
Abstract
Small GTPases, including the proto-oncoprotein Ras and Rho GTPases, are involved in various cellular signaling events. Some of these small GTPases are redox sensitive, including Ras, Rho, Ran, Dexras1, and Rhes GTPases. Thus, the redox-mediated regulation of these GTPases often determines the course of their cellular signaling cascades. This article takes into consideration the application of Marcus theory to potential redox-based molecular mechanisms in the regulation of these redox-sensitive GTPases and the relevance of such mechanisms to a specific redox-sensitive motif. The discussion also takes into account various diseases, including cancers, heart, and neuronal disorders, that are often linked with the dysregulation of the redox signaling cascades associated with these redox-sensitive GTPases. Antioxid. Redox Signal. 14, 689–724.
Mechanistic Properties of Redox-Sensitive Small GTPases
Molecular mechanisms of the redox-mediated modulation of the activity of small GTPases
Chemical reaction process-based mechanism for NKCD or GXXXXGK(S/T)C motif-containing GTPases
Potential mechanism for the action of redox agents on GTPases that possess the CGNKXD motif
I. Introduction
A. Ras superfamily of small GTPases and their activity regulation
The Ras GTPase superfamily includes at least 14 subfamilies of small GTP-binding proteins (Fig. 1). These include Ras, Rho, and Ras-related nuclear protein (Ran) proteins that exist in eukaryotes found in all life forms from yeast to humans (59, 182, 231, 254) (Fig. 1).

1. Protein regulator-mediated activity modulation of Ras superfamily GTPases
These small GTPases function by cycling between active guanine nucleoside diphosphate (GDP)-bound and inactive GTP-bound states (Fig. 2) (74). As illustrated in Figure 2, guanine nucleotide-exchange factors increase the slow intrinsic rate of guanine-nucleotide exchange (GNE) to populate the proteins in their biologically active GTP-bound states (74, 135, 258, 259). The activated GTPases, in turn, interact with a variety of downstream effector proteins that modulate numerous cellular signaling processes, including cellular proliferation, actin cytoskeletal organization, and gene expression (74, 97). The signal is downregulated through the action of GTPase-activating proteins that increase the low intrinsic rate of GTP hydrolysis for most Ras family GTPases and thereby terminate downstream signaling events (Fig. 2) (74, 201, 259). The combined action of these regulators determines the relative proportion of active and inactive GTPases present in the cell. Protein regulators for Ras, Rab, Rho, and Ran proteins are known, but there are no known protein regulator(s) specific to dexamethasone-induced Ras protein1 (Dexras1) or Ras homolog enriched in striatum (Rhes).

2. Redox-mediated activity modulation of Ras superfamily GTPases
Many of these small GTPases are redox sensitive, and their known conserved redox-sensitive sequences have been termed the NKCD, GXXXXGK(S/T)C, and CGNKXD motifs (vide infra). The action of redox agents on these redox-sensitive GTPases is similar to that of guanine nucleotide-exchange factors in that they perturb GTPase nucleotide-binding interactions that result in the enhancement of the GNE of small GTPases.
a. Ras and repressor activator protein 1A GTPases and the NKCD motif
The product of the Harvey rat sarcoma virus oncogene (HRas) belongs to the Ras family of GTPases (Fig. 1). Other well-known members of Ras family GTPases include neuroblastoma Ras (NRas) and Kirsten Ras (KRas) (KRas4A/4B) (Fig. 1). These two KRas isomers are produced from the same gene by alternative splicing. These H, N, and KRas are closely related isoforms (Fig. 1). A subset of the Ras family GTPases also includes related Ras viral, Muscle Ras, and embryonic Ras (ERas) (Fig. 1). Although muscle Ras is closely related to H, N, and KRas (185), related Ras viral merely shares ∼55% sequence homology with H, K, and NRas (49, 248). ERas, sharing a 43% sequence homology with H, K, and NRas, is expressed in mice embryonic cells (272) as well as in certain human tumors (133, 308, 309). Small GTPases include several isoforms such as repressor activator protein 1 (Rap1)A and B as well as Rap2A and B that also belong to the Ras family GTPases (Fig. 1).
Rap1 (as well as its isomers Rap1A and Rap1B) belongs to the Ras family of GTPases. Rap1 has been shown in several cellular models to be involved in the regulation of integrin-mediated cell adhesion (146, 151, 230, 231). Notably, Rap1A is found to be colocalized in phagocytic cells with the superoxide anion radical (O2 •−)-producing enzyme NAD(P)H oxidase (33, 71, 172, 275).
Nearly all Ras superfamily of GTPases have a conserved sequence (I/L/V)(I/V)GNKXD in which two hydrophobic residues I (L or V) and I (or V) followed by a glycine precede the nucleotide base-binding NKXD motif. This glycine residue is conserved in all Ras superfamily GTPases (157). This redox-sensitive NKCD motif is contained within the Ras subfamily of GTPases such as H, N, K, and ERas as well as in Rap1A (Fig. 1).
b. Rho and Rab GTPases and the GXXXXGK(S/T)C motif
The Ras homology (Rho) family of small GTPases makes up a large branch of the Ras superfamily (Fig. 1) (93). The well-characterized family members are RhoA, Rac (Rac1 and its isoforms Rac2 and Rac3), and Cdc42 (290). The sequence homology scores within, but not between, the subfamily of GTPases are significantly high (>55%) (97).
Ras-associated binding (Rab) family GTPases, a subset of Ras superfamily GTPases, includes Rab1 (its isoforms: Rab1A and Rab1B), Rab3 (its isoforms: Rab3A, Rab3B, Rab3C, and Rab3D), Rab5, Rab7, and Rab25 (Fig. 1). Degrees of sequence homologies between Rab GTPases vary. Rab GTPases function via their specific effectors (128, 237, 264, 265) as regulators of distinct steps in membrane traffic pathways, including regulation of vesicle formation and movement. As with H, K, and NRas GTPases, misregulation of Rab GTPases results in development of a variety of cancers (40, 262).
Heo et al. have identified a new class of the redox-sensitive motif, the GXXXXGK(S/T)C motif, in certain Ras superfamily of GTPases (100). This redox-sensitive motif is an alteration of the glycine-rich phosphate-binding GXXXXGK(S/T) loop (P-loop; see Section II) in which an additional redox-sensitive cysteine (i.e., Cys18; Rac1 numbering) is present at the end of the P-loop C-terminus. The GXXXXGK(S/T)C motif is conserved in almost half of Rho family GTPases such as Rac1 (and its isoforms Rac2 and 3), Cdc42, and RhoA (and its isoforms RhoB and C) (100). A number of Rab proteins also have the GXXXXGK(S/T)C motif (e.g., Rab1B, Rab2, Rab2A/B, Rab4, Rab4A/B, Rab14, Rab15, Rab19, and Sec4) (Fig. 1). Intriguingly, many Rab GTPases (e.g., Rab1, Rab1A, Rab8, Rab8A/B, Rab10, and Rab13) possess both the NKCD and GXXXXGK(S/T)C motifs, whereas some Rab proteins—including Rab3 (e.g., Rab3, Rab3A/B/C/D, Rab7, Rab22, and Rab38)—possess only the NKCD motif (Fig. 1).
c. Ran, Dexras1, and Rhes GTPases and the CGNKXD motif
The Ran (or TC4) is a member of the Ras superfamily GTPases that is involved in nuclear transport, cell cycle control, mitotic spindle formation, and postmitotic nuclear assembly (45, 168). The distribution of active GTP-bound Ran provides important spatial information that directs cellular activities during different parts of the cell cycle (45). Ran regulates importin proteins (e.g., importin-α and -β) that bind nuclear localization and export signals to mediate nucleo-cytoplasmic trafficking (32).
Dexras1, a novel Ras-like GTPases, was named for its rapid induction after its treatment with the steroid dexamethasone (a glucocorticoid) in corticotroph cells (83). A physiological study suggests that Dexras1 may be involved in neuronal iron homeostasis (37) and may also participate in signal transduction pathways that in these cells govern the rapid regulatory effects of glucocorticoids on peptide hormone secretion (83). An in vivo study using Rhes (or Dexras2) knockout mice demonstrated that the Rhes GTPase is involved in selected striatal competencies, mainly in locomotor activity and motor coordination (257).
Ckless et al. first showed that Ran GTPase in mouse lung alveolar type II epithelial cells (C10) was S-nitrosated by S-nitrosoglutathione or S-nitroso-N-acetylpenicillamine (SNAP); however, this S-nitrosation site has not been identified (44). A follow-up in vitro study by Heo has identified an unprecedented CGNKXD motif that contains a redox-sensitive cysteine Cys120 in Ran protein (98). This study also shows that, in addition to this CGNKXD motif, Ran possesses an additional redox-sensitive cysteine Cys85 (Ran numbering) (98). This new type of redox center, the CGNKXD motif, also is conserved in Dexras1 and Rhes proteins (98) as well as in some Rab GTPases. Intriguingly, Dexras1 is redox sensitive, and its redox-sensitive residue has been identified as Cys11 (37, 83). This cysteine also is conserved in Rhes, but not in Ran GTPase (98).
B. Biologically relevant redox agents
Reactive nitrogen species (RNS) and reactive oxygen species (ROS) are known to serve as cellular redox signaling agents.
The protein cysteine side chain could exist in either thiol or thiolate (RS−). Thiol includes a cysteine (Cys-SH) or a glutathione (GSH), and RS− includes a cysteinate (Cys-S−) or a glutathiolate (GS−). One-electron oxidized forms of both Cys-SH and Cys-S− or GSH and GS− are a cysteinyl radical (Cys-S•) or a glutathionyl radical (GS•), respectively. The standard midpoint redox potential at pH 7 (E m 7) of Cys-SH or GSH, coupled with Cys-S• or GS•, is known to be ∼1.3 V (vs. normal hydrogen electrode [NHE]) (267). However, E m 7 of Cys-S− or GS−, coupled with Cys-S• or GS•, is known to be ∼0.8 V (vs. NHE) (267).
1. Reactive nitrogen species
The heme-containing enzyme nitric oxide synthase (NOS) catalyzes the oxidation of L-arginine to NO and L-citrulline via the intermediate N-hydroxyl-L-arginine (5, 70, 199). Several isoforms of NOS, such as neuronal NOS (nNOS or NOS1), inducible NOS (iNOS or NOS2), and endothelial NOS (eNOS or NOS3), are known (189, 205). Despite the overwhelming current interest in NOS proteins as NO-producing enzymes, several studies implicate xanthine oxidase in the production of NO (75, 191, 192, 315, 316).
a. Properties of RNS
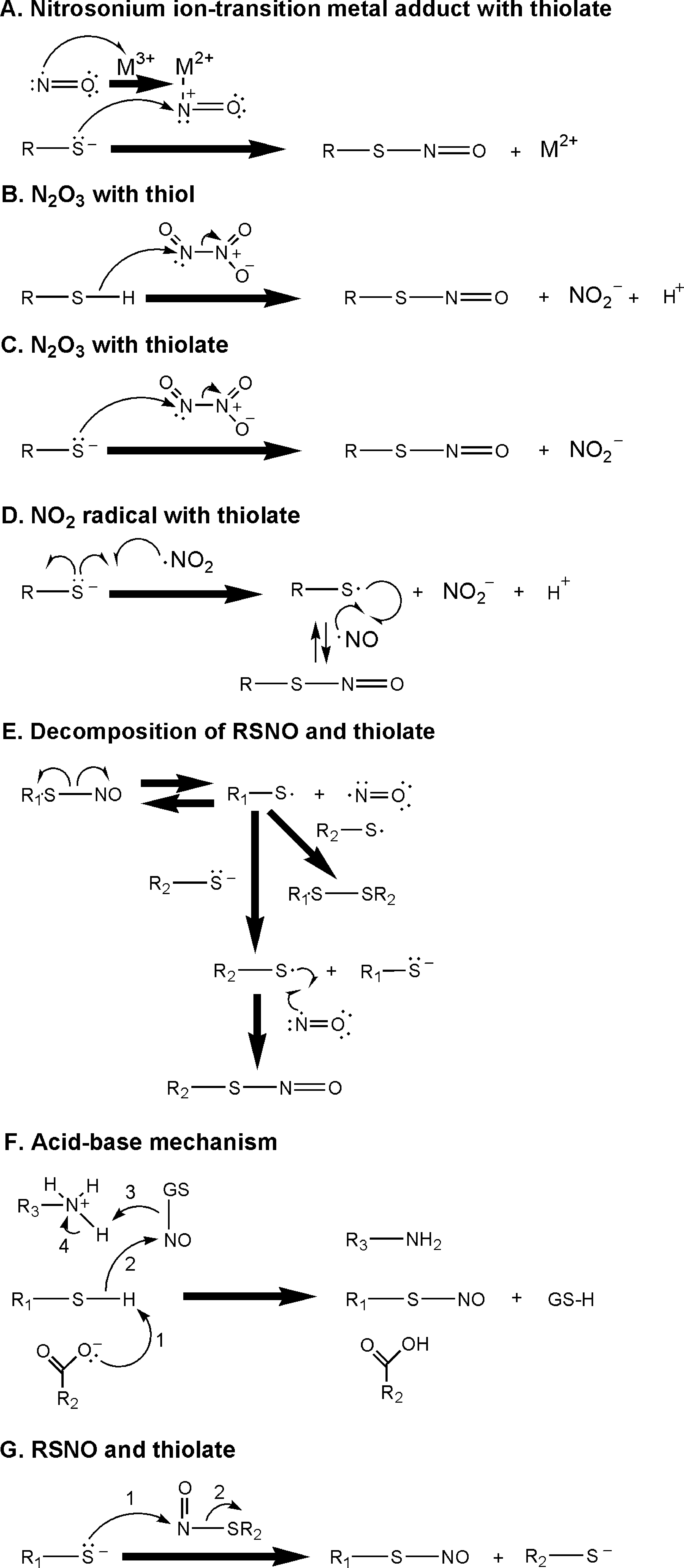
NO has an E m 7 value of ∼−1.7 V and ∼−0.8 V (vs. NHE), respectively, versus the one-electron reduced singlet excited and triplet ground state of the nitroxyl anion (NO−) (14). Other biologically significant RNS include a radical nitrogen dioxide (•NO2; E m 7 = ∼1.0 V vs. NHE) and nonradical higher oxides (e.g., dinitrogen trioxide [N2O3]) (6, 267). NO is a precursor of these various RNS because it can react with O2 to produce •NO2; the reaction of •NO2 with another NO results in production of higher oxides such as N2O3 (6). This N2O3 formation reaction is reversible because the formed N2O3 can be degraded into NO and •NO2 (6). Given that cysteine is the key and common residue of all of the redox-sensitive motifs found in these small GTPases, the mechanisms of the reactivities and chemical reactions of RNS with thiol/thiolate (Fig. 3) are particularly important to comprehending the RNS-mediated regulation of the activity of these GTPases.
