factor OxyR. The active site cysteine of OxyR-RD (C199) readily reacts with H2O2 to form a cysteine sulfenic acid (S-OH) which subsequently condenses with a second cysteine in the same domain (C208) to form an intramolecular disulfide bridge. In the reduced state, C199 and C208 are separated by 17 Å and a substantial conformational change, predominantly affecting peptide segment 205-222, takes place upon oxidation (20). To create HyPer, Belousov and colleagues inserted cpYFP between residues 205 and 206 of OxyR-RD. The cpYFP moiety was further optimized for folding and chromophore maturation, and the Y203F mutation was introduced to visualize the protonated form of the cpYFP chromophore. The resulting cpYFP–OxyR-RD chimera, named HyPer, displays a single emission maximum at 516 nm and two excitation maxima at 420 (protonated chromophore) and 500 nm (deprotonated chromophore). Importantly, the conformational influence of disulfide bond formation leads to a ratiometric shift with the 420 nm peak decreasing and the 500 nm peak increasing. HyPer was shown to detect nanomolar H2O2 in vitro and, when expressed in cells, to respond to micromolar H2O2 added externally. HyPer also allowed detection of oxidative changes induced by growth factors (6, 70). The maximal ratiometric dynamic range in vitro was reported as 3- to 4-fold. However, ratiometric changes in live cell imaging after physiological stimuli were more moderate (i.e., a 15% ratio increase after NGF stimulation), indicating that accurate measurements of physiologically relevant events can be technically demanding.
2. How specific is HyPer as a probe for H2O2?
How accurately does the HyPer signal reflect dynamic variations in endogenous H2O2 levels? On the one hand, there is little doubt that the OxyR regulatory domain is highly selective in its reactivity towards H2O2. On the other hand, the cpYFP moiety of HyPer does not sense H2O2 directly. Instead, the fluorescence response reflects the thiol redox state of OxyR-RD. The thiol redox state of OxyR-RD is the result of the dynamic interplay between disulfide formation and disulfide reduction. This leads to the question of how the HyPer disulfide bond is reduced within eukaryotic cells. In E. coli the disulfide form of OxyR is reduced by the glutathione system, specifically by GSH under catalysis of Grx1 (136). It is likely that reduction of HyPer in eukaryotic cells also depends on the glutathione system. Therefore, in principle, events curtailing the disulfide reducing function of the glutathione system (e.g., limiting NADPH levels or loss of GSH) may well shift the balance towards increased HyPer oxidation, even if H2O2 levels remain the same.
Regarding the role of the glutathione system, there is an additional concern: If Grx mediates reduction of the HyPer disulfide it may also catalyze the reverse reaction, namely disulfide formation, based on the Nernst relationship between the glutathione redox couple and the dithiol/disulfide couple of HyPer. Of note, in vitro, when purified OxyR is incubated with a combination of GSSG and Grx1 (rather than GSSG alone), it is, on a molar basis, as efficiently oxidized as with H2O2 (136). Is it conceivable that Grx-catalyzed oxidation of HyPer by GSSG occurs in intact cells? It is unlikely to be a major concern, at least for the cytosolic compartment. First, oxidation of OxyR-RD by GSSG becomes thermodynamically favorable only at relatively high GSSG concentrations. The midpoint potential of the OxyR dithiol/disulfide couple has been reported as −185 mV (136). Thus, to oxidize HyPer (OxyR) by 10%, an EGSH of −214 mV is required (at GSHtotal = 5 mM this would correspond to [GSSG] = 165 μM). As discussed before, measurements with rxYFP, roGFPs, and Grx1-roGFP2 strongly indicate that such a high EGSH is unlikely to occur in the cytosol of living cells under physiological conditions. Second, it is not clear if intracellular Grx levels would be sufficient to support equilibration. Most likely, GSSG-mediated oxidation of HyPer is limited by the local availability of Grx and therefore a slow process. It appears safe to assume that under most circumstances H2O2 is the predominant oxidant for HyPer. It should be noted, however, that the midpoint potential of the 2GSH/GSSG redox couple depends on GSHtotal. The lower the concentration of the total GSH pool, the more favored is HyPer (OxyR) oxidation by GSSG.
