
Abstract
Heme oxygenase-1 (HO-1) plays a crucial role in tissue pathological changes such as brain injuries. Our previous studies have demonstrated that bradykinin (BK) induces the expression of several inflammatory proteins, including matrix metalloproteinase-9 and COX-2, via mitogen-activated protein kinases and nuclear factor-κB (NF-κB) in rat brain astrocytes (RBA-1). However, the molecular mechanisms underlying BK-induced HO-1 expression in RBA-1 cells remain poorly defined. Here we demonstrated that BK induced HO-1 expression and enzymatic activity via a B2 BK receptor-activated reactive oxygen species (ROS)-dependent signaling pathway. NADPH oxidase (Nox)-dependent ROS generation led to activation of extracellular signal-regulated kinase 1/2 (ERK1/2) and c-Jun-N-terminal kinase (JNK) and then activated the downstream molecules NF-κB and c-Jun, respectively. The c-Fos, an activator protein 1 (AP-1) subunit, was upregulated by activation of NF-κB and c-Jun, which bound to HO-1 promoter and thereby turned on transcription of HO-1 gene. The rat HO-1 promoter containing a putative AP-1 cis-binding site was identified as a crucial domain linking to BK action. Taken together, these results suggested that in RBA-1 cells, activation of ERK/NF-κB and JNK/c-Jun cascades by a Nox/ROS-dependent event enhancing c-Fos/AP-1 activity is essential for HO-1 upregulation and activation induced by BK. Moreover, ROS-dependent NF-E2-related factor 2 activation also contributes to HO-1 induction by BK in astrocytes. Antioxid. Redox Signal. 13, 1829–1844.
Introduction
Reactive oxygen species (ROS) are produced by various enzymatic reactions and chemical processes or directly inhaled, including O2 −, OH · , and H2O2. The ROS are essential for many physiological functions and the killing of invading microorganisms (10, 49). However, several lines of evidence have suggested the pathogenesis of human diseases related to increased oxidative stress (10, 49). It has been shown that ROS can damage DNA, lipids, proteins, and carbohydrates, leading to impairment of cellular functions and enhancement of inflammatory reactions (19, 28, 49). In the brain, the physiological role of ROS (along with O2 − and NO) also extends to the control of vascular tone, which is tightly modulated by metabolic activity within neurons (14, 19). Under pathological conditions, increasing ROS production by several stimuli such as bradykinin (BK) can regulate expression of several inflammatory genes during brain injury (5, 6, 19). Recently, increasing evidence attributes the cellular damage in neurodegenerative disorders such as AD to oxidative stress, which leads to generation of free radicals being responsible for brain inflammatory disorders, causing deleterious effects during CNS pathogenesis (6, 19, 32). Moreover, many reports have shown that ROS are major signaling molecules that mediate microglial activation induced by several inflammatory mediators, including Aβ and lipopolysaccharide (LPS) (30, 39). Although the effects of BK associated with ROS have been reported in cerebral arteriolar effects and cardiac and renal diseases (5, 9), BK-induced astrocytic responses through the generation of ROS are not well characterized.
BK and related peptides are elevated during brain trauma, stroke, and neurogenic inflammation (29). Among brain cells, astrocytes possess a G protein-coupled receptor B2 BK receptor (B2BKR), which couples to a Gq protein to activate phospholipase Cβ, phosphoinositide breakdown, protein kinase C (PKC), and intracellular Ca2+ mobilization in several cell types including astrocytes (7, 21). These signaling pathways contribute to several cellular responses including proliferation, migration, or gene expression through activation of different mitogen-activated protein kinase (MAPK) cascades such as extracellular signal-regulated kinase 1/2 (ERK1/2) (11, 23). Several studies have demonstrated that induction of HO-1 is mediated through activation of several signal molecules including PI3K/Akt and MAPKs (8, 40, 42). Moreover, a variety of putative regulatory elements, including antioxidant response elements (AREs), NF-E2-related factor 2 (Nrf2), activator protein 1 (AP-1), and nuclear factor-κB (NF-κB) sites, in the 5′ region of HO-1 promoters have been reported in various animal species (2, 3). Recently, BK has been shown to regulate expression of several genes through different transcription factors including AP-1 (53) and NF-κB (24).
Therefore, BK may play a potential role in the regulation of specific gene expression, such as HO-1, and thereby prevent inflammatory responses. However, the mechanisms of intracellular signaling pathways involved in BK-induced HO-1 expression in rat brain astrocytes (RBA-1) are unclear. Here, we found that BK induces HO-1 gene expression via a ROS-dependent pathway in RBA-1. This upregulation is mediated via an AP-1 element of the rat HO-1 promoter that is a target of the c-Fos/AP-1 subunit. Two independent pathways, c-Jun-N-terminal kinase (JNK)/c-Jun and ERK/NF-κB pathways, are involved in c-Fos/AP-1 induction, which both require the generation of ROS, and in B2BKR-dependent induction of HO-1 gene expression and activity in RBA-1 cells.
Materials and Methods
Materials
Dulbecco's modified Eagle's medium (DMEM)/Ham's nutrient mixture F-12 (F-12) medium and fetal bovine serum were purchased from Invitrogen. Hybond C membrane and enhanced chemiluminescence (ECL) Western blotting detection system were obtained from GE Healthcare Bio-sciences. HO-1 antibody was from Stressgen (No. SPA895). Phospho-(Thr202/Tyr204)-ERK1/2, phospho-(Thr180/Tyr182)-p38 MAPK, phospho-(Thr183/Tyr185)-JNK1/2, and phospho-p65 antibody kits were from Cell Signaling. NADPH oxidase 1 (Nox1), Nox2, Nox4, phospho-c-Jun, and c-Fos antibodies were from Santa Cruz. All primary antibodies were diluted at 1:1000 in phosphate-buffered saline (PBS) with 1% bovine serum albumin (BSA) (Calbiochem).
Cell cultures and treatment
A RBA-1 cell line was used throughout this study. This cell line originated from a primary astrocyte culture of neonatal rat cerebrum and was naturally developed through successive cell passages (27). Cells were cultured and treated as previously described (24). Primary astrocyte cultures were prepared from the cortex of 6-day-old Sprague–Dawley rat pups as previously described (23) with modifications. The purity of primary astrocyte cultures was assessed with the astrocyte-specific marker, GFAP, showing nearly 95% GFAP-positive astrocytes (Fig. 1E, upper part). Cells were plated onto 12-well culture plates and made quiescent at confluence by incubation in serum-free DMEM/F-12 for 24 h. Growth-arrested cells were incubated with or without different concentrations (1 nM to 1 μM) of BK at 37°C for the indicated time intervals. When the inhibitors were used, cells were pretreated with the inhibitor for 1 h before exposure to BK.
Total RNA extraction and reverse transcription–polymerase chain reaction analysis
Total RNA was extracted from RBA-1 cells as previously described (24). The cDNA obtained from 0.5 μg total RNA was used as a template for polymerase chain reaction (PCR) amplification. Oligonucleotide primers were designed based on GenBank entries for rat HO-1 and β-actin. The following primers were used for amplification reaction: for HO-1 (8), forward primer 5′-CACGCATATACCCGCTACCT-3′ and reverse primer 5′-TCTGTCACCCTGTGCTTGAC-3′; for c-Fos, forward primer 5′-AGACGAAGGAAGACGTGTAAGCACTGCAGCT-3′ and reverse primer 5′-AAGGAGAATCCGAAGGGAAAGGAATAAGATG-3′; for β-actin, forward primer 5′-GAACCCTAAGGCCAACCGTG-3′ and reverse primer 5′-TGGCATAGAGGTCTTTACGG-3′. The amplification was performed in 30 cycles at 55°C for 30 s, 72°C for 1 min, and 94°C for 30 s. PCR fragments were analyzed on 2% agarose 1× TAE gel containing ethidium bromide and their size was compared with a molecular weight marker. Amplification of β-actin, a relatively invariant internal reference RNA, was performed in parallel, and cDNA amounts were standardized to equivalent β-actin mRNA levels. These primer sets specifically recognize only the genes of interest as indicated by amplification of a single band of the expected size (209 bp for HO-1, 600 bp for c-Fos, and 514 bp for β-actin) and direct sequence analysis of the PCR product.