2. Reactive oxygen species
ROS include the O2 •−, a hydroxyl radical (OH•), and a hydrogen peroxide (H2O2) (6). All of these ROS are produced by various metabolic processes as well as by cellular enzymes, including NAD(P)H oxidases, xanthine oxidase, and cyclooxygenases (6, 21, 108, 116, 234).
The O2 •−-generating multimeric enzyme complex NAD(P)H oxidase consists of a membrane-bound heterodimer of cytochrome b 558 with a large glycoprotein gp91 phox and a small protein p22 phox in complex with Rap1A (8, 26, 38, 73, 142, 238). Homologs of the gp91 phox subunit have been termed Nox (Nox isomers, including Nox1 and Nox2) and their occurrences differ in organs (155). The other cytosolic components of the NAD(P)H oxidase include p67 phox , p47 phox , p40 phox , and Rac1 (1). Xanthine oxidase is primarily considered the terminal enzyme of purine (e.g., hypoxanthine and xanthine) catabolism in the cell, catalyzing the hydroxylation of hypoxanthine to xanthine and xanthine to urate; the processes are coupled with reduction of O2 to O2 •− (94, 108, 187). Cyclooxygenases (also known as prostaglandin-endoperoxide synthases), including isoforms of a constitutively active Cox1 as well as inducible forms of Cox2 and Cox3, generate O2 •− (43, 68, 234). In addition, when the substrate L-arginine is not present, NOS produces O2 •− (222, 223, 301, 302, 311).
OH• has been shown to be formed in various subcellular organelles, including endoplasmic reticulum, mitochondria, nucleus, lysosomes, and peroxisomes (81, 140). H2O2 associated with Fenton chemistry is considered to be a main source of OH• in these organelles (vide infra).
a. Properties of ROS
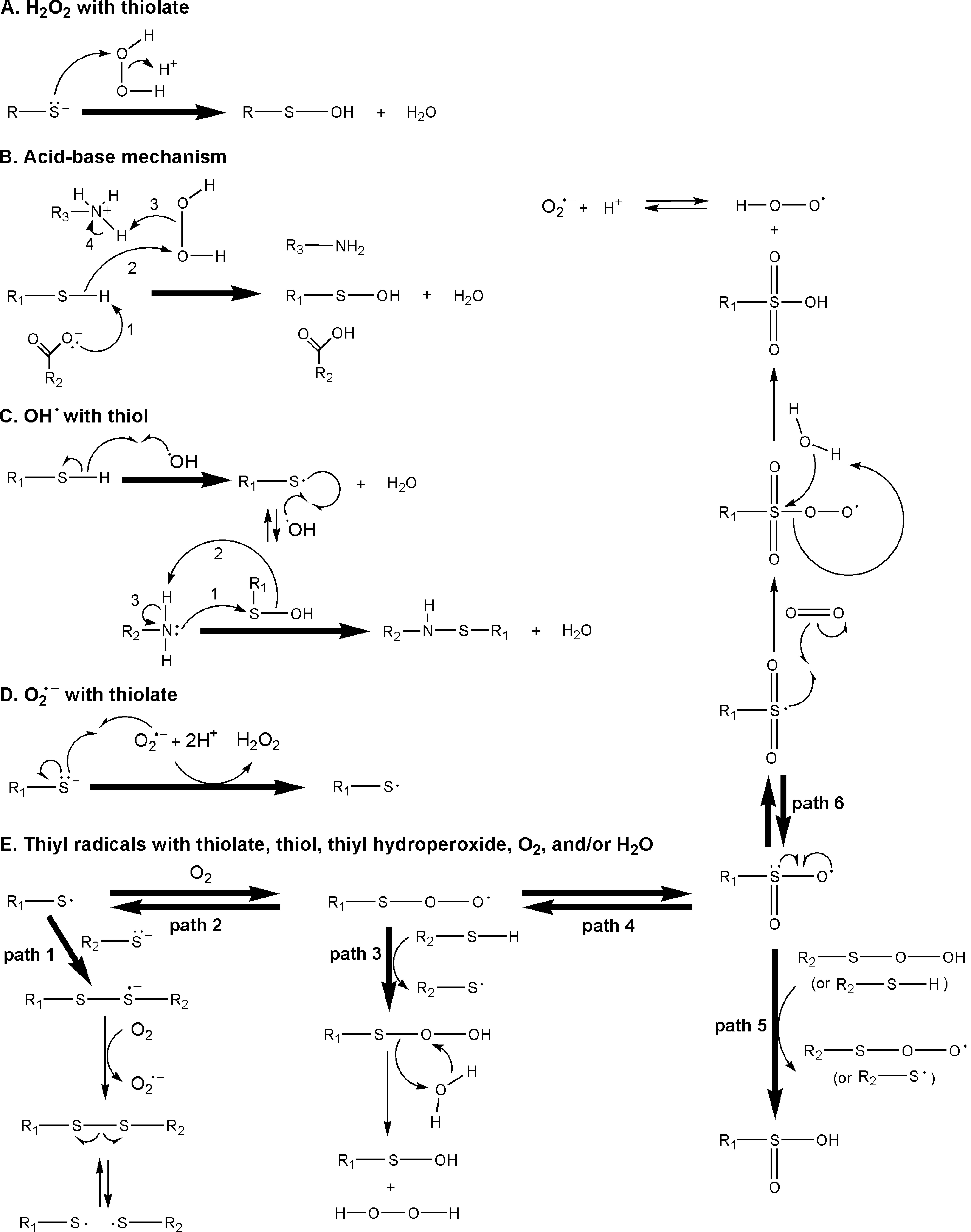
The E m 7 of O2 •− (∼0.9 V vs. NHE) is close to that of •NO2. However, the E m 7 of OH• (∼2.3 V vs. NHE) is much higher than other RNS and ROS (6). Some ROS are interconvertible; for example, H2O2 can be converted into OH− and into OH• when coupled with oxidation of a transition metal ion (e.g., Fe2+ to Fe3+) (91). Although this reaction is commonly referred to as a type of Fenton reaction, it was first proposed by Harber and Weiss (287). H2O2 can also be decomposed into H+ and a perhydroxyl radical (•OOH; a protonated form of O2 •−) when coupled with reduction of a transition metal ion (e.g., Fe3+ to Fe2+), and this process is termed a Fenton reaction (287). Given that the pK a of •OOH is ∼4.8 (20), •OOH can be deprotonated to O2 •− at a physiological pH (i.e., pH 7.4). A reverse process, generation of H2O2 from O2 •−, can be facilitated by superoxide dismutase (186) or the transition metal-catalyzed Harber-Weiss reaction (137). As with RNS, the reactivities and mechanisms of the reaction between ROS and thiol/thiolate (Fig. 4) must be comprehended to understand the redox-sensitive, motif-targeting, and ROS-mediated regulation of the activity of these GTPases.
3. Peroxynitrite
Although peroxynitrite (ONOO−) can be classified as an RNS, ONOO− is classified in a separate section for convenience. The nonradical RNS, ONOO−, can be formed by the reaction of NO with O2 •− (77). This is an example of the reactivity between RNS and ROS to produce another set of RNS and/or ROS. ONOO− is known to be a powerful and destructive oxidant (152, 233), most likely because of its high reduction potential (294). ONOO− can be decomposed into •NO2 and OH• (77). ONOO− can also react with CO2 to produce •NO2 and carbonate radical (CO3 •−) (E m 7 = ∼1.8 V vs. NHE) (22).
The E m 7 of CO3 •− predicts that CO3 •− can react with Cys-SH/GSH or Cys-S−/GS− to produce Cys-S• or GS•. This fuels speculation that CO3 •− has a role in cellular redox signaling via targeting cysteine-based redox-sensitive proteins. However, because the distribution of CO2 is limited in tissues and organs, the role of CO3 •− may be restricted or localized. Additional studies are required to examine what, if any, role CO3 •− has in cellular redox signaling cascades.
4. Cellular redox conditions associated with oxygen content
Because O2 often plays a role in the generation of and interconversion of biologically relevant RNS and ROS, the in vivo and in vitro studies of redox-mediated cellular signaling events also should take O2 content into account.
Hypoxia is a pathophysiological condition in which the whole body or a part of it is deprived of adequate O2. Hypoxia occurs because of a mismatch between the O2 supply and the demand for it at the cellular level (291). Unlike under normal conditions—the nonhypoxic condition—a typical experimental hypoxic condition is a low O2 concentration (typically ∼1–5% O2) in solution. O2 is still present in cells under a hypoxic condition, though in relatively low concentrations. However, although there is some debate about it and a possibility as well that what transpires depends on cellular compartments (e.g., endoplasmic reticulum, mitochondria, lysosomes, and peroxisomes) and cell types, hypoxic cells do not necessarily generate lesser ROS or RNS than cells at normal O2 concentrations. Anoxia, an extreme form of hypoxia, also is a pathophysiological condition in which an organ's tissues are deprived of O2. Therefore, the term “anoxic condition” is theoretically matched with the term “anaerobic condition,” which is defined as an absence of O2 during the course of in vitro experiments (99). The term “aerobic condition” refers to in vitro experimental conditions in an open atmosphere, which corresponds to a “nonhypoxic condition” in pathophysiology. Nevertheless, anoxia and hypoxia are often used interchangeably—without regard to their specific meanings—in pathophysiological conditions to describe a situation that occurs when there is a diminished supply of O2 to an organ's tissues. In this article, if necessary, usage of the terms “hypoxic” and “anoxic” is confined to in vivo experiments; the terms “anaerobic” and “aerobic” are used exclusively for in vitro experimental conditions.
II. Mechanistic Properties of Redox-Sensitive Small GTPases
Structural- and chemistry-based potential molecular mechanisms have been implicated in the activity modulation of the GTPases that contain the redox-sensitive NKCD, GXXXXGK(S/T)C, and CGNKXD motifs.
A. Structure of the redox-sensitive motifs of small GTPases
The nucleotide-binding regions of Ras superfamily GTPases include Switch I (residues 25–40; Ras numbering), which interacts with the nucleotide base, ribose, and γ-phosphate (in the GTP-bound form) (29, 66, 109, 115, 119, 190, 211, 276). Both the P-loop GXXXXGK(S/T) (residues 10–17; Ras numbering) and the Switch II-DXXG motif (residues 57–60; Ras numbering) of these proteins interact with the phosphate group of the bound nucleotide (Fig. 5) (29, 66, 109, 115, 119, 190, 211, 276). The NKXD motif (116 –119) (Ras numbering) of these small GTPases interacts with the guanine base of the bound nucleotide (29, 66, 109, 115, 119, 190, 211, 276).

1. Structural architecture of the bound nucleotide base and chemistry
Because of the presence of C6 oxygen on the purine ring, the guanine base has the highest number of conjugations among the purine (including adenine, guanine, and inosine) and pyrimidine (including cytosine, uracil, and thymine) bases. This ring conjugation complexity of the guanine base could endow it with the lowest redox potential of the bases. The E m 7 of the free guanine base (∼1.3 V vs. NHE) is lower than that of CO3 •− and OH• but higher than that of •NO2 and O2 •− (261). Also, because the stacking interactions of nucleotide bases in DNA are minimal, the redox potential of the guanine base in DNA is unperturbed (125). Thus, the DNA guanine base can be a target of CO3 •− and of OH• but not of •NO2 or O2 •−.
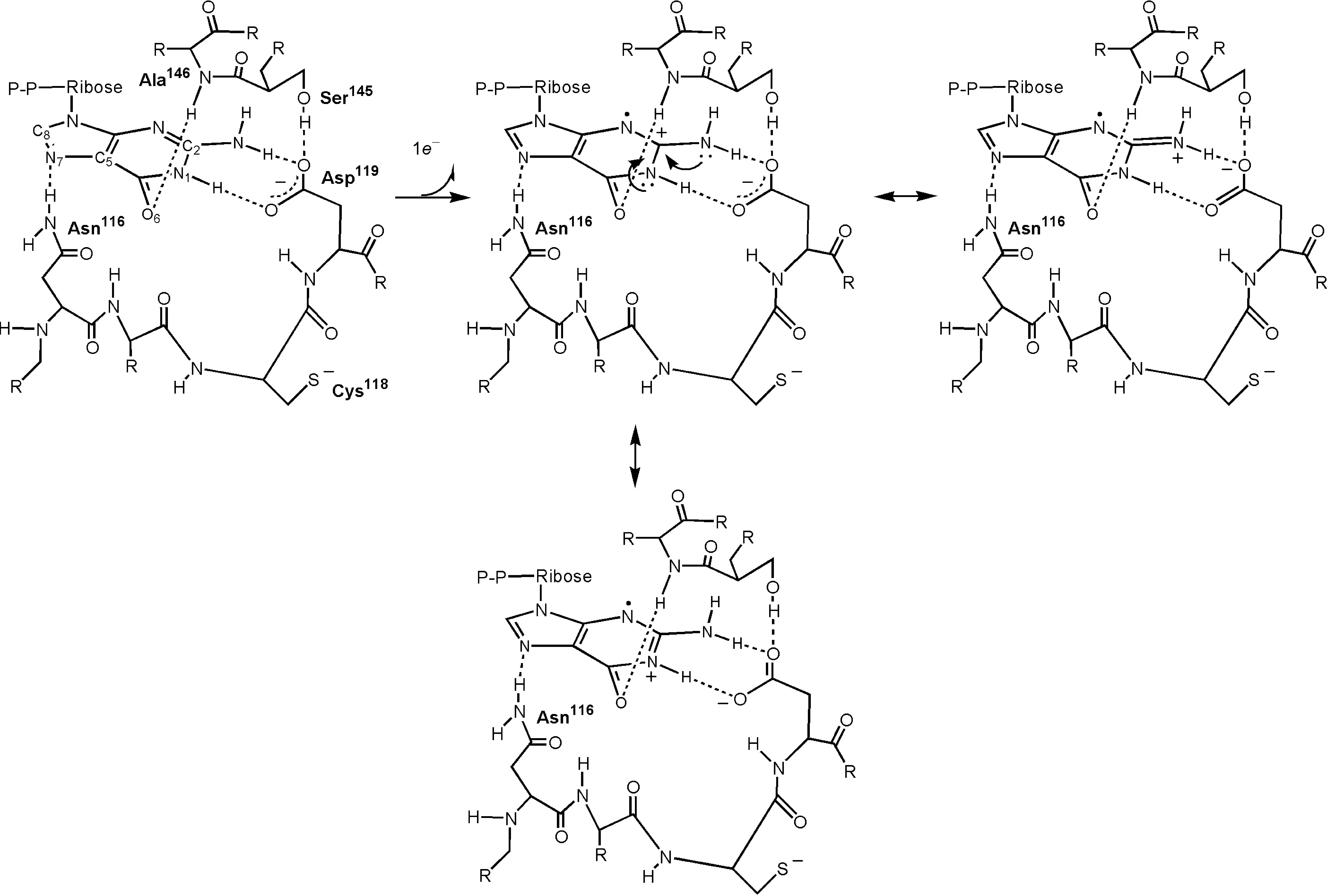
A guanine nucleotide (GDP or GTP) binds to small GTPases with a high affinity (Fig. 5) (106, 126, 127, 171, 229). The pH-dependent amide (NH) backbone chemical shift of Ras Asp119 in the NKXD motif was negligible, and the solvent exchange rates of the Ras Asp119 NH chemical shift associated with the pH change (from pH 5.9 to 8.0) were slow (103). The results suggest the presence of robust high affinity-binding interactions between the guanine nucleotide base N1-H and C2-NH2 and the Ras Asp119 carboxyl side chain. The mutagenesis of these residues consistently and significantly decreases the guanine nucleotide-binding affinity of Ras (47, 57, 65, 317). In addition, the hydrogen-bonding interaction between the bound nucleotide base C6-oxygen and the Ala146 NH of the SAK motif also plays a role in high affinity-binding interactions between Ras and guanine nucleotide (190, 211). An equivalent interaction also is observed in Rho GTPases (66, 109, 115).