With regard to reversibility, HyPer differs fundamentally from Grx1-roGFP2. Grx1-roGFP2 measures the redox potential of a single redox pair by equilibrating with the same. In contrast, HyPer (i.e., OxyR-RD) communicates with two redox pairs simultaneously: 2H2O/H2O2 and 2GSH/GSSG, thus displaying a fundamental asymmetry between the oxidation and reduction reactions. Obviously, roGFP2-Orp1 is subject to the same mechanistic asymmetry as HyPer. Thus, it is probably more appropriate to consider HyPer a probe for the balance between H2O2-mediated disulfide formation and glutathione-mediated disulfide reduction and, correspondingly, roGFP2-Orp1 a probe for the balance between H2O2-mediated disulfide formation and (probably Trx-mediated) disulfide reduction. This kind of probe behavior should not be considered a shortcoming, as measuring the thiol oxidation-reduction balance is highly relevant for the investigation of cellular redox signaling.
Finally, another concern that deserves attention is the influence of pH on HyPer fluorescence. From the pH titration curve (6) it appears that the 500/420 excitation ratio is significantly shifted by pH and that a shift of 0.2 pH units can be sufficient to change the ratio as much as full reduction or oxidation. Thus, for practical purposes it seems important to monitor pH changes in the same experiment.
3. Applications of HyPer
HyPer measurements have been successfully applied to study the wound response of zebra fish larvae (90). Cytoplasmic sensor expression was achieved in whole animals by injecting HyPer mRNA into embryos. Local injury of the tail fin produced a localized, graded, and transient HyPer signal. The interpretation of the observed HyPer response as a H2O2 signal was supported by the fact that pH changes could not be detected. A fluorogenic dye also indicated oxidant formation under the same conditions, and the signal was dependent on the expression of the NADPH oxidase Duox. Thus, it appears very likely that the observed HyPer response was directly caused by H2O2.
B. FRET-based redox probes
Similar to other FRET-based probes (36), conformational changes associated with protein redox states can be exploited to influence the distance between a FRET donor-acceptor pair. Only very few publications investigated the direction of genetically encoded FRET-based redox probes. In general, the FRET strategy requires a redox-active peptide or protein domain. A rather simple concept is to use a peptide linker harboring two cysteines to form an intramolecular disulfide bond under oxidative conditions. In a recent study (61), artificial peptide linkers containing cysteine pairs were placed between a FRET pair of enhanced cyan and yellow fluorescent proteins. However, the maximal changes in FRET efficiency were modest and the detection of physiological redox changes remains to be demonstrated. Kinetics and sensitivity may be limiting factors for the peptide linker approach. It is also not clear which cellular redox couples or enzymes would actually interact with peptide linker cysteines, raising questions about specificity. At the present stage, FRET redox probes with short peptide linkers lag behind the other strategies described before. A more sophisticated FRET redox sensor is HSP-FRET (100) in which the redox-sensitive domain of bacterial hsp33 is placed between ECFP and YFP. Expressed in cardiomyocytes, HSP-FRET detected oxidative changes associated with hypoxia. Future FRET approaches based on protein domains with defined redox behavior and specificity are likely to contribute to the field.
V. Future Directions and Challenges
A. Further development of redox biosensor methodology
The future understanding of interlinked processes in the context of whole organisms will depend on our ability to acquire quantitative data with high resolution in time and space. Generally, quantitative live cell imaging with genetically encoded probes has the potential of fulfilling these demands, albeit only for one or a few physiological parameters at a time. While current redox-sensitive FPs have already shown their potential to deliver quantitative and dynamic information, there is still much room for further improvements and extensions. We here address different options for further tuning of current sensors and for the development of novel probes with altered properties and specificities.