Preparation of cell extracts and Western blotting analysis
Growth-arrested RBA-1 cells were incubated with or without BK at 37°C for the indicated times. The cell lysates were collected and the protein concentration was determined by using the BCA reagents according to the instructions of the manufacturer. Samples from these cell lysates (30 μg protein) were denatured and subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis using a 10% (w/v) running gel. The phosphorylation of MAPKs was identified and quantified using Western blotting as previously described (24). The immunoreactive bands detected using ECL reagents were developed by Hyperfilm-ECL.
Determination of HO activity
HO activity was determined as previously described (8) with some modifications. Briefly, cells were incubated with BK, washed twice with PBS, gently scraped off the dish, and centrifuged (1000 g, 10 min, 4°C). The cell pellet was suspended in MgCl2 (2 mM) phosphate (100 mM) buffer (pH 7.4), frozen at −70°C, thawed three times, and finally sonicated on ice before centrifugation at 18,000 g for 10 min at 4°C. The supernatant (400 μl) was added to a reaction mixture (200 μl final volume, pH 7.4) containing NADPH (0.8 mM), glucose-6-phosphate (2 mM), glucose-6-phosphate-1-dehydrogenase (0.2 U), 2 mg of rat liver cytosol as a source of biliverdin reductase, PBS (100 mM), and the substrate hemin (10 μM). The reaction was conducted for 1 h at 37°C in the dark and terminated by addition of 1 ml chloroform. The extracted bilirubin was measured by the difference in absorption between 464 and 530 nm (extinction coefficient = 40 mM −1 cm−1). HO activity was expressed as picomoles of bilirubin per milligram of protein per hour.
Measurement of intracellular ROS generation
The peroxide-sensitive fluorescent probe 2′,7′-dichlorofluorescein diacetate (DCF-DA) was used to assess the generation of intracellular ROS (31) with minor modifications. RBA-1 cells in monolayers were incubated with RPMI-1640 supplemented with 5 μM DCF-DA for 45 min at 37°C. The supernatant was removed and replaced with fresh RPMI-1640 media before stimulation with BK (10 nM). Relative fluorescence intensity was recorded over time (3–120 min) by using a fluorescent plate reader (Thermo; Appliskan) at an excitation wavelength of 485 nm and emission was measured at a wavelength of 530 nm. Fluorescent images were also obtained by using fluorescence microscopy (Axiovert 200M; Zeiss).
Plasmid construction, transfection, and luciferase reporter gene assays
The plasmids encoding short hairpin RNA of c-Fos (c-Fos shRNA) was kindly provided by Dr. C.P. Tseng (Chang Gung University, Taiwan). The rat HO-1 promoter (accession No. J02722.1; −766 to +20) was constructed (sense primer: GGTACCCAGGAAGTCACAGTGTGGCC; antisense primer: CCCGAGCTCGTCGAGCTGTGGGCGCTCCAT; 63°C) and cloned to the pGL3-basic vector containing the luciferase reporter system. All plasmids were prepared by using Qiagen plasmid DNA preparation kits. RBA-1 cells were transfected with plasmid using the DNA PLUS-Lipofectamine reagent. Transfection with pGal encoding for β-galactosidase was used for control of transfection efficiencies. To assess promoter activity, cells were collected and disrupted by sonication in lysis buffer (25 mM Tris phosphate [pH 7.8], 2 mM ethylenediaminetetraacetic acid, 1% Triton X-100, and 10% glycerol). After centrifugation, aliquots of the supernatants were tested for luciferase activity using a luciferase assay system (Promega). Firefly luciferase activities were standardized for β-galactosidase activity.
Immunofluorescence staining
Growth-arrested cells were treated with 10 nM BK, washed twice with ice-cold PBS, fixed with 4% (w/v) paraformaldehyde in PBS for 30 min, and then permeabilized with 0.3% Triton X-100 in PBS for 15 min. The staining was performed by incubating with 10% normal goat serum in PBS for 30 min, followed by incubating with an anti-c-Fos polyclonal antibody (1:200 dilution) for 1 h in PBS with 1% BSA, washing three times with PBS, incubating for 1 h with fluorescein isothiocyanate-conjugated goat anti-rabbit antibody (1:200 dilution) in PBS with 1% BSA, washing three times with PBS, and finally mounting with aqueous mounting medium. The images were observed under a fluorescence microscope (Axiovert 200M; Zeiss).
Chromatin immunoprecipitation assay
The primers were designed by referencing nucleotide sequence of the rat HO gene as previously described (36). Briefly, RBA-1 cells were crosslinked with 1% formaldehyde for 10 min at 37°C and washed thrice with ice-cold PBS containing 1 mM phenylmethylsulfonyl fluoride and 1% aprotinin. Soluble chromatin was prepared using a chromatin immunoprecipitation (ChIP) assay kit (Upstate) according to the manufacturer's recommendations and immunoprecipitated without (control) or with anti-c-Fos antibody (sc-7202; Santa Cruz) and normal goat immunoglobulin G. Following washes and elution, precipitates were heated overnight at 65°C to reverse crosslinking of DNA and protein. DNA fragments were purified using phenol–chloroform extraction and ethanol precipitation. The purified DNA was subjected to PCR amplification using the primers specific for the region (−460 to −2) containing a putative AP-1-binding site present in the HO-1 promoter region (sense primer: 5′-CAGTCTAGGCAGGTGTTAGT-3′; antisense primer: 5′-AGCGAGCAGCTGCCCGCGCC-3′). PCR fragments were analyzed on 2% agarose 1× TAE gel containing ethidium bromide and the size (458 bp) was compared with a molecular weight marker.
Analysis of data
Data were estimated using a GraphPad Prism Program (GraphPad). Quantitative data were analyzed using analysis of variance followed by Tukey's honestly significant difference tests between individual groups. Data were expressed as mean ± standard error of the mean. A p-value of <0.05 was considered significant.