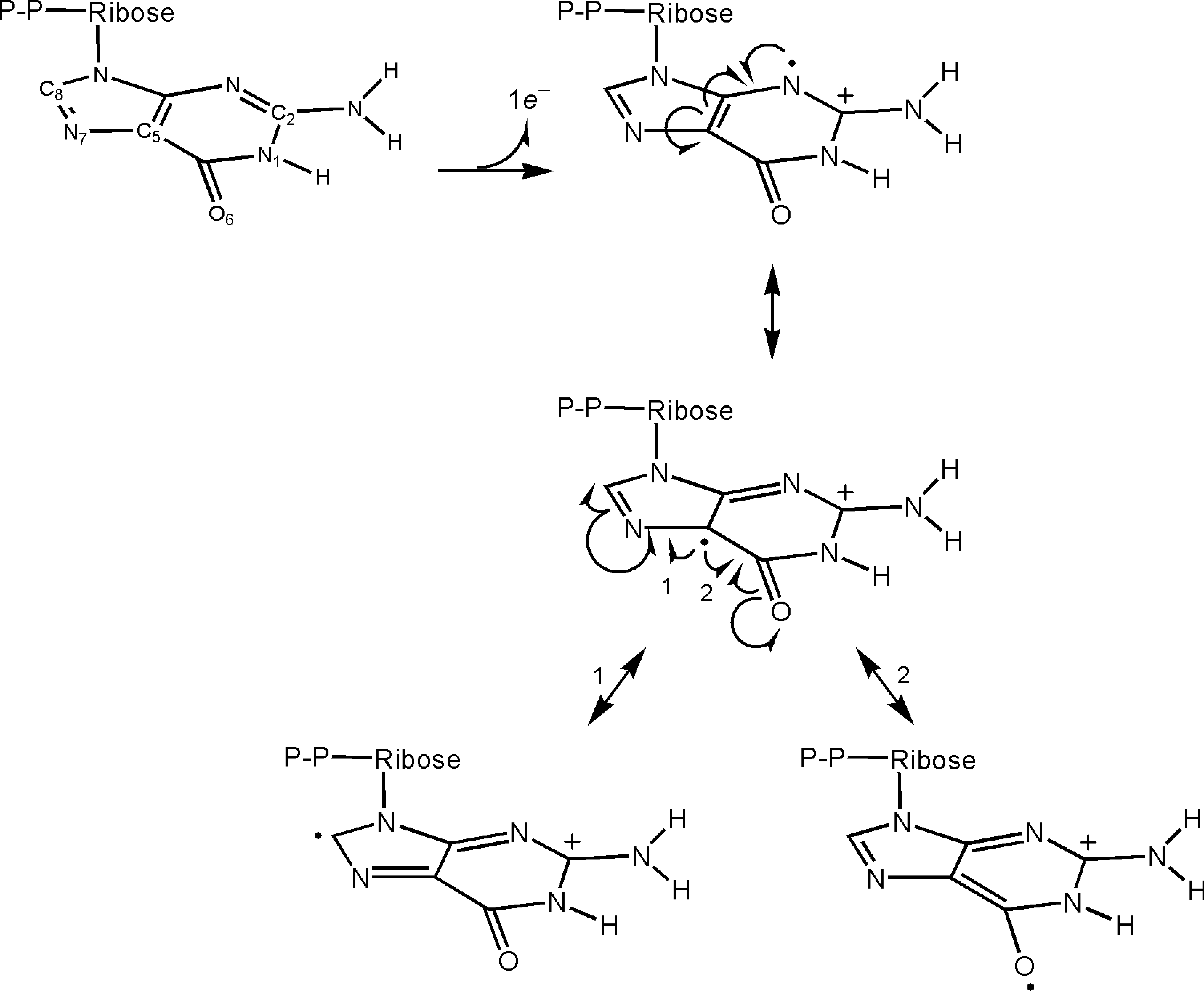
The high affinity-binding interactions between the guanine nucleotide base and the NKXD/SAK motif predict resonance states for one-electron oxidized form of the GDP base (a guanine nucleotide cationic radical [G•+-DP]) bound to small GTPases, including Ras, Rho, and Rab. When these high affinity-binding interactions are absent, the existence of a number of resonance states of G•+-DP can be expected (Fig. 6). However, introduction of the hydrogen bonds between the N1-H/C2-NH2 of the GDP base and the carboxy side chain of the aspartic acid of the NKCD motif (i.e., Asp119 Ras numbering) stabilizes several resonance forms of the bound GDP base (Fig. 7). Resonance stabilization carries the possibility of lowering the redox potential of molecules (i.e., the G•+-DP/GDP couple). These analyses suggest that the redox potential of the small GTPase-bound GDP coupled with G•+-DP is lower than that of the free GDP form or of DNA. The resonance state analysis with GDP can be immediately applicable to the one-electron oxidized form of GTP (i.e., G•+-TP). This is because the γ nucleotide phosphate with the p-loop binding interactions is unlikely to influence the resonance state of the nucleotide base.
2. Structural architecture of the region of the redox-sensitive motif of small GTPases and redox chemistry
Because the P-loop GXXXXGK(S/T) and the NKXD motifs play key roles in binding interactions between small GTPases and their ligand nucleotide, a mechanical perturbation of the binding interactions between these small GTPase motifs and a nucleotide is likely necessary to release the bound nucleotide (i.e., GDP).
a. Structural architecture of the region of the NKCD motif and its chemistry
For convenience, unless otherwise noted, Ras numbering is used solely for structural-based motif analyses. The spatial arrangement of the residues of the NKCD motif associated with the bound nucleotide is virtually identical in all NKCD motif-containing proteins such as Ras and Rap1 GTPases (Fig. 5) (39, 190, 211). Also, a certain aromatic residue, such as phenylalanine (i.e., Phe28), does not belong to the NKCD motif, but nevertheless plays a role in the redox-mediated modulation of Ras activity (104). This Phe28 is largely conserved in the NKCD motif-containing proteins. Although a tyrosine near the site (Tyr141) is not well conserved, it also has been thought to be involved in the redox response of Ras GTPases. The potential redox role and function of Tyr141 remain unclear and thus require investigation.
1. Redox-sensitive cysteine in the NKCD motif
Because the solvent-exposed sulfur atom of the Cys118 side chain in the NKC118D motif is vicinal to several electron-withdrawing oxygen atoms of protein residues such as Glu143 and Thr144 (190, 211), the pK a value of the Cys118 side chain is likely to be smaller than that of Cys-SH and GSH (pK a = ∼8.3). This analysis suggests that under the physiological pH (e.g., pH 7.4), the Cys118 side chain is likely to be presented, at least in part, as a form of RS−. If so, because the redox potential of RS•/RS− is lower than that of the •NO2/NO2 − couple (267), the Ras Cys118 thiolate side chain (Ras Cys-S−) is a suitable target for •NO2. Because the redox potential of O2 •− is similar to that of •NO2 (267), this Ras Cys-S− also can be a target of O2 •−. Also, the nucleophilicity of the sulfur atom of Ras Cys-S− is likely to be diminished because the lone pairs of electrons on the sulfur atom are likely to be occupied by the interaction with these multiple electron-withdrawing groups (vide supra). This analysis does not support the potential Ras S-nitrosation (Ras-SNO) mechanisms suggested in Figure 3A and C, in which a nucleophilic attack by a lone pair of electrons of the sulfur atom of a cysteine residue toward the nitrogen atom either of a metal nitrosonium ion adduct or of N2O3 will be blocked. This analysis also excludes the possibility of nonradical-based formation of Ras Cys118 sulfenic acid via a reaction between Ras Cys-S− and H2O2, because the lone pair of electrons of the sulfur atom that is necessary for a nucleophilic attack toward H2O2 to produce Ras Cys118 sulfenic acid is unavailable (Fig. 4A).
2. Phenylalanine vicinal to the NKCD motif
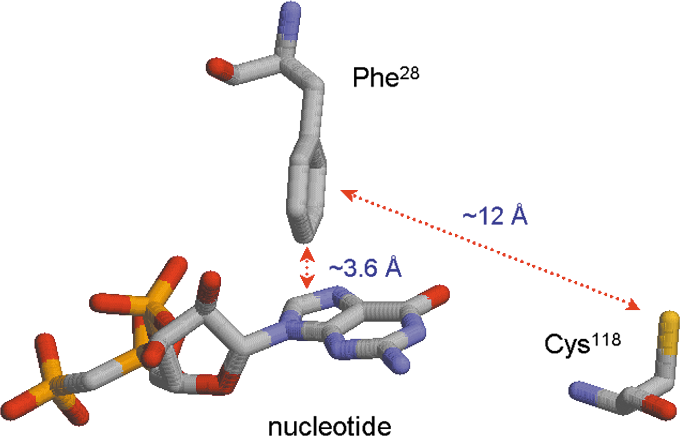
A Ras Switch I residue, Phe28, is conserved in nearly all other small GTPases. The distance between the phenyl group para position (the C4 carbon atom) of Phe28 side chain and the center of Ras-bound GDP guanine base is ∼3.6 Å (Fig. 5) (29, 119, 190, 211, 244, 276). The perpendicular interface between the Phe28 phenyl side chain and the guanine base results in a n-π stacking interaction (Fig. 5) (29, 39, 119, 188, 190, 211, 244, 276, 318).
The close n-π interaction leads to a suspicion that the plus charge on the para position of the phenyl side chain is close to the π electron-enriched guanine side chain, thereby stabilizing one of the cationic radical resonance states of the Phe28 phenyl side chain (Fig. 8A). Moreover, because of this cationic radical resonance stabilization, a reduction can be expected in the redox potential of the Phe28 phenyl side chain (coupled with the cationic radical form of Phe28 side chain). Alternatively, a resonance state with a positive charge at the meta position, and thus a radical at the para position, was postulated as present (Fig. 8B) (104). However, a radical at the para position of the Phe28 side chain could clash with the π electrons of the guanine base. This electronic state is therefore likely to be less favorable than that of a plus charge at the para position (Fig. 8A). This analysis corrects the misstatement in the previous suggestion (104) as following: Because the radical on the para position of the Phe28 side chain (the C4 atom of the phenyl side chain) associated with the bound guanine base is in an unfavorable state, the proposed spin transition (sp2 → sp3) of the C4 atom Phe28 side chain is likely to be forbidden.

The spatial arrangement among the Phe28 side chain, the redox-sensitive Cys118 side chain, and the bound nucleotide guanine base also is intriguing (Fig. 9). The phenyl side chain of the Phe28 faces the sulfur atom of the Cys118 side chain at an angle of 0°–5°, and no residue is present between these two side chains. The distance between the center of the Phe28 phenyl side chain and the sulfur atom of the Cys118 is ∼12 Å (Fig. 9) (190, 211), which minimizes ionic, hydrogen-bonding, and hydrophobic interactions. However, this distance (∼12 Å) does not limit electron transport between two atoms (210).
b. Structural architecture of the GXXXXGK(S/T)C motif and chemistry
The geometry of the nucleotide-binding site of Rho and Rab family proteins that have the GXXXXGK(S/T)C motif is all but identical to that of the Ras family GTPases. This suggests that a high affinity binding interaction also is present between Rho GTPases and the bound guanine nucleotide. Therefore, as with the Ras-bound guanine nucleotide, the redox potential of the bound guanine nucleotide base to small GTPases that possess the GXXXXGK(S/T)C motif is likely to be lowered to that of the free nucleotide base. One exception to their similarity is that an additional redox-sensitive cysteine is located in the nucleotide-binding site of these Rho and Rab family GTPases. However, as noted elsewhere, certain Rab proteins possess both the NKCD and GXXXXGK(S/T)C motifs. It is also noticeable that in addition to the redox-sensitive cysteine in the GXXXXGK(S/T)C motif, a secondary redox-sensitive cysteine is present in certain Rho family GTPases (vide infra).
Besides the GXXXXGK(S/T)C motif, Ras Phe28 is also conserved in GTPases that contain the GXXXXGK(S/T)C motif in the forms of Rac1 Phe28 (same as Ras numbering).
1. Redox-sensitive cysteine(s) associated with the GXXXXGK(S/T)C motif
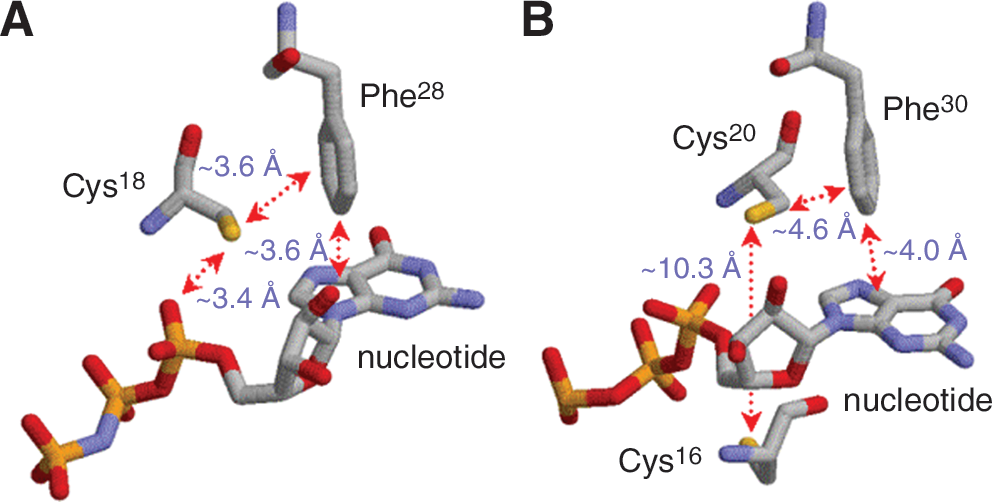
Similar to the redox-sensitive Cys118 side chain in Ras proteins, the sulfur atom of the side chain of the redox-sensitive cysteine (Cys18; Rac1 numbering) in the GXXXXGK(S/T)C motif is accessible by solvent and located in an acidic environment (109). With only slight distance variations, all of these interactions with the sulfur atom of the cysteine in the GXXXXGK(S/T)C motif are well conserved in other Rho GTPases such as RhoA and Cdc42 (66, 115). The structural architecture suggests that, as with the sulfur atom of the Ras Cys118 side chain, the sulfur atom of the side chain of the cysteine in the GXXXXGK(S/T)C motif is likely present as an anionic thiolate form (Cys18 thiolate). Hence, this acidic environment makes the sulfur atom of the Rac1 Cys18 side chain susceptible to redox agents such as •NO2 and O2 •−. A subset of Rho family GTPases, such as RhoA, RhoB, and RhoC, has additional cysteines, Cys16, Cys63, and Cys159 (RhoA numbering; they are not present in Rac1 and Cdc42), close to the redox-sensitive Cys20 (RhoA numbering; equivalent to Rac1 Cys18) in the GXXXXGK(S/T)C motif (115). Among them, the action of one cysteine residue, Cys16 (not present in Rac1 and Cdc42), is characterized as a redox sensitive residue in vitro (105).