1. Improvements and new modalities
a. Redox probes with different colors
Current ratiometric redox probes based on roGFPs all emit green light of the same wavelength. Thus, comparison of steady state EGSH
in different compartments and investigation of interorganellar redox communication often requires separate measurements for each compartment. Simultaneous measurements in two or more subcellular compartments would be more convenient with differentially colored redox probes. A first step in this direction may be the use of rxYFP in combination with roGFPs. It should be possible to employ rxYFP as a bona fide ratiometric probe by fusing it to a redox-invariant FP. Previously, the Cl--sensitivity of YFP has been exploited to generate the Cl- sensor
Following the initial discovery of GFP (111) and dsRed (74), more than 150 distinct fluorescent or colored proteins have been identified from a whole array of different species. Together with the large number of spectral varieties of GFP generated through mutagenesis (135), it can be envisaged that combinatorial approaches may allow for virtually infinite possibilities for further modifications of the current probes. The problem, however, is that it is still extremely difficult to rationally predict sensor properties and design a probe accordingly. In fact, engineering of redox-sensitive disulfides into YFP and GFP are two of only very few examples of rational probe design.
b. Redox probes enabling higher temporal resolution
Grx1-roGFP2 is already highly responsive to changes in EGSH and equilibrates on a second-to-minute time scale. Nevertheless, it may be possible to further shorten the response time of chimeric roGFP-based probes, potentially allowing the detection of highly transient deflections on even shorter time scales. Further kinetic improvements of roGFP-based probes can be envisaged through a combination of approaches previously used to establish second and third generation redox probes (Fig. 4). It is likely that oxidation of roGFP2 within a fusion protein is kinetically limited by the high pK a (> 9) of its cysteines. Thus, the engineering of chimeric roGFP2 to increase cysteine reactivity may result in even faster probes. The pK a of thiols can be tuned by amino acid residues in close proximity (38). Substitution of residues close to one or both cysteines by arginine or lysine was already shown to increase the reactivity of nonchimeric rxYFP (47) and roGFP1 (13). In addition, it is also conceivable to tune the catalytic properties of the redox enzyme employed within chimeric fusion proteins. For example, a single cysteine variant of yeast Grx1p (with a CPYS active site motif ) was more efficient in catalyzing the oxidation of rxYFP than Grx1p with the native CPYC motif (7). Also, a side-by-side comparison of Grx enzymes may identify paralogues or orthologues capable of catalyzing the oxidation and reduction of roGFPs even more efficiently. Finally, the catalytic efficiency of fusion proteins like Grx1-roGFP2 and roGFP2-Orp1 may also be affected by the length of the linker between the two protein domains. The influence of linker length on redox relay efficiency has not been systematically investigated. Obviously, a linker too short would prevent productive inter-domain interactions. On the other side, a very long linker would effectively lower the local Grx concentration experienced by roGFP and thus may also diminish the frequency of productive collisions. As the C-terminus of roGFP emerges from the protein on the same face as the catalytic cysteines, it is likely that the positioning of roGFP2 at the N-terminus of a chimeric construct allows for more flexible interactions between the two domains. Correspondingly, it may be predicted that a linker on the C-terminus of roGFP/rxYFP can be shorter than one at the N-terminus. In fact, rxYFP-Grx1p was created with a linker of just eight amino acids (7), which may approximate the shortest possible linker to allow productive domain interactions.
c. Redox probes with different midpoint potentials
The creation of probes with modified midpoint potential and measuring range is another important topic. roGFPs have been targeted to the ER lumen in human (2, 106), plant (81), and yeast cells (79). In all species, the OxDroGFP in this compartment was found to be beyond the useful dynamic range of the probe (i.e., the redox potential of roGFP2 in the ER was higher than −240 mV). A range of novel roGFP versions with redox potentials of up to −229 mV have been generated (67), but it remains to be seen if these midpoint potentials are sufficiently high to dynamically resolve the actual EGSH in the ER lumen. Although it is possible that roGFPs in the ER reliably equilibrate with 2GSH/GSSG, it cannot be excluded at this stage that roGFPs also interact with Ero1 and members of the PDI family that catalyze the formation of protein disulfide bonds in the ER (15,109). To ensure preferential equilibration of roGFP with 2GSH/GSSG, it may thus be advisable to use Grx fusion proteins for the ER as well.