Results
BK induces HO-1 expression and HO activity
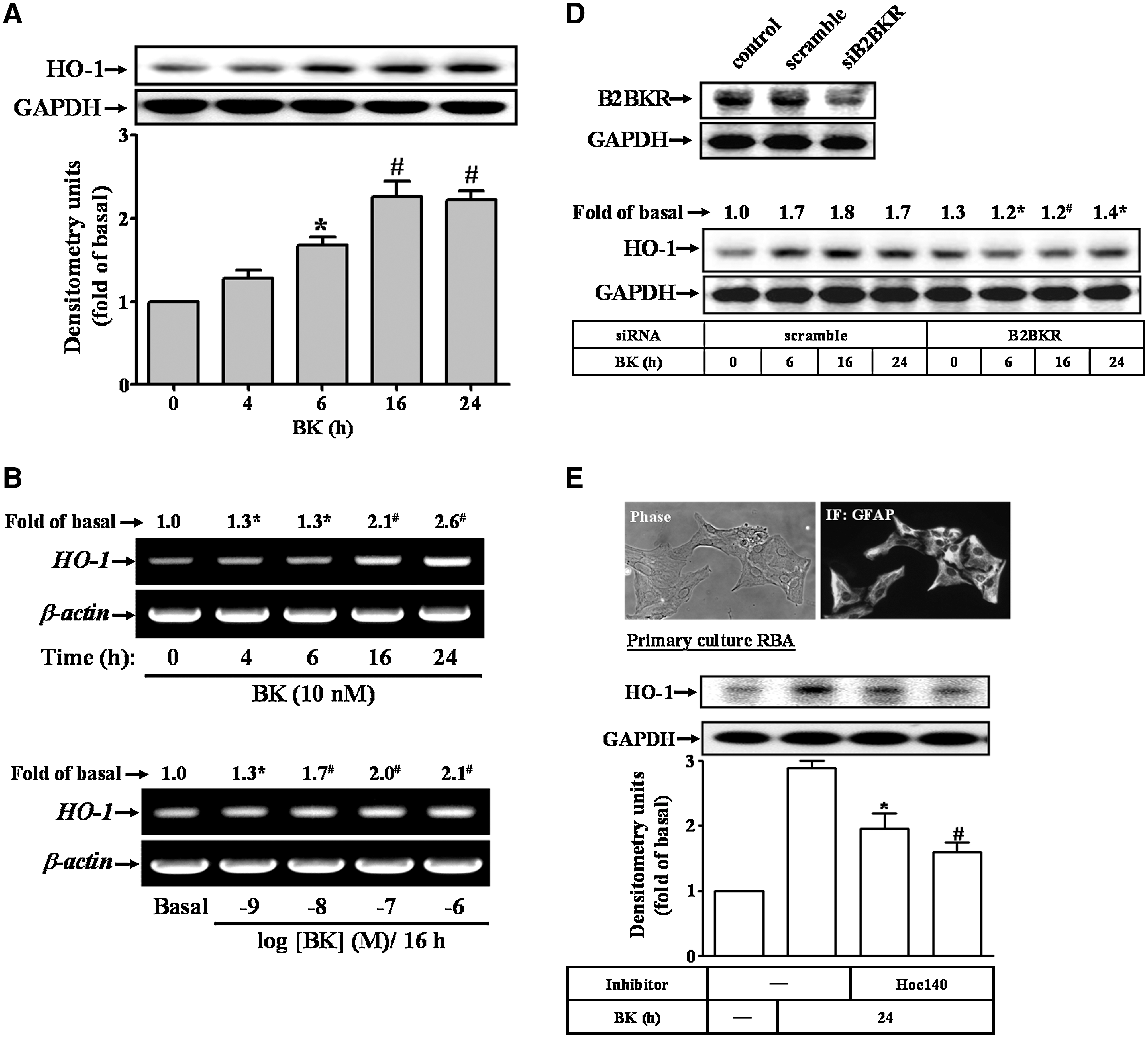
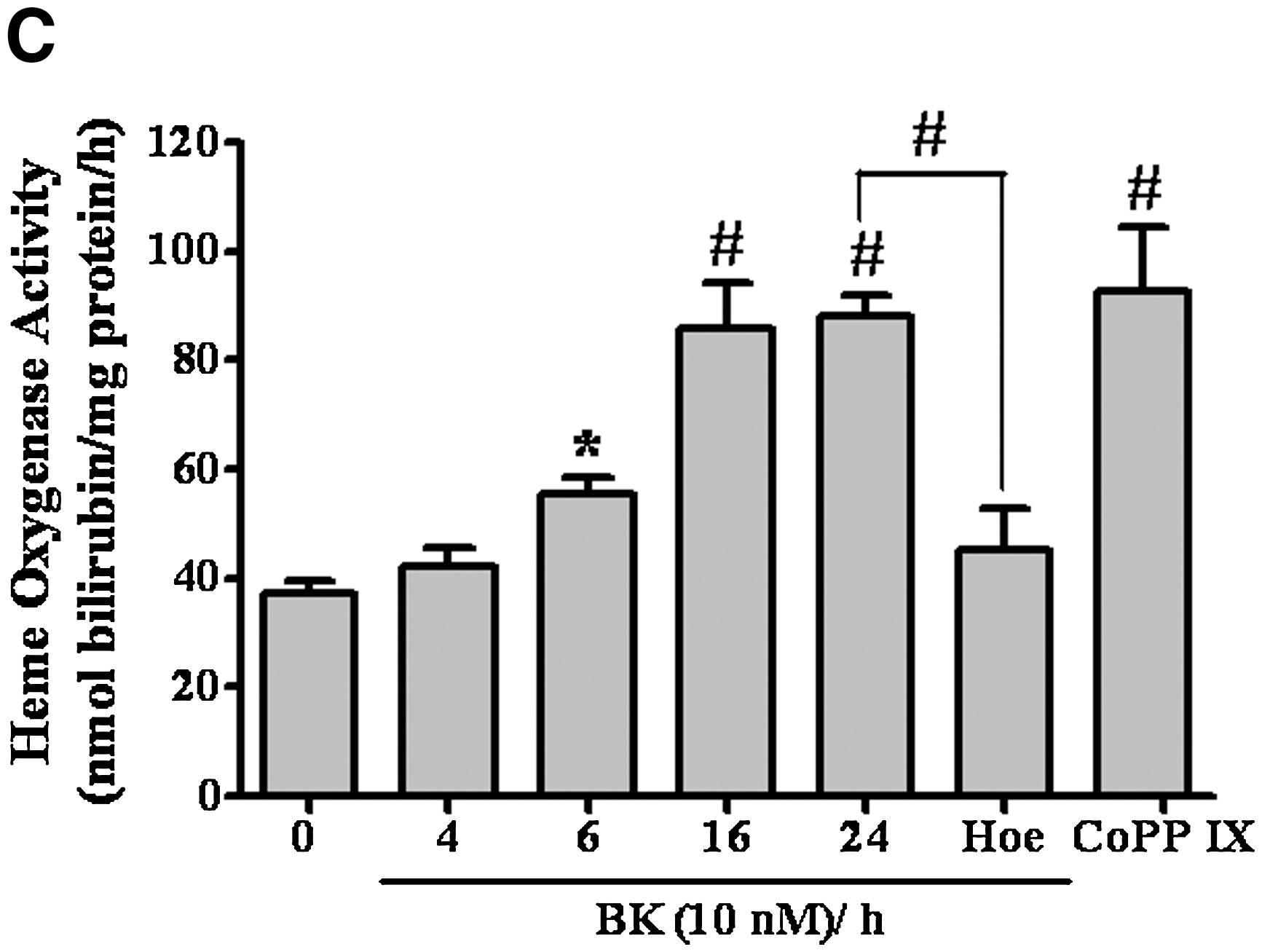
To investigate the effects of BK on HO-1 gene expression and activity, BK-induced HO-1 protein expression in a time-dependent manner was determined using Western blot (Fig. 1A). There was a significant increase within 6 h, which sustained up to 24 h. Next, BK-induced HO-1 mRNA expression in a time-dependent (Fig. 1B, upper panel) and concentration-dependent (Fig. 1B, lower panel) manner was detected using reverse transcription (RT)–PCR. To further determine whether BK-induced HO-1 expression accompanied with an increase in HO activity, the HO enzymatic activity was assessed by measuring the heme metabolite bilirubin in the cell lysates. The data showed that BK induced HO enzymatic activity in a time-dependent manner and a significant increase occurred at 6 h and sustained up to 24 h (Fig. 1C). Cells treated with cobalt protoporphyrin IX (a HO-1 inducer) for 24 h significantly induced HO activity (as a positive control). Moreover, pretreatment with Hoe140 (10 μM), a selective B2BKR antagonist, markedly attenuated BK-induced HO activity, suggesting that BK induces HO activity via a B2BKR in RBA-1 cells. Therefore, we further confirmed the involvement of B2BKR in BK-induced HO-1 expression, using cells transfected with a B2BKR siRNA. As shown in Figure 1D, transfection with B2BKR siRNA significantly knocked down the B2BKR protein expression and attenuated BK-induced HO-1 expression in RBA-1 cells.