2. Phenylalanine vicinal to the GXXXXGK(S/T)C motif
Phe28 (Rac1 numbering is the same as Ras numbering) is well conserved throughout all GXXXXGK(S/T)C motif-containing proteins. The Rac1 Phe28 phenyl side chain also is poised perpendicularly with the bound guanine nucleotide base at a distance of ∼3.6 Å (Fig. 10A). The sulfur atom of the Rac1 Cys18 side chain faces the center of the Phe28 side chain but lies a remote ∼3.6 Å from it (Fig. 10A) (109), whereas the Ras Cys118 side chain is a remote ∼12 Å from the center of the Phe28 side chain (Fig. 9).
c. Structural architecture of the CGNKXD motif and chemistry
To date, the structures of Dexras1 and Rhes GTPases are not available. However, a Ran X-ray crystal structure has been reported (266). As with the NKCD motif, the CGNKXD motif also is a subset of the small GTPases (I/L/V)(I/V)GNKXD motif. However, the structural architecture of the CGNKXD motif (266) differs completely from that of the NKCD motif (190, 211). In addition, unusually stacked phenylalanines are found in GTPases that contain the CGNKXD motif and may play a redox role in Ran, Dexras1, and Rhes proteins (98).
1. Redox-sensitive cysteines associated with the CGNKXD motif
The Ran crystal structure (266) has revealed that the sulfur atom of the Cys120 side chain in the C120GNKXD motif can be accessed from the outside. This also suggests that the redox-sensitive Cys120 side chain in the C120GNKXD motif can be a direct target of redox agents. Although remote from the CGNKXD motif, Ran has another redox-sensitive cysteine Cys85 (98). The sulfur atom of the Cys85 side chain is exposed to solvent and close to several oxygen atoms of Ran residues, including Lys12 and Gln84 (266). However, only a few functional groups interact with the Cys120 side chain (266). Hence, the sulfur atom of the Cys85 side chain is likely to be poised in a relatively acidic environment. This environment, in turn, encourages speculation that the redox potential of the Cys85 side chain is less than that of the Cys120 side chain. Also, the sulfur atom of the cysteine Cys120 side chain is ∼11.8 Å from the sulfur atom of the side chain of Cys85 (266). Intriguingly, no residue is present between these sulfur atoms of the cysteine Cys120 and Cys85 side chains.
2. Multiple packed phenylalanines
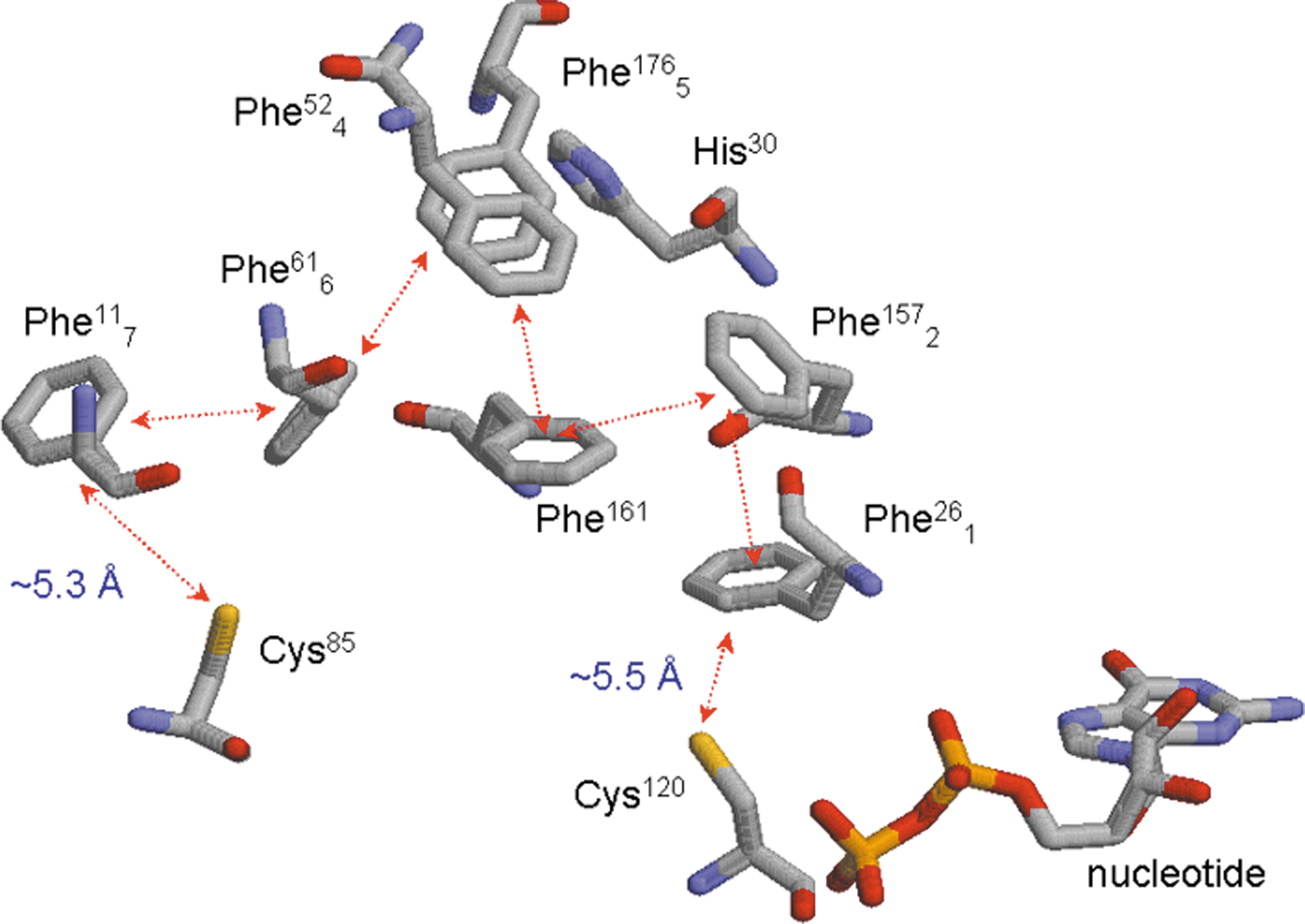
Notably, the Ras and Rho GTPase Phe28 side chain interacts perpendicularly with the bound-GDP guanine base that plays a role in Ras nucleotide-binding interactions (104). However, the Ran Phe26 (sequentially equivalent to the Ras Phe28) phenyl side chain is located away from the bound-GDP guanine base (Fig. 11) (266). Unlike the Ras, Rho, and Rab proteins, the Ran GTPase has an unusually high number of phenylalanines (Fig. 11). Uniquely, in an arrangement that also includes this Ran Phe26 side chain, a series of Ran phenylalanine residues are stacked one with another (Fig. 11). These stacked Ran phenylalanine residues are present between Ran Cys120 and Cys85 side chains: (i) The Phe26 side chain is vicinal to the redox-sensitive Cys120 side chain in the CGNKXD motif at a distance of ∼5.5 Å; (ii) this Phe26 side chain also packs against the Phe157 side chain in a distorted coplanar arrangement; (iii) the Phe157 side chain packs against the side chain of Phe161, which then faces another Ran phenylalanine residue, the Phe52 side chain; (iv) this Phe52 side chain stacks with the side chain of Phe176, which also faces the side chain of Phe61; (v) the Phe61 side chain packs against the Phe11 side chain; and (vi) finally the Phe11 side chain faces a sulfur atom of the side chain of Cys85, another redox-sensitive Ran cysteine. Including Ran Phe26, the multiple-stacked Ran phenylalanines are termed herein as a Ran aromatic cluster, which consists of seven phenylalanines. For convenience, the seven phenylalanines in the Ran aromatic cluster are numbered from 1 to 7 as Phe26 1, Phe157 2, Phe161 3, Phe52 4, Phe176 5, Phe61 6, and Phe11 7 (Fig. 11). The average distance between each Ran phenylalanine residue side chain is ∼5 Å. A sequence analysis suggests that although some Ran phenylalanines are matched to tyrosines, the residues assigned for the Ran aromatic cluster are virtually conserved in Dexras1 and Rhes proteins (98). Considering the structural features of the Ran aromatic-stacking cluster (Fig. 11) in conjunction with the packing distances, the main force held in the stacked-aromatic cluster is likely to be a stacked π − π interaction. In fact, such a π − π stacking interaction has been observed in hemoglobin in which the Tyr24 and His20 side chains are contained in a coplanar arrangement within an hemoglobin α chain pack at a distance of 4.8 Å (58).
3. Structural relevance of Ran GTPases redox motifs with respect to Dexras1 and Rhes GTPases
Studies have identified a redox-sensitive cysteine, Cys11 (Dexras1 numbering; equivalent to Rhes Cys10), in Dexras1 (63, 120). A sequence analysis shows that both Dexras1 and Rhes GTPases possess the CGNKXD motif (98). However, the composition of the aromatic cluster of Dexras1 and Rhes proteins differs slightly from that of the Ran aromatic cluster (98): Many tyrosines, in lieu of phenylalanines, are present in the Dexras1 and Rhes GTPases. The structures of Dexras1 and Rhes are unknown. However, given that the Dexras1 and Rhes protein sequences are similar to that of Ran GTPase (98), the structure of Dexras1 and Rhes can be predicted by using SWISS-MODEL (88, 246) with the Ran PDB coordinate 1BYU as a reference (266). A comparison of the Ran crystal structure and the SWISS-MODEL analysis for Dexras1/Rhes further suggests that: (i) The putative redox-sensitive Dexras1 Cys11 side chain (same as the Rhes Cys10 side chain) is structurally equivalent to the Ran Cys85 side chain; (ii) the Ran aromatic-stacked cluster also is conserved, with some variations, in Dexras1 and Rhes GTPases; and (iii) the order of the packed-aromatic cluster of Dexras1 and Rhes proteins does not completely match with Ran.
B. Molecular mechanisms of the redox-mediated modulation of the activity of small GTPases
One potentially attractive model mechanism to explain the action of redox agents on redox-sensitive small GTPases is that the conformation of the nucleotide-binding site of small GTPase is altered by the end product of the reaction between a redox agent and the redox-sensitive residue of GTPases; the result is the release of the bound nucleotide from small GTPases. Another possible mechanism is that the chemical reaction process between a redox agent and the redox-sensitive residue of GTPase perturbs Ras nucleotide binding interactions. In this explanation, this perturbation causes dissociation of the bound nucleotide from small GTPases.
1. Redox-induced chemical modification-based protein conformational change mechanisms for the NKCD motif-containing GTPases
The redox-sensitive Ras NKCD motif was discovered before identification of the GXXXXGK(S/T)C and CGNKXD motifs. Consequently, the first attempt to suggest a redox regulation mechanism for GTPases was made for a Ras protein that contains the NKCD motif. Because the redox architecture of GTPases is virtually identical within its motif type (i.e., the NKCD motif-containing GTPases), the mechanism proposed for the Ras protein can be applied to other proteins that contain the NKCD motif.
a. Conformational change-induced activation mechanisms for the NKCD motif-containing GTPases
Although the biologically relevant chemical reaction mechanism responsible for the formation of Ras-SNO is still being debated (see Section II), the basic idea for the mechanism of redox-dependent Ras activation is rooted in detection of the Ras-SNO adduct coupled with observation of the Ras-dependent upregulation of cellular signaling events (156, 161).
The key implication of the redox-induced protein conformational change mechanism is that a reaction between the redox-sensitive cysteine of Ras and NO forms a protein-chemical adduct such as Ras-SNO. This adduct then changes the conformation of the GTPase protein. This protein possessing the SNO moiety is thought to have, at a minimum, an altered local structural feature that enhances its preference for binding with a ligand GTP, but not GDP, to populate an active form of GTPase or interact favorably with its effector proteins. However, several factors of this potential mechanism are matters of concern and should be considered (vide infra).
b. Kinetic and structural features of Ras-SNO
Ras-SNO and non-S-nitrosated Ras have equal binding affinities for guanine nucleotide (101). It is also possible that the NO moiety of Ras-SNO mimics an active form of Ras in cells so that Ras downstream effector proteins recognize Ras-SNO as an active Ras. However, the Ras-SNO binding interaction with the Ras binding domain of Raf does not differ significantly from that of the non-S-nitrosated Ras (101). These analytical results indicate that the kinetics of Ras-SNO associated with its ligand and binding partner do not differ from that of unmodified Ras.
Under aerobic conditions, NO induces change in Ras circular dichroism (CD) spectra that couples with activation of Ras and formation of Ras SNO (158). This result opens the possibility that the state of the Ras-SNO formation via an unknown chemical process perturbs the Ras structure, which in turn results in enhancement of Ras GNE. However, a comparative structural analysis using NMR spectroscopy finds no significant structural difference between Ras-SNO and unmodified Ras (293), eliminating the possibility that the change in Ras conformation associated with the formation of Ras-SNO is a driving force for modulation of Ras activity. However, the NO/O2-mediated change in Ras CD spectra remains to be explained. One possible explanation for this Ras CD result could be that, without alteration of the Ras structure, the process of the Ras-SNO formation somehow perturbs the Ras nucleotide-binding interactions, resulting in release of the nucleotide from Ras. Although the nucleotide-deficient Ras is unstable, the structure of the nucleotide-deficient Ras differs from that of the nucleotide-bound Ras. Hence, although temporal, the NO/O2-mediated dissociation of the bound nucleotide results in the alteration of the Ras structure that was detected in the CD analysis.
c. Kinetic properties of Ras-SSG, as well as Ras-SOH or Ras-SO2H
Intriguingly, ROS-mediated formation of Ras cysteine-glutathione disulfide (Cys-SSG), a distinct form of modification of the Ras NKCD motif, can be coupled with Ras activation in cells (see Section III). Also, oxidized forms of Ras, Ras-SOH or Ras-SO2H, also can be found “as purified Ras from the E. coli expression” (101). However, the binding properties of Ras Cys-SSG, Ras-SOH, or Ras-SO2H with a guanine nucleotide or a Ras effector protein Raf do not differ significantly from that of the reaction of unmodified Ras with a guanine nucleotide or Raf (unpublished data). The results suggest that kinetic properties of Ras with different chemical adducts, such as Ras Cys-SSG, do not significantly differ from that of the unmodified Ras.