d. Redox probes with improved targeting properties
RoGFPs can be specifically targeted to mitochondria in HeLa (40, 49) and plant cells (87, 107) by using appropriate N-terminal targeting sequences. Similarly, Grx1–roGFP2 was successfully targeted to mitochondria in HeLa cells (40). Surprisingly, however, the same Grx1–roGFP2 fusion protein was not imported into mitochondria of Drosophila and different plant species despite the use of established species-specific mitochondrial targeting sequences (own unpublished observations). These problems indicated that human Grx1 may prevent correct mitochondrial targeting of the chimeric probe when expressed in heterologous systems. This restriction may be overcome in two different ways, either by using species-specific Grxs for the fusion protein, or alternatively, by inverting the order of domains and fusing Grx1 C-terminally of roGFP2. Indeed, recent results demonstrate that an inverted probe with human Grx1 fused to the C-terminus of roGFP2 (roGFP2-Grx1) with the 30 aa spacer (GGSGG)6 is successfully targeted to mitochondria in transgenic plants (Bausewein and Meyer, unpublished data) and flies (Albrecht and Dick, unpublished data).
2. Novel probes for key redox couples
Redox-sensitive FPs provide a probe scaffold that is amenable to improvement, reconstruction, and expansion. Numerous opportunities for the creation of novel probes can be envisaged. In the following paragraphs we give just a few examples for the possible deployment of redox-sensitive FPs in the design of novel probes.
First of all, peroxidase-roGFP redox relays clearly offer opportunities for further probe development. The finding that the thiol-based peroxidase Orp1 efficiently mediates electron flow between H2O2 and roGFP2 (41) suggests that the same principle may also be applicable to other peroxidases. In fact, the selenocysteine-based glutathione peroxidase Gpx4 (also known as phospholipid hydroperoxidase, PHGPx) also mediates roGFP oxidation in a live cell context (41). It is now conceivable that a number of roGFP-peroxidase probes can be created to visualize the intracellular oxidative response of individual peroxidases. In this regard, a potentially attractive goal may be the design of a genetically encoded probe for lipid peroxidation.
Another important goal for the future is the development of the hitherto unavailable Trx-specific redox biosensor. As discussed before, existing roGFPs and rxYFP do not interact with Trxs (see Section III.B). Thus, a different strategy is needed to generate a Trx-sensitive probe. It may be possible to introduce novel disulfide switches into GFP, designed from the outset to be accessible to the Trx active site. It may also be possible to create a chimeric redox relay between a Trx target protein and a roGFP. Yet another possibility may be to insert a Trx-sensitive protein domain directly into the barrel of a redox-insensitive FP. A precedent for the latter strategy is the Ca2 +-sensor
A probe for the redox pair AA/DHA is also high on the wish list. After disproportionation of two molecules monodehydroascorbate (MDHA) to AA and DHA the reduction of DHA to AA is catalyzed by dehydroascorbate reductase (DHAR) and leads to a mixed glutathione-enzyme disulfide (26, 110). Normally this mixed disulfide would be reduced by GSH to form GSSG, but in analogy to the catalytic mechanism of Grx1–roGFP2 it may also be transferable to roGFP2 where it may trigger disulfide formation and a corresponding change in fluorescence. It remains to be seen if a specific probe for AA/DHA can be build upon these or similar principles.