To demonstrate whether BK-induced HO-1 expression also occurred in primary cultured astrocytes, as shown in Figure 1E, we first identified the primary cultured astrocytes by GFAP immunofluorescent staining, showing nearly 95% GFAP-positive astrocytes (upper panel). BK also induced HO-1 protein expression, which was inhibited by pretreatment with Hoe140 (lower panel). These results indicated that BK-induced responses in rat primary cultured astrocytes are similar to those of RBA-1 cells. Thus, the following experiments were performed using RBA-1 cells, which can be applied as a model throughout this study.
BK induces HO-1 expression through Nox-derived ROS generation
Several studies have demonstrated that ROS contributes to HO-1 expression in various cell types (42). To determine whether ROS participated in HO-1 induction, a ROS scavenger NAC and Nox (a major source of ROS) activity inhibitor diphenyleneiodonium (DPI) were used. Pretreatment with NAC (10 mM) or DPI (1 μM) attenuated the BK-induced HO-1 protein and mRNA expression (Fig. 2A, B), suggesting that Nox-derived ROS may play a potential role in BK-induced HO-1 expression in RBA-1 cells. To explore whether BK induced ROS generation, the cells were loaded with DCF-DA (a ROS indicator) and then stimulated with BK (10 nM) for the indicated time intervals. Our data showed that BK increased the ROS generation in a time-dependent manner with a maximal response within 3 min and sustained over 120 min (Fig. 2C). The results were further supported by the data of fluorescence images obtained using a fluorescent microscopy (Fig. 2C, inset panel). As shown in Figure 2D, BK-induced ROS generation was markedly attenuated by pretreatment with Hoe140, NAC, or DPI, suggesting that BK-stimulated ROS generation led to HO-1 expression via a B2BKR-dependent activation of Nox in RBA-1 cells.

To further determine which Nox isotype was involved in these responses, a variety of Nox siRNA were used. As shown in Figure 2E, transfection with Nox1 or Nox2 siRNA, but not Nox4, knocked down the expression of respective Nox protein and significantly attenuated BK-induced ROS generation and HO-1 expression (Fig. 2F) in both RBA-1 cells and primary culture astrocytes. These results suggested that BK-induced HO-1 expression is mediated through Nox-dependent ROS generation in brain astrocytes.
Involvement of ROS-dependent ERK1/2 and JNK1/2 pathways in the induction of HO-1 by BK
HO-1 expression is regulated via the activation of MAPKs in many cell types (8, 42). Recently, we have demonstrated that MAPKs participate in the expression of several inflammatory genes in RBA-1 cells (22, 21). Therefore, we further determined whether MAPKs also involve in BK-induced HO-1 expression in RBA-1 cells. First, we found that pretreatment with the inhibitors of MEK1/2 (PD98059, 30 μM) and JNK (SP600125, 1 μM) but not p38 MAPK (SB202190, 10 μM) significantly blocked BK-induced HO-1 protein and mRNA expression (Fig. 3A, B), indicating that ERK1/2 and JNK1/2 were involved in BK-induced HO-1 expression in RBA-1 cells.

To determine whether phosphorylation of these MAPKs was necessary for BK-induced HO-1 expression, activation of these kinases was assayed using Western blot with an antibody specific for the phosphorylated form of ERK1/2, p38 MAPK, or JNK1/2. As shown in Figure 3C, BK stimulated phosphorylation of ERK1/2 and JNK1/2 in a time-dependent manner, but failed to stimulate p38 MAPK phosphorylation. Moreover, phosphorylation of ERK1/2 and JNK1/2 as well as c-Jun stimulated by BK was blocked by pretreatment with PD98059 and SP600125, respectively. Next, we examined whether BK-stimulated phosphorylation of ERK1/2 and JNK1/2 was mediated through ROS generation. As shown in Figure 3D, pretreatment with NAC significantly attenuated BK-stimulated phosphorylation of ERK1/2, JNK1/2, and c-Jun. These results indicated that ROS plays a critical role in BK-stimulated ERK1/2 and JNK1/2 phosphorylation, leading to HO-1 expression in RBA-1 cells.
BK induces HO-1 expression via ROS/ERK cascade and NF-κB activation
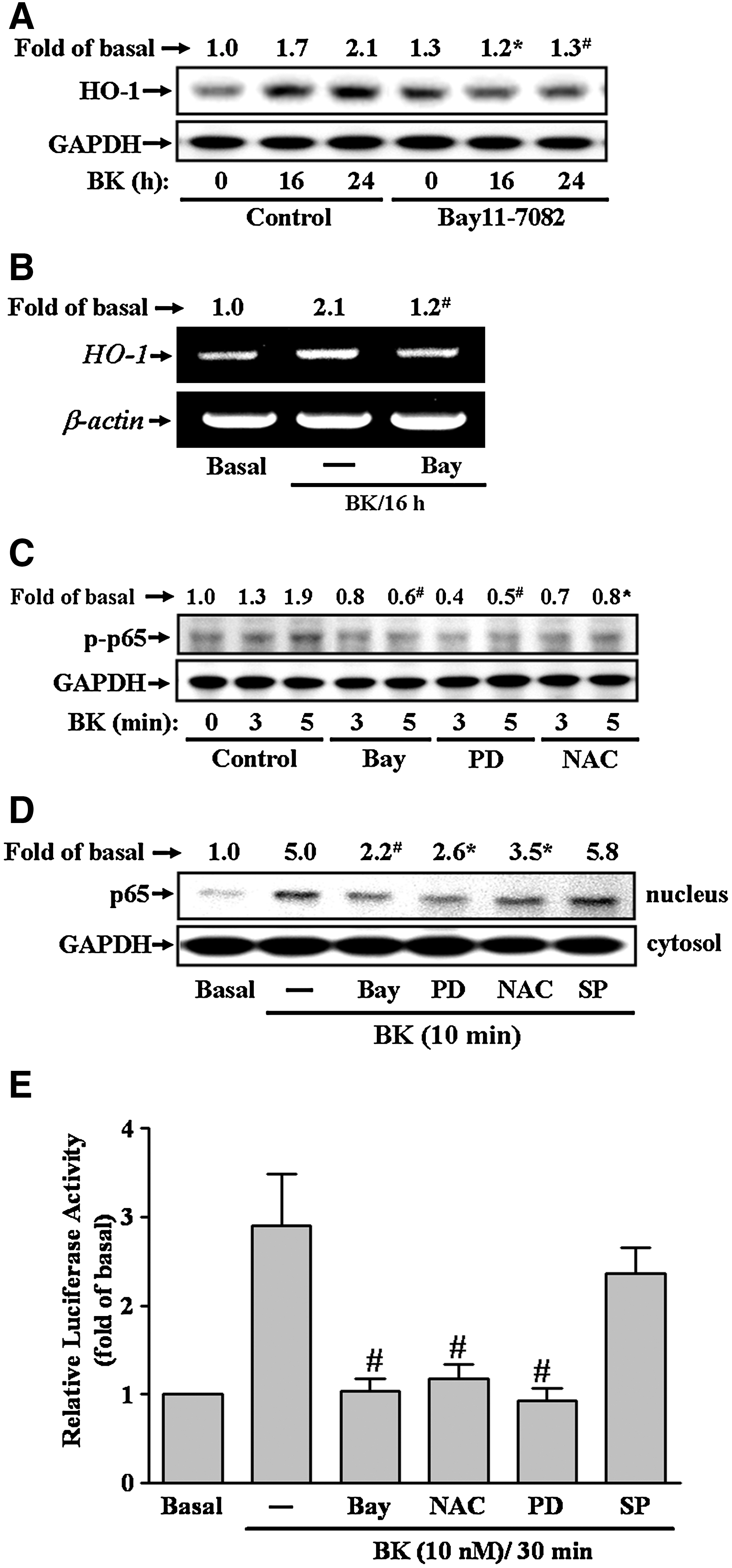
To investigate whether NF-κB was involved in BK-induced HO-1 expression, an NF-κB inhibitor Bay11-7082 (38) was used. Pretreatment with Bay11-7082 (1 μM) inhibited BK-induced HO-1 protein and mRNA expression (Fig. 4A, B). It has been shown that phosphorylation and translocation of NF-κB, such as p65 subunit, is required for induction of NF-κB-targeted genes (50). To determine whether p65 was phosphorylated and translocated into nucleus stimulated by BK in RBA-1 cells, the whole cell lysate and nuclear fraction were prepared and analyzed using Western blotting. As shown in Figure 4C and D, BK stimulated a rapid phosphorylation and translocation of p65 in RBA-1 cells, which was significantly blocked by pretreatment with Bay11-7082, PD98059, or NAC, but not with SP600125. NF-κB activation by BK was further confirmed by reporter gene activity assay. RBA-1 cells were transfected with an NF-κB luciferase reporter gene (pκB-Luc). As shown in Figure 4E, BK-stimulated NF-κB transcription activity was inhibited by pretreatment with Bay11-7082, PD98059, or NAC, but not SP600125. These results demonstrated that NF-κB was involved in HO-1 expression mediated through ROS generation followed by the activation of ERK in RBA-1 cells.