Although further studies of these mechanisms are required, these kinetic and structural analysis results devalue the potential role and function of the state of Ras-SNO or the state of Ras Cys-SOH/Cys-SO2H in redox-mediated Ras activation.
2. Chemical reaction process-based mechanism for NKCD or GXXXXGK(S/T)C motif-containing GTPases
An in vitro kinetic analysis shows that O2 is essentially required for NO-mediated Ras nucleotide dissociation (104). Because NO reacts with O2 to produce •NO2, this result was interpreted to mean that •NO2 is the active redox agent for the facilitation of the dissociation of nucleotide from Ras. This conclusion was further confirmed by using authentic •NO2 gas that facilitates Ras nucleotide dissociation in the absence of O2 (101). N2O3, which is another NO reaction product with O2, was eliminated from the list of possibly active Ras-targeting redox agents because N2O3-mediated Ras nucleotide dissociation, under anaerobic conditions, was minimal (99, 104). The action of the NO/O2 mixture (containing the active redox agent •NO2) on the Ras mutant C118S was ineffective, suggesting that Ras Cys118 in an NKCD motif is the target site for •NO2. Notably, •NO2 reacts with the sulfur atom of Cys-S− or GS−, but not with Cys-SH or GSH, respectively, to produce Cys-S• or GS• (see Section I). Hence, identification of •NO2 as an agent that targets the Ras NKCD motif suggests that Ras Cys118 thiyl radical (Cys118-S•) is likely to be formed by the reaction between •NO2 and Ras Cys118-S−. A prediction on the formation of Ras Cys118-S• also is consistent with the Ras structural analysis and suggests a hypothesis that the Ras Cys118 side chain is in a deprotonated state (see Section II). However, this analysis leads one to correct the previous statement that the state of Cys118-S−, but not the Cys118-SH state, is a target of •NO2 (99, 104). This analysis also leads to a hypothesis that the action of the •NO2-mediated Ras nucleotide dissociation is associated with the formation of Ras Cys118-S• and thus is a radical-based process.
An unexpected but important factor to consider for the redox regulation mechanism of Ras activity is that a chemically modified nucleotide 5-guanidino-4-nitroimidazole ribose diphosphate (NIm-DP) or a product of 5-oxo-GDP degradation, but not an intact nucleotide, was detected from the Ras sample treated with NO/O2 or O2 •−, respectively (101, 102, 104). The result indicates that the bound nucleotide base is involved in the dissociation process of the redox-mediated Ras nucleotide. The redox potential of a free guanine base, coupled with G•+-DP, is ∼1.3 V (vs. NHE) (261). Only if the redox potential of the bound GDP base is lower than that of Ras Cys118-S•/Ras Cys118-S−, can Ras Cys118-S• abstract an electron from the bound GDP base to produce Ras Cys118-S− and G•+-DP. Therefore, as analyzed in Section II, it can be postulated that the tight hydrogen bonding interactions between the bound nucleotide base and the NKCD/SAK residues lower the redox potential of the bound nucleotide base below the redox potential of the sulfur atom of the Cys118 side chain.
The sulfur atom of the Cys118 side chain does not face the nucleotide base (Fig. 9) (29, 119, 211, 244, 276). Moreover, the Asp119 carboxy side chain lies between the sulfur atom of the Cys118 side chain and the bound nucleotide base (Fig. 9). These structural architectures suggest that, although oxidation of the bound nucleotide base coupled with a reduction of Ras Cys118-S• is feasible in terms of redox potential, a direct electron transport from the bound nucleotide base to the sulfur atom of the Cys118 side chain is impossible. Hence, an indirect interaction between the bound nucleotide base and the formed Ras Cys118-S• also requires consideration. As analyzed in Section II, the Phe28 phenyl side chain interacts perpendicularly with the bound nucleotide base and faces the sulfur atom of the Cys118 side chain (Fig. 9). Therefore, the Phe28 phenyl side chain is likely a mediator of a possible electronic interaction between the bound nucleotide base and Ras Cys118-S•. Ras-released NIm-DP was not detected from the NO/O2-treated mutant Ras F28L, which lacks the Phe28 side chain (Ras F28L) (104). The result of the mutant study, in conjunction with the structure of the Phe28 phenyl side chain in the NKCD motif-containing GTPase, suggests a hypothesis that the π electron-enriched Phe28 phenyl side chain serves as a conduit for transfer of an electron from the bound nucleotide base to Ras Cys118-S• to produce G•+-DP and Ras Cys118-S−.
a. RNS-mediated molecular mechanism of the NKCD motif-containing GTPases
On the basis of the results of these several studies and analyses (vide supra), Heo et al. have proposed a distinctive radical-based molecular mechanism to explain RNS-mediated Ras activity (Fig. 12): A reaction of the solvent-exposed sulfur atom of Ras Cys118-S− with •NO2 produces Ras Cys118-S• (Fig. 12, Step 1). The Ras Cys118-S• formed in this reaction abstracts an electron from the bound nucleotide base via the Phe28 side chain to produce Ras Cys118-S− and G•+-DP (Fig. 12, Steps 2 and 3). On the basis of the resonance stabilization analysis in Section II, a cationic radical intermediate state is proposed for the Phe28 side chain in which a plus charge and a radical, respectively, are localized at the para and meta positions of the Phe28 side chain. As with the DNA cation radical G•+ (125, 194, 251), release of a proton leads to production of a relatively stable form of G•-DP (Fig. 12, Steps 4, 5, and 6). Another •NO2 reacts with G•-DP to produce a GDP-NO2 adduct (Fig. 12, Step 7). Formation of G•-DP perturbs the hydrogen bonding interaction between the Ras NKCD/SAK motif and the bound nucleotide base. This perturbation results in release of the bound nucleotide as a form of GDP-NO2 and production of the nucleotide-deficient Ras that has Cys118-S−. As with the guanine-NO2 adduct in DNA (87, 206), the free GDP-NO2 is hydrated and then decarboxylated to produce NIm-DP (Fig. 12, Steps 8, 9, and 10). The nucleotide-deficient Ras that is formed, in turn, binds with abundant GTP to produce a GTP-bound active Ras in cells. However, if GDP, instead of GTP, is populated, the active form of Ras will not be formed. Hence, this radical-based molecular mechanism does not dictate the state of active or inactive Ras but only facilitates nucleotide dissociation from Ras.

A certain ROS, O2 •−, also enhances wild-type (wt) Ras nucleotide dissociation (101, 102). Like •NO2, O2 •− can react with the sulfur atom of Cys-S− or GS−, but not with either Cys-SH or GSH, to produce Cys-S• or GS• (see Section I). As with •NO2, the O2 •−-mediated Ras C118S nucleotide dissociation was limited, suggesting that the Ras Cys118-S− also is a target of O2 •− to produce Ras Cys118-S•. Because this Ras Cys118-S• is the same radical species that is generated by •NO2 with Ras, the O2 •−-mediated Ras activity modulation also is likely to be based on the radical Ras Cys118-S•.
b. Radical-based chemical modification of the redox-sensitive cysteine in the NKCD motif of Ras proteins
The presence of a radical-based molecular mechanism leads to a prediction that a nucleotide-deficient Ras possessing Cys118-S− and free GDP-NO2 will be formed upon treatment of Ras with •NO2 (100). There is a possibility that •NO2 targets Cys118-S− of the nucleotide-deficient Ras before it binds with the nucleotide; however, the factors that make such an event appear possible may hinge on the Ras GNE rate. Also, if GDP is depleted but an excess amount of •NO2 is present, •NO2 reacts with Cys118-S− of the nucleotide-deficient Ras to produce a nucleotide-deficient Ras Cys118-S•. The Cys118-S• on the nucleotide-deficient Ras cannot be reduced immediately because the electron donor, which is the bound nucleotide base, is absent from the nucleotide-deficient Ras. The Cys118-S• on the nucleotide-deficient Ras may then react with NO, if NO is present, to produce a nucleotide-deficient Ras-SNO. This radical-based Ras-SNO formation mechanism is a process analogous to the formation of RSNO by the reaction between an RS• and NO (see Section I). When a nucleotide (e.g., GTP) is present, the nucleotide-deficient Ras-SNO then binds with a nucleotide to produce a stable nucleotide-bound Ras-SNO. However, this O2-dependent (if NO is the initial starting redox agent) radical-based S-nitrosation process (99, 104) may not be applicable for other cellular proteins. The doubt surrounding this applicability exists because the O2-independent transition metal-mediated process of transnitration (Fig. 3A) is suspected of dominating protein S-nitrosations in cells (24).
Similar to what one expects to occur with the formation of Ras SNO, if O2 is present instead of NO, the nucleotide-deficient Ras Cys118-S• may react with O2 to produce a nucleotide-deficient oxidized Ras (e.g., Ras Cys-SOH, Cys-SO2H or Cys-SO3H) (see Section I). In a separate reaction, GS• or Cys-S•, respectively, can be formed by a reaction of O2 •− (or •NO2) with GS− or with Cys-S− (Fig. 4D). This GS• or Cys-S• also can react with the nucleotide-deficient Ras Cys118-S• to produce a nucleotide-deficient Ras Cys-SSG or Ras cysteine disulfide. As with the nucleotide-deficient Ras-SNO, the nucleotide-deficient oxidized Ras or the nucleotide-deficient Ras Cys-SSG/Cys118-SS-Cys binds a nucleotide to yield a stable form of chemically modified Ras GTPases.
The proposed radical-based formation of Ras Cys118 side chain modification (e.g., Ras-SNO and Ras Cys-SSG) suggests that the chemical modification of the Ras Cys118 side chain is not directly linked to modulation of Ras activity but occurs as an end product of the redox process. This would explain why either Ras-SNO or Ras Cys-SSG was detected at the same time the Ras downstream effector proteins were activated.
c. Implication of the role of the chemically modified redox-sensitive cysteine in the NKCD motif of Ras proteins
Notably, Ras-SNO no longer reacts with •NO2 to produce any radical species (101). Similarly, Ras-SNO cannot be a target of O2 •−. As with the formation of Ras-SNO, the oxidized Ras (e.g., Ras-SOH) does not react with either O2 •− or •NO2. Hence, it is tempting to conclude that the formation of the chemical modification of the Ras Cys118 side chain such as Ras-SNO and Ras-SOH results in termination of the redox signaling associated with Ras. However, the chemical modification of the Ras Cys118 side chain into a form of Ras Cys-SSG may not terminate redox signaling because Ras Cys-SSG can be homolytically cleaved into Ras Cys118-S• and GS• (Fig. 4E, Path 1). The GS moiety on the Ras Cys-SSG may be displaced with another cysteine or its analogs. Yet, Ras Cys-SSG per se does not react with either •NO2 or O2 •− to produce any protein radical species.
The cellular role or function of the chemically modified Ras Cys118 side chain, such as Ras-SNO or Ras Cys-SSG, is unknown. For example, no upstream or downstream protein of Ras has been identified as a specific Ras-SNO or a Ras Cys-SSG targeting protein. Despite research into the denitrosation of other redox-sensitive cellular proteins (16), the fate of Ras-SNO in cells has not been investigated. However, glutaredoxin-1 and thioredoxin-1 (Trx1) have been implicated separately in the deglutathionylation of Ras Cys-SSG (2, 154, 306).
d. Beyond the proposed mechanism for the action of redox agents on Ras GTPases that contain the NKCD motif
Although the proposed radical-based molecular mechanism would explain the results of a number of studies (2, 9, 27, 53, 78, 99, 156 –160, 209, 253, 284, 312), several questions about the mechanism itself must be resolved before this explanation can be accepted. Perhaps the most important of these questions is, how does the Phe28 side chain mediate electron transfer from the bound nucleotide base to Ras Cys118-S•? Addressing this question requires consideration of Marcus theory (129, 183, 184) and of the local electronic state of the Phe28 side chain associated with the bound nucleotide base. To date, Marcus theory has been applied only to radical chemistry associated with DNA oxidative stress (121, 255, 263).
1. Relative redox potential of the Phe28 phenyl side chain as well as the bound nucleotide base and Ras Cys118-S−
Although the redox potential of a free phenylalanine side chain is unknown, this potential has been predicted to be close to or less than 2.1 V (vs. NHE) (145, 283). The redox potentials of Ras Cys118-S− and the bound nucleotide base have been predicted to be Ras Cys118-S•/Ras Cys118-S− (∼0.8 V vs. NHE) >G •+-DP/GDP (<∼0.8 V vs. NHE; vide supra). Hence, it is apparent that electron transfer through the Phe28 side chain is kinetically unfavorable. As analyzed in Section II, the close n-π interaction between the Phe28 phenyl side chain and the bound nucleotide base could lower the redox potential of the Phe28 phenyl side chain via stabilization and localization of one of the cationic radical states of the Phe28 phenyl side chain (plus a charge on the C4 atom of the phenyl side chain). However, it is unclear how much the redox potential of the Phe28 phenyl side chain declines because of the close n-π interaction with the bound nucleotide base. Nevertheless, it is predictable that the redox potential of the Phe28 side chain of the GTPase-bound NKCD motif-containing GTPases may not be as high as in a free phenylalanine phenyl side chain (>∼2.1 V vs. NHE) (145, 283). Even if the redox potential of a Phe28 side chain were lower than that of a free phenylalanine side chain, the redox potential of the Phe28 side chain still could exceed that of a Cys118 side chain and thus be higher than that of the bound nucleotide base. If so, an electron transfer from the bound GDP base to Ras Cys118-S• via the Phe28 side chain includes an uphill step of Phe28 to Ras Cys118-S• to produce a Phe28 side chain cationic radical (Phe28•+) and Ras Cys118-S− (Phe28 → Ras Cys118-S•), respectively, followed by a downhill process from the bound GDP base to Phe28•+ to produce Phe28 and G •+-DP (GDP base → Phe28•+). This brings up the question of how this uphill electron transfer proceeds.