Another highly desired probe is one for the real-time observation of NO•. NO• can undergo nitrosylation reactions with protein and low molecular weight thiols. It is conceivable that a trans-nitrosylation reaction targeted to roGFP could trigger formation of the roGFP disulfide bridge. A fusion between roGFP and a primary NO• acceptor protein may thus allow NO• imaging. Recently, proteomic profiling of S-nitrosylation identified proteins with unexpected stability of the nitrosothiol group (95). The authors suggest that stable S-nitrosylation reflects a protein conformational change that shields the nitrosothiol group. Whether such NO•-dependent conformational changes can be exploited for the generation of future NO• probes remains to be tested.
3. Redox biosensor transgenic model organisms
Arguably, one of the ultimate methodological goals of redox biology is the ability to study clearly defined redox processes as they occur in the intact physiological context of the living organism. Genetically encoded redox probes now offer an opportunity to make progress into this direction. Much initial work on the characterization and application of roGFP-based probes has been done in transgenic Arabidopsis plants (57, 71, 81, 107). For the near future, it can be expected that the yeast S. cerevisiae will also play a major role in this context. Although rxYFP has been initially characterized in yeast (92, 93), the ratiometric behavior of roGFP provides a principle advantage over nonratiometric rxYFP and future work in yeast will likely build on roGFP variants. For example, yeast cells expressing roGFP-based probes may be genetically screened for deviations from steady state redox state and thus help identifying novel regulators of redox homeostasis. Similarly, a variety of genetically tractable model organisms expressing roGFP-based probes or HyPer are currently being created and studied to address diverse biological questions. To our knowledge, the list of transgenic species currently generated in various laboratories includes mice, zebra fish, C. elegans, D. melanogaster, barley, tobacco, trypanosomes, Chlamydomonas rheinhardtii, and Synechocystis. It remains to be seen what can be achieved with these transgenic models. In this regard, the expression and application of HyPer in zebra fish larvae is a promising first step (90). To unlock the potential of redox imaging in non-transparent organisms, various technical challenges need to be overcome. For example, the potential use of roGFP-based probes in two-photon imaging will have to be investigated. Another potential direction of technical investigation is the in situ chemical conservation of the biosensor redox state and its analysis in tissue sections. An attractive long-term perspective for redox biosensor transgenic mouse models is the comparison between healthy and diseased states and the testing of drugs for their actual in vivo influence on defined redox states in defined target tissues.
B. Applications
Independent of future probe developments, many questions in basic research can be addressed with currently available redox-sensitive FPs. Given the selective equilibration of Grx1–roGFP2 with 2GSH/GSSG, this is especially true with regard to the investigation of glutathione homeostasis and glutathione-dependent transmission of redox signals (80). In addition to applications described in Sections II.B.6 and III.A.2.e, we here exemplarily highlight another application of redox-sensitive FPs, namely to dissect cellular glutathione compartmentalization and interorganellar transport (Section V.B.1). We will then briefly address future probe applications in high throughput screening (Section V.B.2).
1. Exemplary applications in cell biology: Glutathione compartmentalization and transport
Glutathione is required in most subcellular compartments of eukaryotic cells. Despite the ubiquitous need for GSH, its biosynthesis is restricted to either the cytosol in mammalian cells or the plastids and the cytosol in plant cells, which highlights the need for membrane transport of GSH into other organelles (Fig. 23). In Arabidopsis, the rate-limiting enzyme GSH1 is exclusively targeted to the plastids while the second enzyme, glutathione synthase (GSH2), is targeted to both plastids and cytosol. This subcellular distribution of the two GSH biosynthetic enzymes implies the need for export of γ-glutamylcysteine (γ-EC) from plastids to supply cytosolic GSH2 with its substrate (97, 123) (Fig. 23A). First candidate proteins for mediating this transport of γ-EC and/or GSH from plastids to the cytosol were identified recently in Arabidopsis. Based on high sequence homology to the chloroquine-resistance transporter from Plasmodium falciparum, PfCRT, these proteins were named CRT-like transporters (CLTs) (75) (Fig. 23A). Lack of all three CLT isoforms, CLT1, CLT2, and CLT3, caused a slight decrease in total GSH content in shoot tissue, but a more pronounced effect in subcellular compartmentation of GSH was resolved with roGFP2 in situ. While plastid-targeted roGFP2 in both wild-type and the triple knockout mutant was almost fully reduced, roGFP2 expressed in the cytosol showed a distinct oxidation in the triple mutant. In contrast to a cytosolic EGSH of about −315 mV in wild-type plants, the cytosolic EGSH in the triple mutant was about 20 mV less reducing. This result is similar to the change in EGSH observed for the partially GSH-deficient cad2 mutant (Fig. 5). In general, targeting of roGFP to organelles like mitochondria and peroxisomes may provide an excellent opportunity to test candidate proteins for their ability to transport GSH in situ and to better understand the dynamics of glutathione compartmentation within eukaryotic cells.