The induction of transcription factor c-Fos/AP-1 is required for BK-induced HO-1 expression
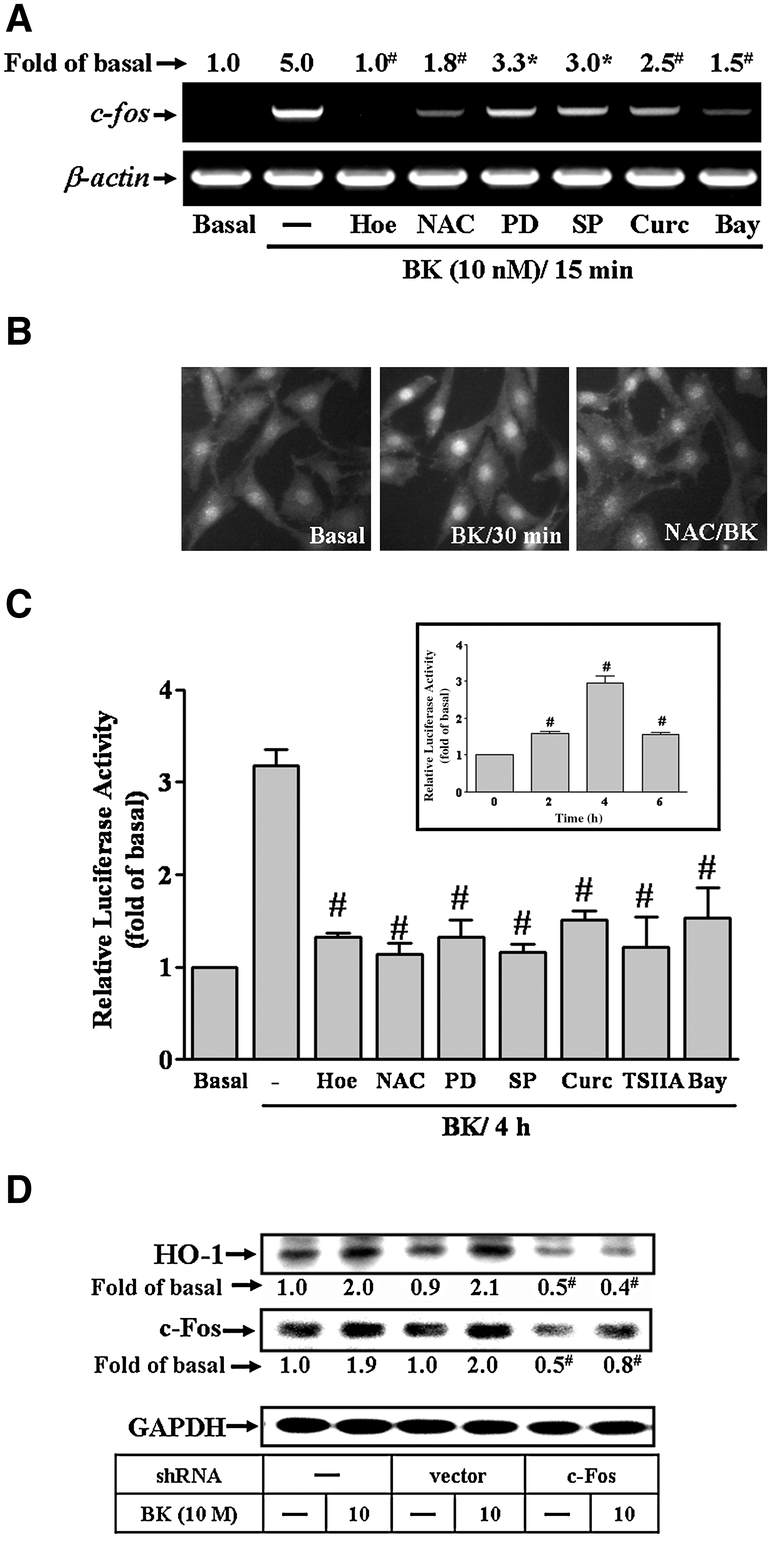
To determine whether BK-induced HO-1 expression was mediated through transcription factor AP-1, two AP-1 inhibitors, that is, curcumin and TSIIA, were used. As shown in Figure 5A and B, pretreatment with AP-1 inhibitor curcumin or TSIIA attenuated BK-induced HO-1 protein and mRNA expression, suggesting that AP-1 is required for BK-induced HO-1 expression in RBA-1. To further investigate whether BK stimulated the expression of AP-1 (containing immediate early gene c-Fos and c-Jun subunits), the levels of protein and mRNA of c-Fos and c-Jun were determined using Western blot and RT-PCR, respectively. BK induced c-Fos protein (Fig. 5C) and mRNA (Fig. 5D) expression in a time-dependent manner, but not c-Jun (data not shown). The c-Fos induction by BK reached a peak within 60 min (protein) and 15 min (mRNA), respectively. BK-induced c-Fos protein expression was attenuated by pretreatment with curcumin or TSIIA (Fig. 5E). These results suggested that BK-induced HO-1 expression is mediated through upregulation of transcription factor AP-1, particularly c-Fos subunit, in RBA-1 cells.