2. Application of Marcus theory to the Phe28 side chain-mediated electron transfer between the bound nucleotide base and thiyl radical of Ras GTPases
A theory has been developed that contends that electron transfer in proteins is regulated by pathways that are optimal combinations of through-bond, hydrogen bond, and through-space (19). The driving force of a reaction, as predicted by this Marcus theory (183, 184), also affects the rate of electron transfer in and between proteins (134). Studies such as these allow evaluation of reorganization energies, which strongly influence reaction rates. Briefly, the redox event associated with GTPases that contain the NKCD motif can be expressed as an energy diagram (Fig. 13). In accordance with Marcus theory (129, 183, 184), the endergonic tunneling steps (1 k et en and 3 k et en; Steps 1 and 3, respectively, in Fig. 13) are concomitant with the exergonic electron transfer events (2 k et ex and 4 k et ex; Steps 2 and 4, respectively, in Fig. 13). In physiological reactions, endergonic tunneling associated with 1 k et en and 3 k et en (particularly 1 k et en in Fig. 13) has come to be considered a biologically relevant process, but only with some reluctance. However, thermally assisted tunneling (quantum probability) up to ∼0.5 eV uphill has been shown in photosynthetic systems (210, 300). As discussed above, the value of the redox potential of the Phe28 side chain is expected to be less than 2.1 V (vs. NHE). Hence, 1 k et en between the Phe28 side chain and Cys118-S• would not be very endergonic. Besides, a previous study has explicitly noted that “although the overall reactions are usually modestly favorable, these chains often include VERY endergonic steps” (210). A number of uphill electron transfer events in various proteins, including azurin, myoglobin, and cytochrome complexes, also have been reported (51, 85). For example, although the redox potential of an aromatic amino acid Trp142 in cytochrome b is expected to be much higher than that of the two redox centers ubiquinol and cytochrome c 1 (267), Trp142 intervenes in electron transfers between these two redox centers (28).
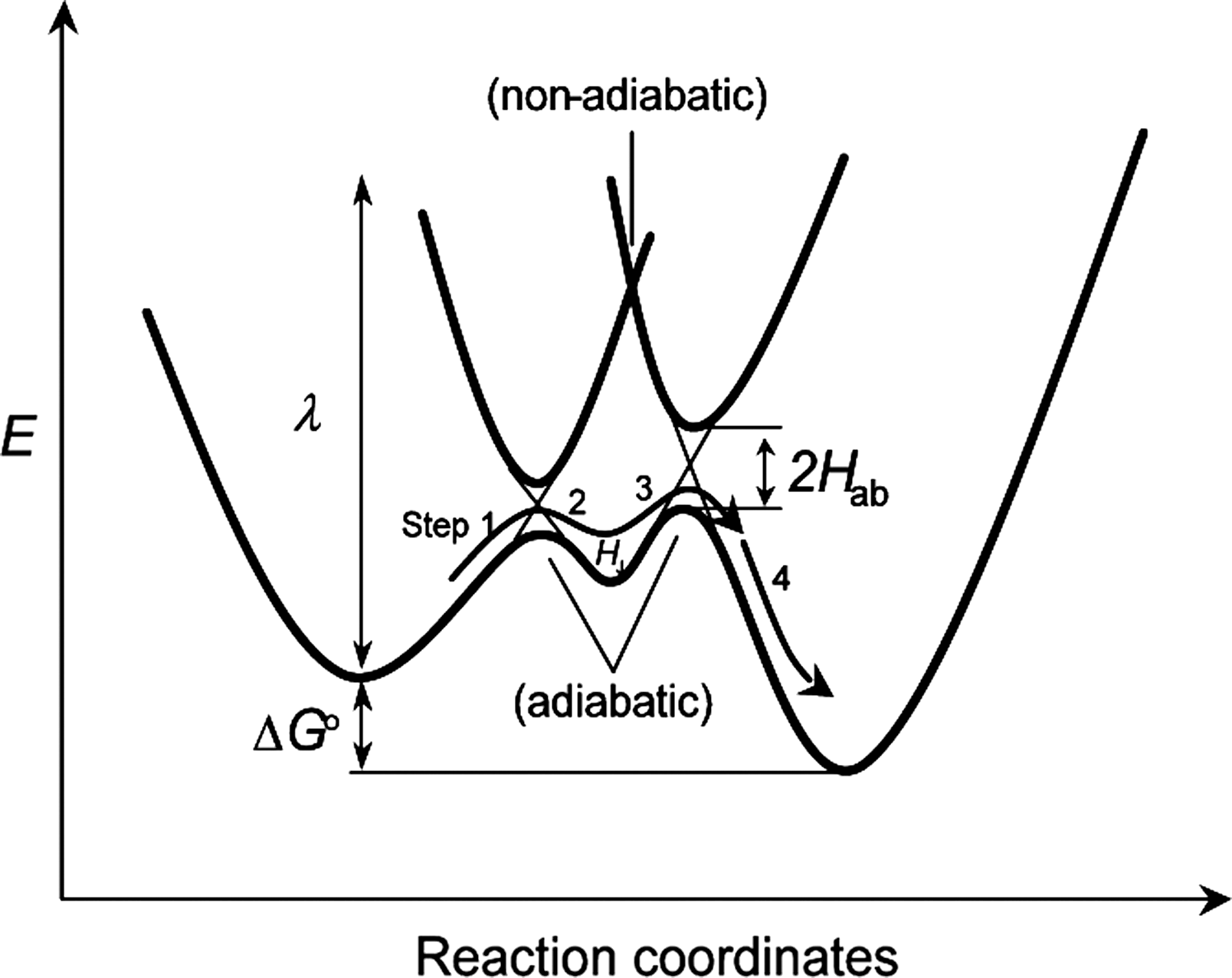
In Marcus theory (183, 184), the electron transfer rate (k et; a combination of 1 k et en, 2 k et ex, 3 k et en, and 4 k et ex) between redox centers is almost inversely proportional to the edge-to-edge distance and roughly proportional to the protein packing density (ρ) in the volume between redox centers; and ρ is nearly inversely proportional to the square root of the barrier height (Fig. 13) (210). A study also has shown that ligands with π systems provide easy passage of electrons for outer-sphere redox reactions, where k et for the [Co(phe)3]3+/[Co(phe)3]2+ system is 40 M/s, which is many orders of magnitude faster than that for [Co(NH3)3]3+/[Co(NH3)3]2+ (64). Electron tunneling between redox centers via aromatic side chains (ρ = 0.77 ± 0.09) is consistently faster than that for electron tunneling through a vacuum (ρ = 0) (85, 210). These results lead one to suggest that the enriched π electrons in the Phe28 side chain function via an electron tunneling mechanism as an electron transfer-intervening medium to mediate highly directional uphill electron transfer between the Cys118 side chain and the bound nucleotide base (Fig. 13).
In summary, the molecular mechanism from the bound nucleotide base to Ras Cys118-S• via a Phe28 side chain (Fig. 12) can be explicitly tethered with the corresponding energy diagram (Fig. 13): (i) Although the first endergonic and exergonic tunneling steps (1 k et en and 2 k et ex; Steps 1 and 2, respectively, in Fig. 13) correlate with the reaction Step 2 in Figure 12, the transient energy minimized state H J located between adiabatic curvatures (Fig. 13) couples with the product of Step 2 in Figure 12; and (ii) the second endergonic and exergonic processes in the energy diagram (3 k et en and 4 k et ex; Steps 3 and 4, respectively, in Fig. 13) correspond to Step 3 in Figure 12.
e. Proposed mechanism for the action of redox agents on GTPases that contain the GXXXXGK(S/T)C motif
RNS and ROS that target the GXXXXGK(S/T)C motif-containing GTPases are identical to those of the NKCD motif-containing GTPases (100, 105). The corresponding end products also are identical to those of proteins that have the NKCD motif (100, 105). Like the NKCD motif-containing GTPases, the GXXXXGK(S/T)C motif-containing GTPases have a redox-sensitive cysteine and a phenylalanine (100, 105). The only difference is that the distance between the redox-sensitive cysteine and phenylalanine in the GXXXXGK(S/T)C motif-containing GTPases is ∼3-fold shorter than the distance in the NKCD motif-containing GTPases (see Section II). Although the effect of this distance has not been investigated, this similarity of the two motifs suggests that when Marcus theory is applied, the redox mechanisms of proteins possessing the NKCD and the GXXXXGK(S/T)C motifs are likely to be essentially identical.
A second redox-sensitive and unique cysteine is often found in certain GXXXXGK(S/T)C motif-containing GTPases (105). These include RhoA, RhoB, and RhoC. In RhoA, the second cysteine (RhoA Cys16) and the primary cysteine (RhoA Cys20) in the GXXXXGK(S/T)C motif face each other, but the bound nucleotide is located between these two cysteines (Fig. 10B) (60, 115). Formation of the nucleotide deficient form of the Rho protein moves the sulfur atoms of the Cys16 and Cys20 side chains closer to each other. Closer proximity allows them, under certain conditions, to form a disulfide bond via a radical-based mechanism (Figs. 3 and 4) (105). A redox agent, •NO2 or O2 •−, facilitates RhoA GDP dissociation to produce a nucleotide-deficient RhoA that has both a RhoA thiolate (RhoA Cys20 thiolate side chain) and a free chemically modified GDP. Before binding with GTP (or GDP), another •NO2 or O2 •− reacts with RhoA Cys20 thiolate side chain and/or RhoA Cys16-S− to produce a RhoA Cys20 thiyl radical (Cys16-S•). This Cys16-S• then reacts with another one to produce a nucleotide-deficient RhoA Cys16-SS-Cys20 disulfide. As discussed in Section I, a quenching process, such as reduction of O2 into O2 •−, might be necessary if the intermediate for disulfide formation is a RhoA Cys16-SS-Cys20•−. This RhoA Cys16-SS-Cys20 disulfide bond blocks access to fresh nucleotide binding, perpetuating a state of nucleotide-deficient RhoA that is likely to be an inactive form of RhoA. Hence, it is possible that the role of the second redox-sensitive cysteine is to block RhoA redox activation.
3. Potential mechanism for the action of redox agents on GTPases that possess the CGNKXD motif
A recent study shows that Ran GTPase can be activated by •NO2, one of the RNS, but not by either NO or N2O3 (98). The target Ran redox sites are identified as cysteine residues (the solvent-exposed Cys85 and Cys120 in the CGNKXD motif ) (105). It must also be taken into account that an intact nucleotide is released as an end product of •NO2-mediated Ran nucleotide dissociation (98). This result suggests that the bound guanine nucleotide may not be involved in the •NO2-mediated Ran nucleotide dissociation. This also strongly implies that the redox-regulation mechanism for Ran is likely to differ from the radical-based GDP modification mechanism for Ras and Rho GTPases.
Intriguingly, although the Ran Phe26 side chain is away from the bound nucleotide (Fig. 11) (266), the Ran Phe26 plays a role in the •NO2-mediated Ran nucleotide dissociation (105). Also, results from CD spectra suggest that, although treatment of wt Ran with •NO2 alters its secondary structure, its treatment with •NO2 had no effect on the CD spectrum of Ran F161L (unpublished data). Given that the side chains of Ras Phe26 and the Phe161 belong to the packed Ran phenylalanine residues (see Section II), it can be hypothesized that the Ran aromatic cluster is involved in the redox response of the Ran protein. Noticeably, the stacked aromatic cluster is placed between these two redox-sensitive cysteines, Cys85 and Cys120 (see Section II).
a. Potential molecular mechanism of the redox regulation of GTPases that contain the CGNKXD motif
These results in conjunction with the structural and sequence analyses of Ran, speculation about the nature of a potential molecular mechanism is possible (Fig. 14): A redox-competent RNS (e.g., •NO2) or ROS (e.g., O2 •−) reacts with the solvent-accessible Ran Cys120 thiolate side chain (Cys120-S−) located in the CGNKXD motif to produce the Ran Cys120 thiyl radical (Cys120-S•). In turn, this Cys120-S• couples with the Ran Cys85 thiolate (Cys85-S−) via the Ran aromatic cluster to produce Cys120-S− and Cys85-S•. The Cys85-S• thus formed can be quenched by NO (if NO is present) to produce an S-nitrosated Ran (44). The electronic coupling between these two Ran redox centers generates a temporal Ran aromatic cluster cationic radical (aromatic cluster•+). The formed transient aromatic•+ subsequently perturbs the π-π stacking interactions in the Ran aromatic cluster. The result is alteration of the Ran guanine nucleotide-binding site. The altered conformation of the Ran guanine nucleotide-binding site perturbs the Ran guanine nucleotide-binding interaction and leads to the release of an unmodified nucleotide that results in formation of a nucleotide-deficient Ran. Cellular abundant GTP binds to the nucleotide-deficient Ran to produce an active Ran (Fig. 14).
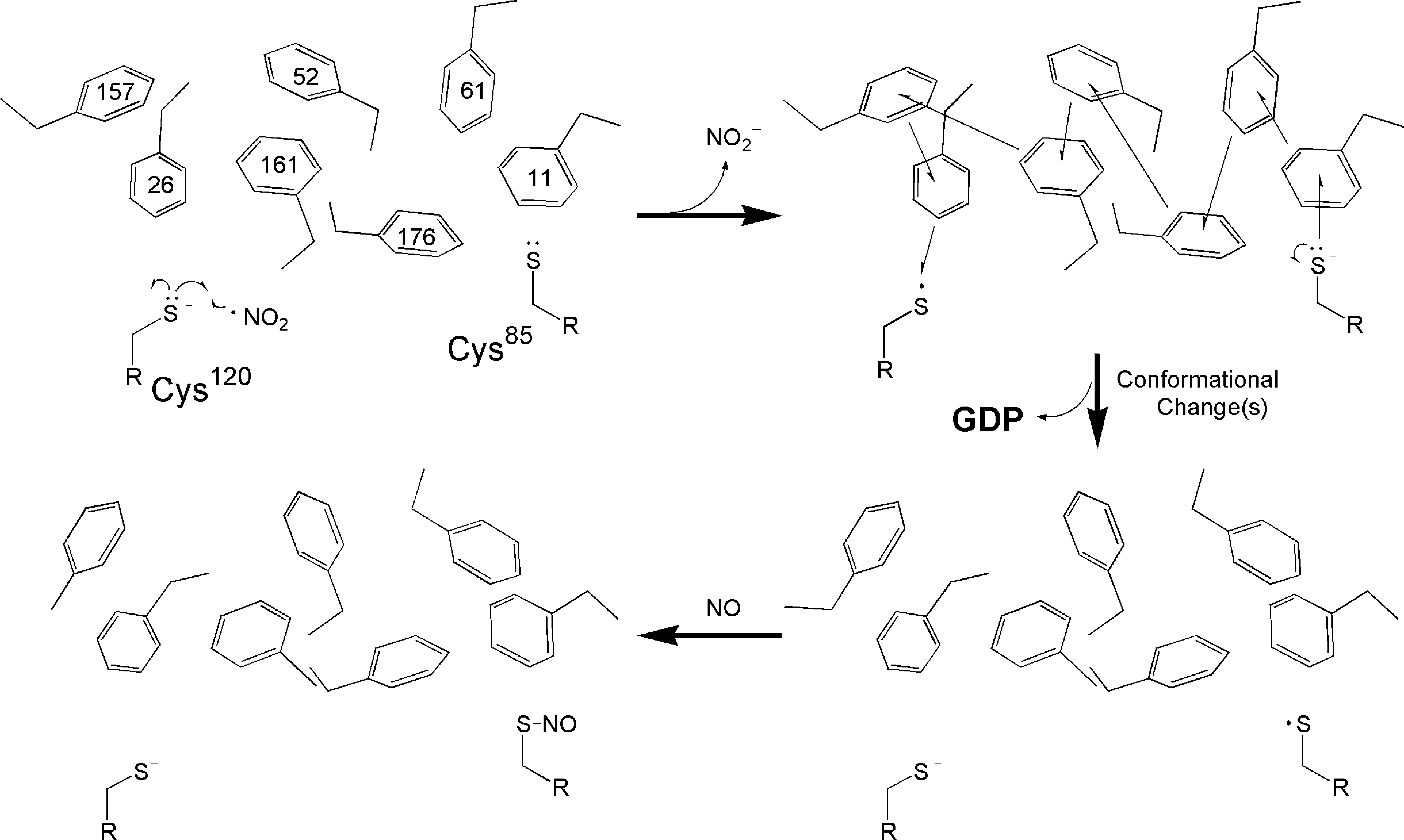
This postulated mechanism requires an assumption that the relative redox potentials among three Ran redox components are ordered as follows: Cys120-S•/Cys120-S− > Cys85-S•/Cys85-S−. This assumption of the relative redox potential between the sulfur atom of Cys120 and the Cys85 side chain is based upon an analysis of the local structural environments of the Ran protein (see Section II).