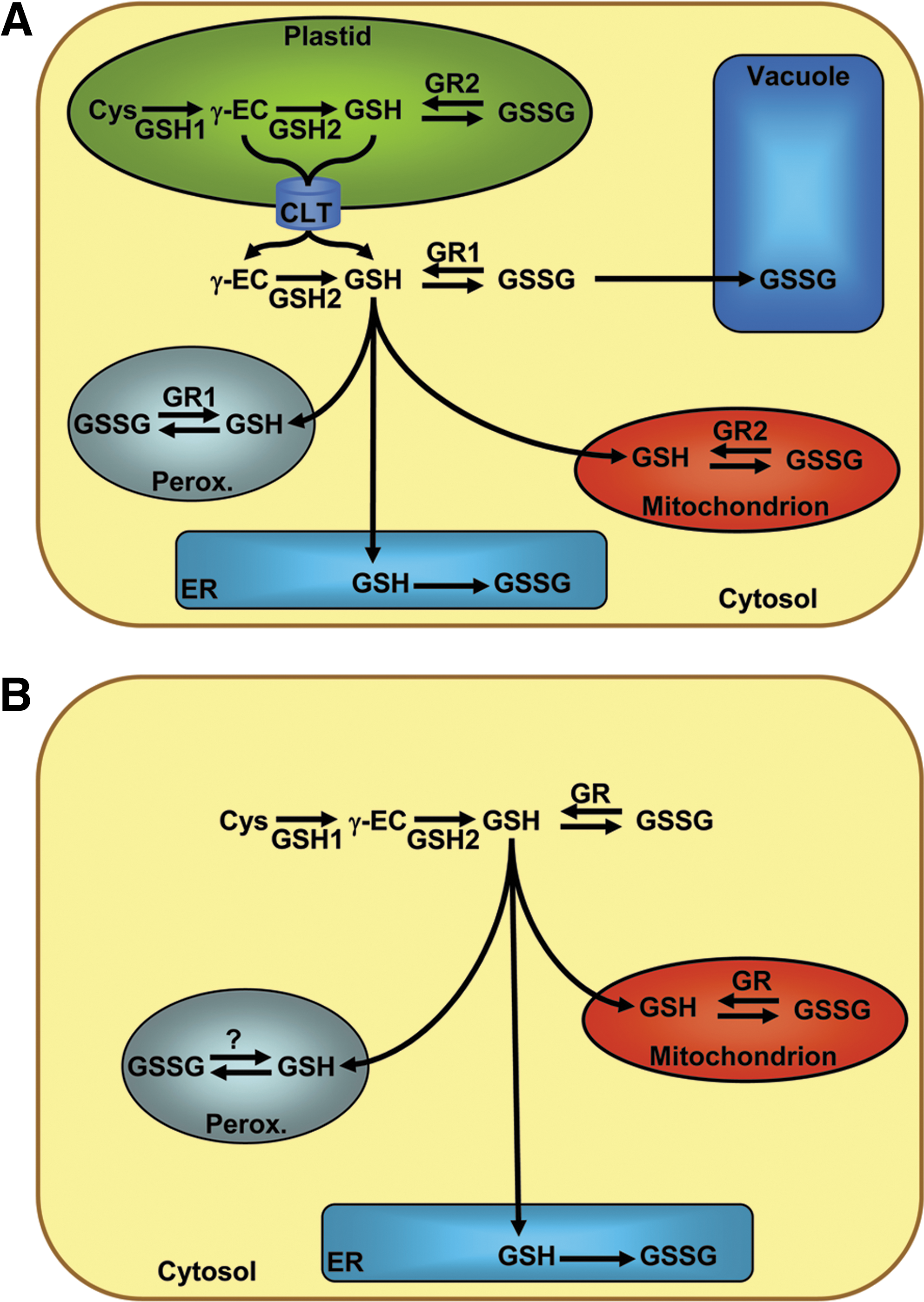
2. High throughput screening
Biosensors on the basis of fluorescent proteins have a huge potential for high throughput screening (HTS) to identify novel drugs affecting specific physiological processes. The use of genetically encoded probes avoids dye loading and washing steps and leads to more robust assays. An example of successful HTS based on GFP-derived sensors is the use of the Cl--sensitive YFP-H148Q to screen for new classes of potent CFTR activators of the cystic fibrosis transmembrane conductance regulator (CFTR) (68). Rapid quantitative screening of a 60,000 compound library on cells grown on 96-well plates led to the identification of a large number of strong and weak activators. Recently, similar HTS to identify proteins that correct the F508del-CFTR defect was done with YFP–H148Q/I152L (116).
Similar screening approaches can be envisaged with roGFP-based probes to identify novel genes involved in cellular redox homeostasis and drugs affecting defined redox processes. Provided that roGFP variants with appropriate midpoint potentials are available, such screens can be conducted for both reductive and pro-oxidative processes. Although a ratiometric analysis for roGFP is technically more demanding than a single wavelength screen like the one described for Cl--sensitive YFP, it seems feasible to do such screens on a plate reader, automated microscopes or by FACS. For identifying fast and transient effects on EGSH in a time frame where correction for protein concentration is less critical, it may also be possible to use only a single wavelength to monitor changes in roGFP2 oxidation (121).
C. Overall perspective: Exciting times ahead