BK-induced c-Fos/AP-1 expression is mediated through ROS-dependent ERK/NF-κB and JNK/c-Jun cascades
To further investigate whether ERK/NF-κB and JNK/c-Jun pathways were involved in BK-induced AP-1 expression, as shown in Figure 6A, BK-induced c-Fos/AP-1 expression was inhibited by pretreatment with Hoe140, NAC, PD98059, SP600125, curcumin, or Bay11-7082. BK-induced c-Fos/AP-1 expression was further supported by the results obtained using immunofluorescence staining against a c-Fos antibody. The result showed that BK-stimulated expression and accumulation (in nucleus) of c-Fos/AP-1 was attenuated by pretreatment with NAC in RBA-1 cells (Fig. 6B). The AP-1 activation by BK was further confirmed by reporter gene activity assay. The AP-1 transcription activity was enhanced by BK (Fig. 6C, inset panel), which was significantly inhibited by pretreatment with Hoe140, NAC, PD98059, SP600125, curcumin, TSIIA, or Bay11-7082 (Fig. 6C). These results suggested that BK-induced AP-1 expression and activation is mediated through ROS-dependent ERK/NF-κB and JNK/c-Jun pathways. To ensure that c-Fos/AP-1 was essential for BK-induced HO-1 expression, transfection of cells with c-Fos shRNA downregulated the expression of total c-Fos protein and markedly abolished BK-induced HO-1 expression (Fig. 6D), indicating that upregulation of c-Fos/AP-1 was required for BK-induced HO-1 expression in RBA-1 cells.
BK-induced recruitment of c-Fos/AP-1 enhances the HO-1 promoter transcriptional activity and HO activity
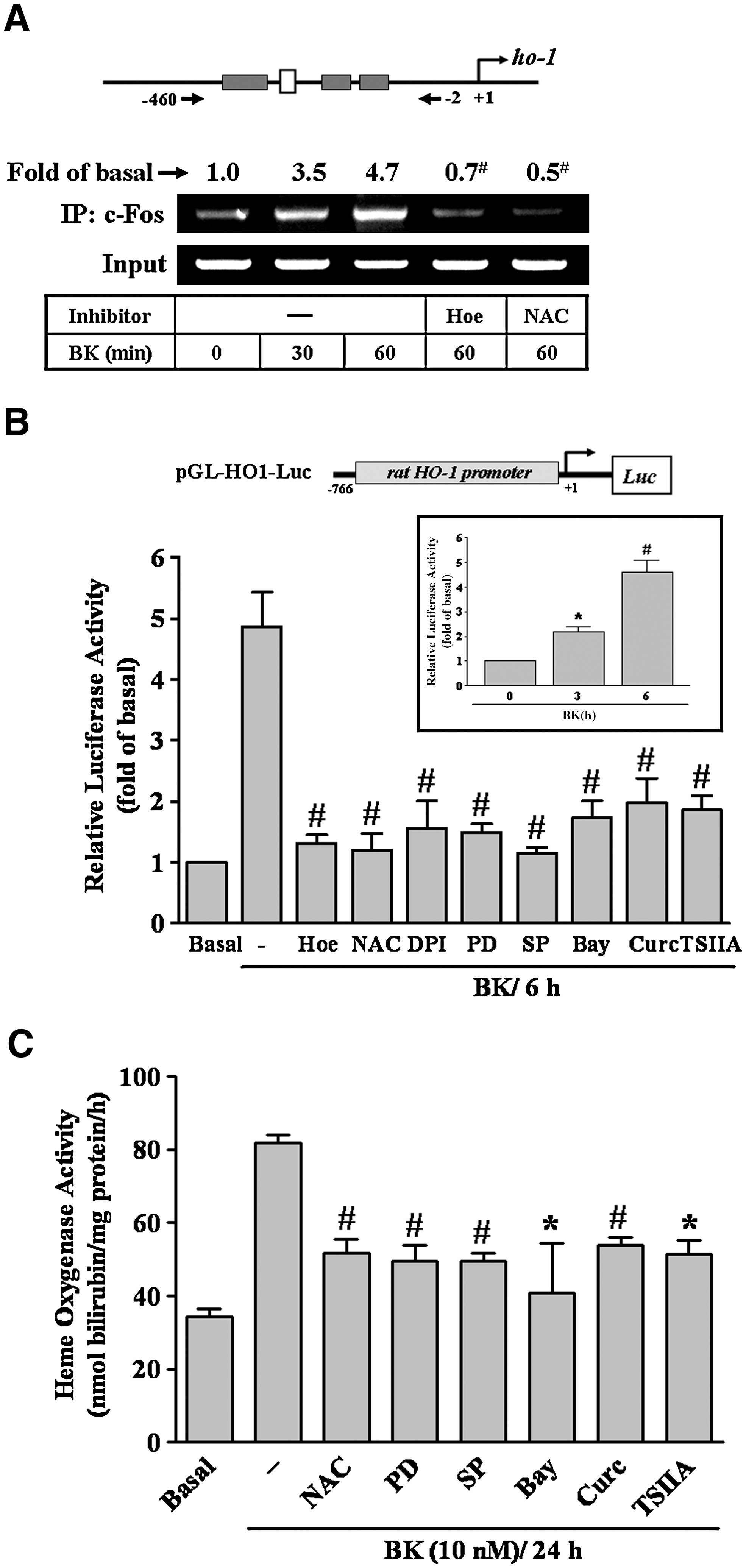
To investigate whether c-Fos bound to the HO-1 promoter region, a ChIP-PCR assay was performed. Chromatin was immunoprecipitated with an anti-c-Fos antibody and the HO-1 promoter region (−460 to −2) was amplified using PCR. As shown in Figure 7A, BK stimulated in vivo binding of c-Fos to the HO-1 promoter in a time-dependent manner with a maximal response within 30 min, which was attenuated by pretreatment with Hoe140 or NAC. The data suggested that BK-stimulated binding of c-Fos to HO-1 promoter is mediated through a B2BKR/ROS-dependent pathway. To further examine whether BK induced HO-1 gene regulation via initiating its promoter activity, the HO-1 promoter was constructed and its activity was evaluated using a promoter-luciferase activity assay. The HO-1 promoter was constructed into a pGL3-basic vector containing a luciferase reporter system (as illustrated in Fig. 7B, upper part), which contains several putative recognition elements for a variety of transcriptional factors. To determine the effect of BK on the HO-1 promoter activity, as shown in Figure 7B, BK increased the HO-1 promoter activity in a time-dependent manner (inset panel) with a maximal response within 6 h, which was inhibited by pretreatment with Hoe140, NAC, DPI, PD98059, SP600125, Bay11-7082, curcumin, or TSIIA. Finally, we determined whether these signaling pathways were involved in BK-induced HO enzymatic activity. As shown in Figure 7C, BK-enhanced HO enzymatic activity was significantly abolished by pretreatment with NAC, PD98059, SP600125, Bay11-7082, curcumin, or TSIIA. These results confirmed that BK induced HO-1 promoter activity and HO enzymatic activity via enhancing c-Fos/AP-1 binding to the HO-1 promoter. The induction of the HO-1 promoter activity was mediated through B2BKR/ROS-dependent ERK/NF-κB and JNK/c-Jun pathways converging to c-Fos/AP-1 upregulation in RBA-1 cells.