Because the sulfur atoms of Ran Cys120 and Cys85 face each other from a distance of ∼11.8 Å, a direct electron transfer from Cys85-S− to Cys120-S• (or vice versa; from Cys120-S− to Cys85-S•) is possible without involvement of the Ran aromatic cluster. However, this direct electron transfer is a less attractive possibility than a transfer via the aromatic cluster because electron transfer through a vacuum is less favorable than through an electron-rich medium (i.e., via a π electron-enriched medium) (48).
1. Mechanistic properties of the Ran aromatic cluster and Marcus theory
Although a detailed experiment should be performed to elucidate a true molecular mechanism and/or test the potential proposed mechanism, Marcus theory (183, 184) and the electron tunneling hypothesis (19, 84, 134, 162, 202, 210) can partly answer a few remaining questions associated with the electron transfer mechanism and the redox-induced Ran protein conformational change. One of the key unanswered questions is, how does the packed aromatic cluster serve as an electron conduit if its redox potential is higher than those of the Cys120 and Cys85 side chains (i.e., aromatic cluster•+/aromatic cluster >Cys120-S•/Cys120-S− > Cys85-S•/Cys85-S−)? Accounting for the postulation of the relative redox potentials of the Ran redox-sensitive cysteines and the aromatic cluster (vide supra), the Ran redox components (i) Cys120 (redox agent target) and (ii) Cys85 (electron donor) can be matched to the redox components in the NKCD motif-containing GTPases. These components are, respectively, (i) Cys118 (the redox agent target) and (ii) the bound nucleotide (the electron donor). Although the electron mediator for the NKCD motif-containing GTPases is a single phenylalanine residue (Fig. 12), Ran, in contrast, has packed multiple phenylalanines. However, an earlier study has clearly noted that tunneling often includes multiple endergonic steps (210). Hence, the Ran aromatic cluster can be considered to be functionally matched to the Ras Phe28 (electron transfer mediator). Thus, the energy diagram for these redox components for the NKCD motif-containing GTPases (Fig. 13) also can be readily applicable to the Ran protein electron transfer. The energy diagram (Fig. 13) illustrates that, overall, the electron transfer process from the Cys85 side chain to the Cys120 side chain is thermodynamically favorable (if Cys120-S•/Cys120-S− < Cys85-S•/Cys85-S−, the reverse is true). However, as with the electron transfer in GTPases that contain the NKCD motif, the predicted path of the electron transfer from the Ran Cys85 side chain to the Cys120 side chain via the aromatic cluster includes a series of endergonic and exergonic electron transfer events (Steps 1, 2, 3, and 4 in Fig. 13). Notably, significantly larger ρ-values can be found in the π systems of aromatic residues (64, 85, 210). For this reason, by taking Marcus theory into account (183, 184), electron tunneling between two Ran cysteine redox centers via aromatic cluster π systems is likely to be feasible.
2. Redox-mediated activity regulation of Dexras1 and Rhes GTPases
The SWISS-MODEL analysis (see Section II), in combination with redox chemistry, predicts that the redox architectures of Dexras1 and Rhes GTPases are similar to that of the Ran protein. Hence, it can be speculated that the mechanism proposed for the Ran protein is applicable to the action of RNS and/or ROS on Dexras1 and Rhes proteins. Further rigorous studies are essential to elucidate the molecular mechanism for the RNS/ROS-mediated activation of Dexras1 and Rhes GTPases.
b. Potential mechanistic reason for the redox-induced conformational change of Ran GTPase
Although Marcus theory explains the electron transfer between these redox-sensitive cysteines via the stacked aromatic cluster, redox-induced conformational change in the aromatic cluster is a matter of speculation. The proposed electron transfer within the Ran redox components points to a transient charge-transfer resonance interaction (Fig. 14). For example, Ran Cys120-S• abstracts an electron from the aromatic cluster to produce Ran Cys120-S− and a potential transient intermediate, the Ran aromatic cluster•+. The formed transient aromatic cluster•+, in turn, abstracts an electron from Ran Cys85-S− to produce the aromatic cluster and Ran Cys85-S•. Studies have suggested that a benzene dimer is quite stable in the uncharged parallel-stacked configuration (coplanar arrangement; i.e., D6h, C2h, C8, C2b, and C2) (144, 165, 235). Moreover, a thermodynamic study showed that the degrees of change in the enthalpy and entropy of uncharged benzene aromatic-aromatic interactions is not altered significantly within a temperature range of −23°C to 137°C (215). However, when the benzene dimer is charged (i.e., cation dimer), an energy difference of only 0.07 eV (235) can easily interconvert the coplanar configuration of the benzene dimer into the eclipsed stacked configuration (perpendicular arrangements; i.e., C2v, Cs, and C1 configuration). In silico, abstraction of an electron from one of the Ran phenylalanine side chains to produce a transient aromatic•+ state results in repulsion of the packed phenylalanine side chains, which is consistent with the previous results (235). Given that the Ran aromatic cluster is well integrated into the entire protein's architecture, the Cys120-S•-driven perturbation of the multiple π-π stacking interactions of the aromatic cluster could result in a Ran protein conformational change. Further, when one end of the Ran aromatic cluster (Phe26 1) becomes rooted in the Ran guanine nucleotide-binding site (consisting of the CGNKXD motif ), it is possible to speculate that the Ran protein conformational change also couples with alteration of the structural architecture of the CGNKXD motif and results in release of the nucleotide from Ran. However, although the bound nucleotide is close to the Phe26 1 side chain, no direct electronic interaction between the Phe26 1 side chain and the Ran-bound nucleotide has been observed (266); this suggests that the Ran-bound nucleotide is likely to be redox incompetent (vide supra). The released nucleotide is, therefore, not expected to be chemically modified, which also is consistent with the previous observation (98).
III. Redox Modulation of the Activity of Small GTPases and Its Physiological and Pathophysiological Relevance
A redox agent, as an upstream and/or downstream regulator of these redox-sensitive small GTPases, plays a key role in various cellular signaling events. Dysregulation of small GTPases by a redox agent(s) or misregulation of redox signaling by small GTPases or vice versa alters cellular signaling pathways. Such alterations often cause various diseases.
A. Diseases associated with dysregulation of the redox signaling of the NKCD motif-containing GTPases
Because Ras plays a key role in many cellular signaling cascades, diseases relevant to dysregulation of redox signaling often result in deregulation of Ras-dependent cellular signaling events. Further, because the redox-sensitive NKCD motif of Ras was identified more than a decade ago, considerable pathophysiological data is available, including some bearing directly on the relevance of redox-mediated misregulation of the Ras NKCD motif to certain diseases. Rap1A, another reprehensive protein that possesses the NKCD-motif, is a regulator of NAD(P)H oxidase. However, a pathophysiological outcome associated with the misregulation of Rap1A redox signaling has not been clearly investigated.
1. Upregulation of Ras GTPases by redox agents
Lander et al. showed that NO activates Ras by enhancing Ras GNE in human T cells (158). Another study has shown that treatment with N-methyl-
Several Ras-dependent cellular signaling cascades are implicated in the redox responses of Ras (Fig. 15). For example, various oxidative agents (i.e., H2O2 and hemin) also could activate Ras by enhancing Ras GNE and thus upregulate the mitogen-activated protein kinase (MAPK) pathway in Jurkat cells (Fig. 15) (159). This study also showed that expression of a dominant negative Ras mutant in Jurkat cells blocked signaling by these oxidative agents. This result, which was assessed by Ras-dependent activation of the nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB), suggests that direct activation of Ras (Fig. 15) may be a central mechanism by which a variety of oxidative redox stress stimuli transmit their signal to the nucleus. Moreover, the NO-mediated activated Ras results in tyrosine phosphorylation of various cellular proteins (i.e., focal adhesion kinase and Src kinase) through stimulation of MAPK pathway (Fig. 15) (200). NO-mediated Ras activation also stimulated extracellular signal-regulated kinases, c-Jun terminal kinase (JNK) and extracellular signal-regulated kinase (ERK), respectively, via the phosphatidylinositol 3′-kinase (PI3K) and Raf signal transduction pathways (Fig. 15) (55, 56). A more recent study has shown that exogenous addition of H2O2 induces chemical modification of the Ras redox site; this modification facilitates activation of the antiapoptotic serine threonine kinase (Akt) but not activation of ERK (2).

a. Cancers
Ras is considered the most prevalent oncogene found in human cancer because 30% of human tumors contain activated Ras mutations (12, 23). Hence, it is not surprising that cancer is one of the most prevalent disorders caused by misregulation of Ras activity by a redox agent. Numerous studies show that cancers, to a large extent, are induced by misregulation of Ras redox signaling combined with an alteration of Ras downstream cellular transduction cascades (Fig. 15).
1. RNS
The hypoxia-inducible transcription factors that bind to hypoxia-responsive elements (HREs) are key players in cellular responses to changes in oxygen tension as well as their responses to ROS (141). One early study suggested that HREs were associated with Ras and MEK1 in human prostate cells (LNCaP and PC3), and NO was a major signaling intermediate responsible for the HRE induction in these cells (253). Overexpression of iNOS has consistently been shown to be directly involved in deregulation of the growth of hepatocellular carcinoma cell specimens from rats, mice, and humans via activation of NF-κB, which couples with the HRas-dependent ERK pathway (Fig. 15) (31). Further, cell compartment-dependent Ras redox regulation coupled with the ERK pathway has been reported (114). This study showed that upregulation of eNOS activates NRas but not KRas on the Golgi complex of T cells. This NRas activation was due to the formation of Ras-SNO (see Section II) at the redox-sensitive Ras NKCD motif residue Cys118, which induces T cell death. In certain human breast cancer cells (MDA-MB-231, MCF-7, and MDA-MB-468), not only the Raf-MEK1/2-ERK1/2 cascade but also the PI3K/Akt pathway was shown to be upregulated via Ras activation (Fig. 15), and this action of NO was independent of guanosine 3′,5′-cyclic monophosphate (cGMP) (217). Lim et al. showed that a constitutively active oncogenic Ras (e.g., KRas G12V) in mice stimulates the PI3K-Akt signaling cascade, which in turn activates eNOS by phosphorylation (174). This activated eNOS then further activates other wt Ras isoforms, such as H and KRas GTPases, via formation of Ras-SNO at their redox-sensitive site Cys118 in the NKCD motif (174). The study suggests that this oncogenic Ras-PI3K-Akt pathway coupled with the eNOS-Ras cascade is, at least in part, responsible for initiation and maintenance of oncogenic Ras-mediated growth of tumors. Moreover, a study using a rat intestinal epithelial cell line (IEC-6) shows that an activating mutation of KRas leads to enhancement of the iNOS expression via various transcription factors, including NF-κB, and the endogenously produced NO contributes to tumorigenesis and continuation of the tumor growth of KRas-transformed cells (273). In addition, the MAPK pathway (Fig. 15) also participates in redox-mediated tumorigenesis associated with Ras activation. A study using human melanoma cell lines (WM793 and WM35) shows that activation of mutations of NRas enhances iNOS expression in human melanoma via activation of the MAPK pathway (62). Intriguingly, a potentially reversible process, induction of Ras-activating mutation by upregulation of NOS, also has been investigated. Patients with acute myeloid leukemia had a high expression of iNOS, but there was no correlation between expression of iNOS and expression of K, H, and NRas mutation and expression (25). However, another study reported that treatment of Sencar mice with a topical application of an NO releasing agent, (±)-(E)-4-methyl-2-[(E)-hydroxyimino]-5-nitro-6-methoxy-3-hexenamide, enhances tumor-initiating activity via induction of mutation in HRas at codon 61 or 13 to produce a constitutively active oncogenic HRas GTPase (241).
2. ROS
Irani et al. shows that NIH 3T3 cells transformed with the constitutively active HRas G12V produce large amounts of ROS (118). This study also shows that the MAPK activity was decreased and JNK activity was unchanged in HRas G12V-transformed cells. This suggests that the mitogenic activity of cells induced by HRas G12V associated with ROS was independent of the MAPK and PI3K-JNK pathways (Fig. 15). The same research group also shows that one ROS, O2 •−, plays a role as a signaling molecule in phagocytic cells, such as modulating the activity of Ras and Rac (117). Another study shows that, in human gastric adenocarcinoma cells, the Ras-dependent ERK1/2 pathway is a common path the response for the response to oxidative stress associated with ROS (Fig. 15) (111). However, given that ROS can activate ERK1/2 signaling cascades via inhibition of protein phosphatases, ROS likely activates the ERK1/2 signaling pathway at multiple points (167, 227, 292). For example, Ras expression is not necessarily required for ROS-mediated activation of the ERK1/2 signaling pathway: ROS induces the activation of the ERK1/2 signaling pathway in cells prepared from ventricles of 1-day-old Wistar rats and overexpressed with a dominant negative Ras mutant (320). This may be due to in part to the c-Src-mediated phosphorylation and activation of phospholipase Cγ (286). Also, blockage of MEK1/2, an upstream protein of ERK, by its inhibitor (U0126 or PD98059) results in inhibition of oxidative stress-induced ERK1/2 activation (166, 169). This result suggests that oxidative stress signaling is not exerted directly on ERK1/2 but is targeted at upstream cascades such as Ras.
ROS are shown to modulate cellular oxidative stress at the level of transcription. A recent study using fibroblast and osteosarcoma cells shows an explicit role in cancer by Ras associated with ROS in which Ras induces expression of an oncogenic transcription factor, the forkhead box protein M1, and the action of Ras was O2 •− dependent (212). The forkhead box protein M1 is overexpressed in most human carcinomas (178, 220, 281). The Ras-dependent protein kinase C (PKC) pathway in gastric cancer (AGS-B) cells also has been shown to be responsible for oxidative stress-mediated activation of the histidine decarboxylase promoter (111, 112).