Within only few years after their introduction, redox-sensitive FPs have proven to be extremely useful for providing new insights into cellular redox biology. Genetically encoded redox probes together with high resolution confocal imaging allow the investigation of redox processes in single cells or even at subcellular level. Thus, the application of these probes provides information that was not accessible by other currently available methodologies. The full potential of roGFP-based probes can now be exploited in targeted approaches testing the function of particular genes in cellular redox homeostasis, and in HTS for novel genes and metabolic regulators of intracellular redox homeostasis. The design of novel redox-sensitive FPs is aimed at detecting and quantifying different ROS and other redox active components with the highest possible resolution in time and space. Vice versa, a better mechanistic understanding of how specificity is achieved and maintained in ROS signaling will certainly inspire novel reporter design strategies.
To be statistically meaningful and comparable between different laboratories, quantitative image analysis requires more rigor in experimental design and methodology than comparable qualitative assessments (35). In this context, the exact understanding of probe behavior and detailed in vitro and in vivo characterization is essential for producing meaningful and interpretable quantitative data. Further expansion of the repertoire of genetically encoded redox-sensitive FPs bears a huge potential for providing new insight into the dynamic behavior of the cellular redox system with minimal perturbation from outside. With the rapidly increasing number of applications based on genetically encoded redox probes and better mechanistic understanding of thiol-dependent redox processes, exciting times are ahead for the growing field of redox biology.
Footnotes
Acknowledgments
The authors gratefully acknowledge funding of their work from the German Research Foundation (DFG), the European Commission and the Landesstiftung Baden-Württemberg.
Abbreviations Used
Reviewing Editors: Shawn C. Burdette, Salvatore Cannistraro, Pier G. Mastroberardino, and S. James Remington