BK-induced HO-1 expression via ROS-dependent Nrf2 signaling
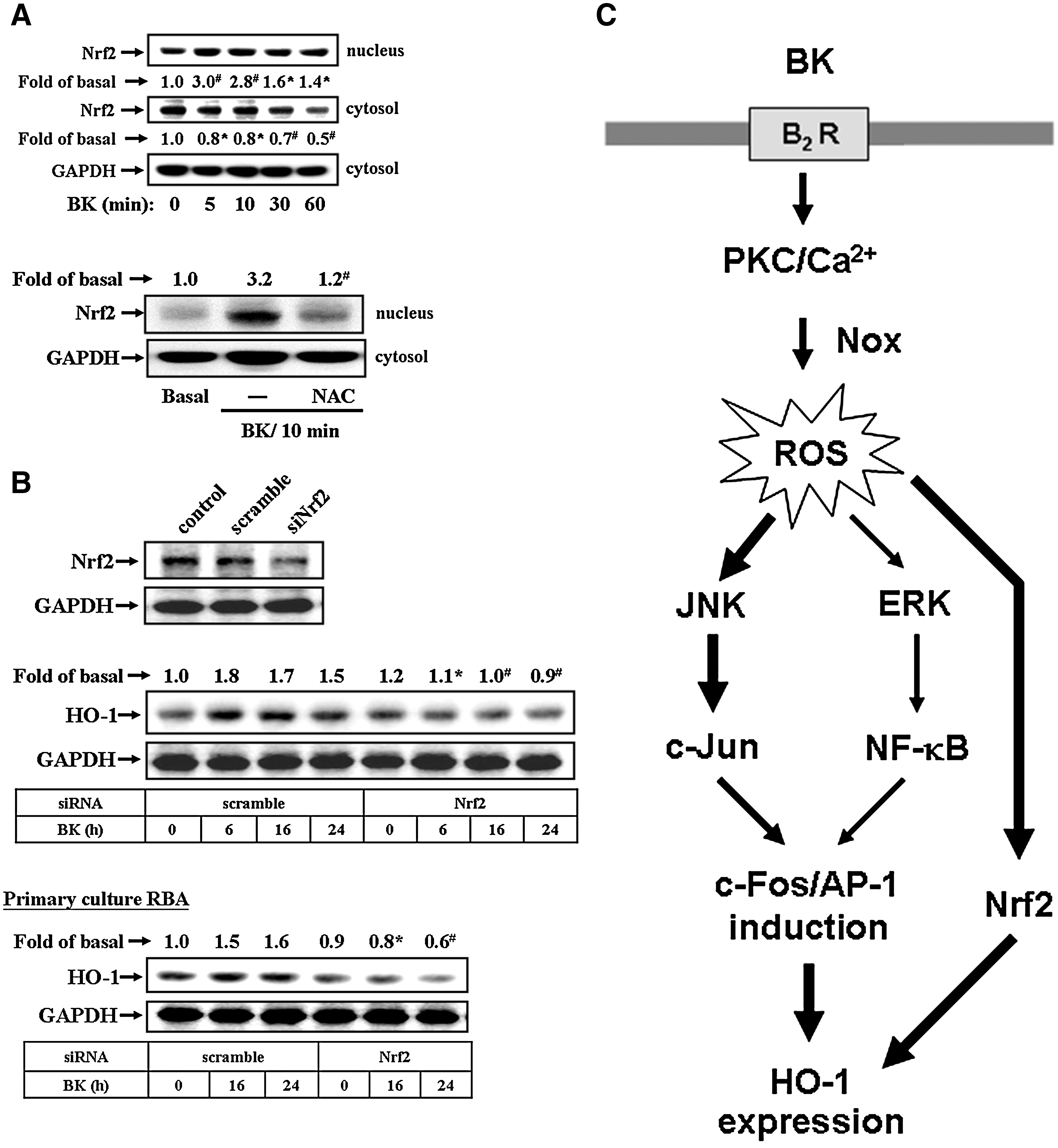
In addition, Nrf2 has been reported to mediate the expression of a wide array of ROS-dependent genes. Thus, we also demonstrated whether ROS-dependent Nrf2 signaling was involved in BK-induced HO-1 expression. As shown in Figure 8A, BK stimulated Nrf2 translocation from cytoplasm into nucleus in a time-dependent manner, with a significant response within 5–10 min (upper panel). Pretreatment with NAC significantly inhibited Nrf2 translocation stimulated by BK (Fig. 8A, lower panel), suggesting that BK stimulated a ROS-dependent Nrf2 translocation in RBA-1 cells. To further demonstrate the role of Nrf2 in BK-induced HO-1 expression, cells were transfected with Nrf2 siRNA and then exposed to BK for the indicated time intervals. We found that transfection with Nrf2 siRNA knocked down the expression of total Nrf2 protein and attenuated BK-induced HO-1 expression in RBA-1 cells (Fig. 8B, upper panel) and primary culture astrocytes (Fig. 8B, lower panel). These results indicated that ROS-dependent Nrf2 signaling may be involved in BK-induced HO-1 expression in RBA-1 cells.
Discussion
The inducible HO-1 is upregulated in response to xenobiotics, oxidative stress, cellular injury, and diseases (41, 51). The role of HO-1 expression in the regulation of inflammation has emerged over the past decade, besides its well-known function as an indicator of oxidative stress (3, 51). HO-1 contributes to a wide range of biological activities in different brain diseases, such as stroke and AD (47, 51). Previous studies on brains from AD patients demonstrate colocalization of HO-1 protein in GFAP-expressing astrocytes and in neurons of the hippocampus (44). Moreover, expression and activity of HO-1 have been shown to be elevated in various neuroinflammatory and neurodegenerative disorders (47, 51), indicating that HO-1 may be a critical molecule in the pathophysiological processes in these diseases. Moreover, BK and related peptides are simultaneously produced and released following injury to the human CNS (29). Despite an obviously important role of BK or related peptides in brain diseases, the processes by which BK was implicated in astrocytic functions are not completely understood. Thus, we used a well-established rat astroglial cell line RBA-1 cells (27) to investigate the mechanisms underlying BK-induced HO-1 expression and its activity. Our results suggest that in RBA-1 cells, activation of ROS-dependent JNK/c-Jun and ERK/NF-κB pathways leading to induction of c-Fos/AP-1, mediated through a B2BKR, is essential for BK-induced HO-1 gene expression and its activity.
ROS concentration-dependently exerts a key role in the normal physiological functions and the inflammatory responses (17, 28, 49). In the brain, ROS also extends to the control of vascular tone, which is tightly modulated by metabolic activity within neurons (14). Moreover, increasing oxidative stress (i.e., ROS production) by diverse stimuli can regulate the expression of inflammatory genes such as COX-2 linked to pathogenesis of human CNS disorders (5, 17, 25, 49). Recently, increasing evidence attributes the cellular damage in neurodegenerative disorders such as AD to oxidative stress, which is due to generation of free radicals implicated in brain inflammatory disorders (6, 19, 32). The effects of BK on ROS generation have been reported in cerebral arterioles and cardiac and renal diseases (5, 9, 13). In this study, we found that BK-induced HO-1 expression was mediated through Nox-dependent ROS generation, because pretreatment with ROS scavenger NAC or Nox activity inhibitor DPI attenuated BK-induced responses (Fig. 2A, B). The involvement of Nox-dependent ROS in BK-induced HO-1 expression was further confirmed by ROS generation, which was also inhibited by pretreatment with Hoe140, NAC, or DPI (Fig. 2C, D), suggesting that B2BKR/Nox-ROS is involved in BK-induced HO-1 expression in RBA-1 cells. Consistently, many reports have also shown that ROS are a major signaling factor that mediates microglial activation induced by inflammatory mediators, including Aβ and LPS (30, 39). Herein we are the first group to establish that intracellular ROS generation contributes to upregulation of HO-1 induced by BK in brain astrocytes.
Moreover, Nox are considered to be a major source of ROS in several physiological and pathological processes (10, 25). To date, five Nox isotypes have been discovered, including Nox1–5 (25). Several reports have shown that Nox1, Nox2, and Nox4 are expressed in brain cells (10, 25), suggesting that these Nox isotypes may be crucial to ROS generation in the brain. However, the role of these enzymes in the upregulation of brain astrocytic HO-1 remains unclear. First, our data have demonstrated that Nox activity is involved in these responses by pretreatment with a Nox inhibitor DPI (Fig. 2). Next, the data of knockdown distinct Nox by transfection with their respective siRNA indicated that BK-induced ROS generation and HO-1 expression were mediated through activation of Nox, including Nox1 and Nox2, in RBA-1 cells and primary culture astrocytes (Fig. 2E, F). These results are consistent with previous studies showing that Nox is expressed in astrocytes and contributes to ROS generation (1, 33). Additionally, we also suggested that BK-stimulated Nox/ROS signaling might be mediated through PKC or Ca2+ signals, confirmed by pretreatment with pan-PKC inhibitor (GF109203X) or intracellular Ca2+ chelator BAPTA/AM (data not shown). The regulatory mechanisms will be investigated in detail in our future research.
Abnormal MAPK regulation might be implicated in CNS inflammation and injury (4, 26). Moreover, BK has been reported to act as an important inflammatory mediator through activation of MAPK cascades in different cell types (21, 29, 53). In the present study, we demonstrated that ERK1/2 and JNK1/2, but not p38 MAPK, are essentially required for HO-1 induction by BK, because BK-induced responses were attenuated by pretreatment with respective MAPK selective pharmacological inhibitors including MEK1/2 (PD98059) or JNK (SP600125) (Fig. 3A, B). The involvement of these two kinases in BK-induced HO-1 expression was further confirmed by BK-stimulated phosphorylation of ERK1/2 and JNK1/2 (Fig. 3C). These results are consistent with the HO-1 expression mediated through ERK1/2 and JNK1/2 induced by ARE in HepG2 cells (52) and heme-mediated neuronal injury (4). Moreover, pretreatment with ROS scavenger NAC significantly attenuated the phosphorylation of ERK1/2 and JNK/c-Jun stimulated by BK (Fig. 3D), suggesting that increased ROS are crucial for BK-stimulated ERK1/2 and JNK1/2 phosphorylation in RBA-1 cells. These results are consistent with the reports showing that ROS-dependent activation of MAPKs, including ERK1/2 and JNK1/2, is involved in the regulation of cellular functions by diverse stimuli in several cell types (8, 37). The distinct groups of MAPKs are activated by a ROS-dependent manner, which is cell type-specific and dependent on the external stimuli.
The progressive increase of oxidative stress during injuries not only causes oxidative damage to cellular macromolecules, but also modulates the pattern of gene expression through functional alterations of transcription factors. Here we focus on the roles of transcription factors (i.e., NF-κB and AP-1), which are well known to be modulated during oxidative stress associated with inflammatory diseases (45). Moreover, several reports have shown that BK-induced gene expression is mediated through several transcription factors such as NF-κB (23) and AP-1 (53). However, the mechanistic connection between the HO-1 expression induced by BK and the ROS-dependent pathway has not been established in RBA-1 cells. Here, we demonstrated that NF-κB was essential for BK-induced HO-1 expression, which was inhibited by pretreatment with an NF-κB inhibitor Bay11-7082 (Fig. 4A, B). Moreover, the involvement of NF-κB in BK-induced responses was further ascertained by NF-κB phosphorylation, translocation into nucleus, and promoter activity determined using Western blot, cell fraction isolation, and promoter reporter analyses (Fig. 4). These responses were significantly attenuated by pretreatment with the inhibitor of NF-κB (Bay11-7082), MEK1/2 (PD98059), or a ROS scavenger (NAC), but not JNK inhibitor (SP600125). These results suggested that the ROS/ERK cascade, but not JNK, is involved in NF-κB signaling, which contributes to BK-induced HO-1 expression in RBA-1 cells. In addition, AP-1 was required for BK-induced HO-1 expression, which was attenuated by pretreatment with two AP-1 inhibitors curcumin or TSIIA (Fig. 5A, B). These results are consistent with the finding that AP-1 plays a key role in the HO-1 upregulation by arsenite in murine embryonic fibroblasts (20).
The involvement of AP-1 in these responses was further supported by the results indicating that BK induced c-Fos, an AP-1 subunit, expression and activation via ROS-dependent ERK/NF-κB and JNK/c-Jun pathways, as determined using RT-PCR and promoter reporter analyses (Fig. 6). Moreover, the role of c-Fos/AP-1 in BK-induced HO-1 expression was confirmed by transfection with c-Fos shRNA to knock down c-Fos protein and to attenuate BK-induced HO-1 expression (Fig. 6D). Several lines of evidence have shown that the 5′ region of HO-1 promoter contains a variety of putative regulatory elements, including AP-1 and NF-κB sites, in several animal species (2, 3). Thus, we also demonstrated that BK-stimulated c-Fos/AP-1 was recruited to an HO-1 promoter by a ChIP-PCR assay, which was reduced by pretreatment with Hoe140 or NAC, suggesting that BK induces c-Fos/AP-1 recruitment via a ROS-dependent manner (Fig. 7A). Next, we found that BK stimulated HO-1 promoter activity, which was attenuated by Hoe140, NAC, PD98059, SP600125, Bay11-7082, curcumin, or TSIIA (Fig. 7B), indicating that BK induced HO-1 promoter activity via a ROS-dependent pathway. Similarly, the HO enzymatic activity is stimulated by BK through the same pathway (Fig. 7C). These results are consistent with the reports indicating that angiotensin II regulates a variety of gene expression including HO-1 through a ROS-dependent activation of NF-κB in the heart (13), and alpha-lipoic acid induces HO-1 expression through the production of ROS and subsequent activation of the p44/42 MAPK pathway and AP-1 in vascular smooth muscle cells (8). In addition to AP-1, a well-known redox-related transcription factor Nrf2 has been shown to contribute to the regulation of several antioxidant and cytoprotective genes including HO-1 (2, 3). In this study, we first demonstrated that BK can stimulate Nrf2 translocation via a ROS-dependent manner (Fig. 8A), which subsequently results in HO-1 upregulation in brain astrocytes (Fig. 8B). These findings are the first evidence to demonstrate that BK induced HO-1 expression via a ROS-dependent Nrf2 pathway in astrocytes.
In conclusion, we have demonstrated that BK directly induces HO-1 expression via B2BKR and ROS-dependent activation of JNK/c-Jun and ERK/NF-κB, linking to upregulation of c-Fos/AP-1, which results in the promotion of HO enzymatic activity in RBA-1 cells. Moreover, ROS-dependent Nrf2 signaling also plays a crucial role in BK-mediated HO-1 expression in astrocytes. On the basis of the observations from literatures and our findings, Figure 8C depicts a model for the molecular mechanisms underlying BK-induced HO-1 expression and activity in RBA-1 cells. These findings concerning BK-induced HO-1 expression and activity in brain astrocytes imply that BK might play a critical role in the modulation of brain injuries and diseases. Pharmacological approaches suggest that targeting HO-1 and their upstream signaling components may provide useful therapeutic strategies for brain injury, tumor, and inflammatory diseases.
Footnotes
Acknowledgments
This work was supported by the National Science Council, Taiwan (grant numbers: NSC97-2321-B-182-007 and NSC98-2321-B182-004 [to C.M.Y.]; NSC96-2320-B-182-009 and NSC98-2320-B-255-001-MY3 [to H.L.H.]), and the Chang Gung Medical Research Foundation (grant numbers: CMRPD150313, CMRPD140253, CMRPD150253, and CMRPD180371 [to C.M.Y.]; CMRPF170022 [to H.L.H.]).
Author Disclosure Statement
No competing financial interests exist.