One study has shown that a constitutively active oncogenic Ras variant upregulates expression of Nox1 in NIH 3T3 cells (268) via the Ras-dependent MAPK pathway (195). Conversely, this study also showed that use of small interfering RNAs to inhibit Nox expression blocked transformation of phenotypes (anchorage-independent growth, morphological changes, and production of tumors in athymic mice) associated with Ras upregulation (195). The result of this study suggests that the production of ROS by Nox is required for oncogenic Ras transformation. Nox1 consistently plays a key role in Ras-induced tumor angiogenesis, possibly via activating the Raf-MEK1/2-ERK1/2 pathway-dependent transcription factor Sp1 in transformed normal rat kidney cells and gastric cancer (150, 243). Suppression of Nox activity has been shown to be able to block the oncogenic HRas-mediated growth of NIH 3T3 cells (42). Transcriptional regulation of Nox1 by Ras also has been investigated with CaCO-2 cells derived from a certain human colon adenocarcinoma (72, 136). The presence of active Ras in CaCO-2 cells also correlates with expression of Nox1 mRNA (3). An in vivo study using transgenic mice shows a correlation between Nox1 mRNA expression and activation of mutations in Gly12 and Gly13 of KRas (164). This correlation is tightly coupled with incidences of colorectal cancer.
b. Heart diseases
As with cancers, many cardiovascular disorders are linked to the dysregulation of redox-sensitive Ras and its downstream signaling transduction pathways.
1. NOS
An in vivo study using rats has shown that blockage of NO synthesis by inhibiting NOS results in inhibition of Ras signaling that attenuates end-organ damage during severe hypertension and endothelial dysfunction (18). Oliveira et al. showed that a Ras-dependent Raf-MEK1/2-ERK1/2 signaling pathway (Fig. 15) in rabbit aortic endothelial cells was stimulated by sodium nitroprusside (SNP) or SNAP as an NO source and a cGMP analog, 8Br-cGMP (208, 209). Importantly, this study also suggested a Ras activation mechanism in which the transamination of the NO moiety of S-nitrosoglutathione or SNAP onto the Cys118 site Ras produces active Ras-SNO. Apurinic/apyrmidinic endonuclease/redox factor-1 (APE1/ref-1) plays a key role in DNA repair and also governs the reductive activation of many redox-sensitive transcription factors (285). Using APE1/ref-1+/–mice, it was shown that APE1/ref-1 also regulates endothelium-dependent NO production and vasomotor tone via HRas (122). Interestingly, the action of ONOO− for the activation of ERK in cardiomyocytes derived from a rat heart is not through Ras but via an unusual signaling cascade involving Raf and MEK1 (218). This study also suggests that PI3K and PKC may be responsible for phosphorylation and activation of Raf (41, 147) because both PI3K and PKC were shown to be targets of ONOO− (10, 11, 90, 193, 319).
2. ROS
H2O2-mediated activation of ERK via Ras plays a key role in protecting cardiac myocytes from apoptotic death in cardiac myocytes prepared from ventricles of 1-day-old Wistar rats (4). This oxidative stress-induced ERK1/2 activation is reported in a variety of cell types. These sources are pleural mesothelial cells isolated from the parietal pleura of Fischer 344 rats (123), astrocytes obtained from a 2-day-old Sprague-Dawley rat brain (277), T cells (86), pulmonary epithelial cell cultures (30), chinese hamster lung fibroblast (V79) cells (143), ventricular myocytes isolated from hearts of adult male Sprague-Dawley rats (304), lung microvascular endothelial cells (280), and hepatocytes derived from rats (46).
In human umbilical vein endothelial cells, Ras has been shown to mediate the Raf-MEK1/2-ERK1/2 pathway (Fig. 15) leading to apoptosis after oxidative injury (50, 214). However, in PC12 cells, H2O2 activates ERK through Ras, and this activation has been shown to play a key role in preventing apoptosis (89, 313). One study shows that Angiotensin II (AII) induces hypertrophy in vascular smooth muscle cells (VMCs) derived from mice via ROS-mediated Ras activation that may contribute to atherosclerosis or restenosis (13). It also shows that ROS from NAD(P)H oxidase induces hypertrophy via activation of the Raf-MEK1/2-ERK1/2 pathway (Fig. 15) via Ras activation in ventricular myocytes derived from the hearts of adult rats (304).
As seen in the action of eNOS on H, N, and KRas (114, 174), the Ras Cys118 site can be chemically modified by ROS but in a different manner. Adachi et al. showed that the Ras Cys118 side chain in the NKCD motif was S-gluathionylated (Ras Cys-SSG) in cultured Rat VMCs poised with exogenous H2O2 or overexpressed with NAD(P)H oxidase (2). Intriguingly, this study also showed that AII-induced hypertrophy is associated with Ras activation by formation of Ras Cys-SSG. An overexpression of glutaredoxin-1 caused effective deglutathionylation of Ras that was coupled with inactivation of Ras and an attenuation of AII-induced hypertrophy (2). Also, overexpression of Trx1 in cells derived from adult rat ventricular myocytes alleviates hypertrophy that is mediated by α-adrenergic receptor-stimulated Ras activation (154) via the Raf-MEK1/2-ERK1/2 signaling pathways (Fig. 5) (303, 304). An in vivo study tested transgenic mice with cardiac-specific overexpression of Trx1 for development of hypertrophy (306). These mice did not develop hypertrophy. However, antisense inhibition of Trx1 results in hypertrophy in cultured cardiac myocytes via activation of the Ras-dependent Raf-MEK1/2-ERK1/2 cascade (306).
c. Neuronal diseases
As with some cancers and heart disorders, various neuronal diseases appear to be the result of dysregulation of various cellular signaling events via the redox-sensitive Ras.
1. RNS
An in vivo study using a postnatal day-7-rat model of perinatal hypoxia-ischemia showed that Ras is activated in both the hippocampus and cortex within 2 h after hypoxia-ischemia (284). This study also shows that, although the Ras activation is NOS-dependent, its contribution to the pathophysiologic NO-dependent mechanisms of neurologic injury was minimal. Importantly, although expression of nNOS was detected in a subtype of the cortical interneurons of the brains of normal humans and those with Alzheimer's disease, nNOS was also detected in a subset of the pyramidal neurons of the patients with Alzheimer's disease, and this nNOS in the pyramidal neurons is highly colocalized with expression of Ras GTPase (180). One study has shown that NO-induced Ras-mediated MAPK activation is required for oxygen-glucose deprivation tolerance in cortical neurons (78). In addition, a study using human embryonic kidney 293 cells has shown that NO stimulates ATP-sensitive potassium (KATP) channels via targeting a redox-sensitive Ras Cys118 site that leads to activation of the MAPK pathway but not activation of the PI3K pathway (175). This NO-Ras-MAPK-KATP pathway in primary hippocampal cells prepared from an embryonic day-19 rat has been shown to be responsible for neuroprotection mediated by neuronal ischemic preconditioning (175). A recent study shows that the Ras-dependent Raf-MEK1/2-ERK1/2 pathway is involved in NO-induced neural protection in cortical neurons derived from the brains of 16-day-old Wistar rat embryos (247).
2. ROS
In PC cells, constitutively active Ras has been shown to enhance ROS production (76) that activates ERK via Ras (249), suggesting that the Ras-dependent Raf-MEK1/2-ERK1/2 pathway route is involved in the neuronal signaling response. Creatine may enhance survival signaling via activation of the Ras-NF-κB cascade in primary cerebrocortical cultures (132). This in turn may be coupled with the protective effects of creatine against glutamate cytotoxicity in neuronal cells and in animal models of neurodegenerative diseases.
d. Other disorders
The misregulation of the redox signaling of Ras with its downstream cascades also has been linked to various disorders linked with immune and embryo developments. The Ras-dependent activation of Raf also leads to stimulation of a phosphorylation of Ets-like protein-1 and tumor necrosis factor-α messenger RNA induction; both actions suggest that NO, through the Ras-dependent Raf-MEK1/2-ERK1/2 pathway, modulates a host's defenses and the inflammation of T lymphocytes (55). ROS-mediated signaling via Ras, NF-κB, and related transducers may link to embryopathies (289).
2. Downregulation of Ras GTPases by redox agents
In some cases, endogenously released NO or an exogenous treatment of an NO donor inactivates Ras, which may link to certain diseases. In N293 cells (human embryonic kidney 293 transfected with nNOS), Ras was inactivated via S-nitrosation at the HRas Cys118 site in the NKCD motif by NO generated by nNOS; the Raf-MEK1/2-ERK1/2 cascade coupled with Ras was blocked (225, 226).
Treatment of an NO donor, SNAP or DETA/NO, inhibits activation of MAPK via inactivation of the Ras and Raf cascades in human pulmonary arterial smooth muscle cells (198). However, one study has reported that expression of iNOS results in upregulation of Ras that blocks VMCs proliferation via MAPK activation and operates independent of cGMP (139).
Intriguingly, in T cells, an NO donor, 2,2-(hydroxynitrosohydrazono)-bis-ethanamine, decreases expression of Ras; this decrease is linked to the role of NO as an inhibitor of lymphocyte cytotoxicity (67). This study also shows that NO neither activates nor inhibits the expressed Ras.
3. Modulation of Rap1A GTPase activity by redox agents
Unlike the Ras proteins, the redox regulation of Rap1A and its signaling consequences has not been studied extensively. RNS- or ROS-mediated inactivation of Rap1A has not been observed. However, several studies using human T-cell Jurkat cells have shown that Rap1A activity is sensitive to NO, because SNP and SNAP can promote Rap1A-GTP loading thereby leading to NO-induced activation of Rap1A (196, 236), which colocalizes with NAD(P)H oxidase (172). Moreover, expression of an activated Rap1A mutant in Wistar rat thyroid cells treated with NO-releasing agent SNP enhances sensitivity to apoptosis, whereas cells expressing wt Rap1A resisted NO-initiated cell death (236). Hence, it is possible that Rap1A activity also may be regulated by NAD(P)H oxidase-produced ROS. These in vivo results support the intriguing possibility that ROS-mediates regulation of the activity of Ras-related proteins.
B. Diseases associated with dysregulation of redox signaling of GTPases that contain GXXXXGK(S/T)C motif
Because the discovery of the redox-sensitive GXXXXGK(S/T)C motif in certain GTPases, including RhoA, Rac, Cdc42, and some Rab GTPases, was relatively recent, no comprehensive explanation has been advanced to explain the link between the misregulation of the redox signaling of these Rho and Rab proteins and the pathophysiological effects that result. However, because the action of RhoA and Rac GTPases is often linked to redox-relevant cardiovascular functions, several redox-relevant pathophysiological studies associated with RhoA and Rac have been conducted.
Cdc42 is downstream of the G-protein coupled receptor (Fig. 16) (113, 239). Rac signaling can be coupled with Ras via PI3K (Fig. 15) (239). As noted elsewhere, Rac1, one of the Rac GTPases, plays an essential role in ROS production from NAD(P)H oxidases. Both Rac and Cdc42 also are downstream of integrin-linked kinases (Fig. 16) (54). Also, there is evidence that Rac is not only downstream of integrin-linked kinases but also downstream of thrombin and hypoxia (79, 80). Functionally, Rac plays a key role in the membrane ruffle formation, whereas Cdc42 modules form of membrane projections (54). Rac is upstream of RhoA and the kinase of MAPK, which is upstream of the MAPK pathway (Fig. 16). RhoA is upstream of the Rho-associated kinase (ROCK) that functions for actin filament stabilization (Fig. 16). Another RhoA path includes G-actin that facilitates actin polymerization. The action of Rho-dependent ROCK with G-action results in stress fiber formation. A Ras-dependent JNK pathway can be a target of RhoA (177). Rac, however, can inhibit this stress fiber formation via blocking the ROCK pathway (Fig. 16) (35). Because Rho GTPases have emerged as key mediators of Wnt signaling pathways (176, 245), it is possible to speculate that the effect of RNS or ROS on Rho GTPases could affect morphological and transcriptional changes as well as embryogenesis and tumorigenesis via Wnt signaling events (216, 232).

1. Upregulation of Rho proteins by redox agents
Although a definite role for redox agents in direct cellular regulation of Rho proteins remains to be established, one study has shown that Caki-1 cells exposed to hypoxia exhibit increased Cdc42, Rac1, and RhoA protein expression and activity (278). This study also found that overexpression and activation of these Rho proteins was downstream of, and dependent on, the production of ROS because specific inhibition of ROS-producing Nox downregulated GTPases activity (Fig. 16). To date, however, unlike with the Ras and Rho proteins, neither an in-cell nor an in vivo study of the redox regulation of Rab GTPases has been made.
a. Cardiovascular diseases
Typical cardiovascular disorders associated with dysregulation of redox modulation of Rho GTPases occur through the RhoA-ROCK or Rac-Nox pathway (Fig. 16). No extensive studies have been made of other potential diseases such as cancer and neuronal disorders associated with misregulation of Rho redox responses.
1. RNS
A body of evidence suggests that RhoA regulates via the ROCK pathway (Fig. 16). NO production from eNOS and that misregulation of this process results in the pathogenesis of several cardiovascular disorders, including hypertension and atherosclerosis. Studies show that RhoA inhibits eNOS expression (163), and inhibition of ROCK in human endothelial cells leads to the activation of the PI3K-Akt-eNOS pathway (Fig. 15) and cardiovascular protection (299). The Rho-ROCK pathway coupled with eNOS is linked significantly to the development of erectile dysfunction and the pelvic atherosclerosis of rats (213). A study using the rat intestinal cell line (IEC-6) shows that NO impairs mucosal healing by inhibiting enterocyte migration via activation of RhoA coupled with the SH2-containing protein tyrosine phosphatase (36). A recent study also shows that de novo nNOS expression was sufficient to induce, via the RhoA-ROCK pathway (Fig. 16), synaptic withdrawal in adult rat motoneurons (271). Isolated rat aortic rings (endothelium denuded) treated with ROS released from an xanthine oxidase and its substrate xanthine mixture were shown to induce Ca2+ sensitization via activation of the RhoA-ROCK signaling pathway but not via the Ca2+-independent PKC (124). Also, short-term air pollution increases hypertension in a rat artery via O2 •−-mediated upregulation of the RhoA-ROCK pathway (270). One study also has shown that NO plays an antagonistic role against O2 •− via Rac GTPase in smooth muscle cells of transgenic mice overexpressed with human cDNA of the constitutively active mutant of Rac1 (96). NO released from 2,2-(hydroxynitrosohydrazono)-bis-ethanamine consistently inhibits the activity of NAD(P)H oxidase in human VMCs via inhibition of Rac1 translocation (203). Conversely, the formation of O2 •− by the NAD(P)H oxidase scavenges endothelial NO and thus is responsible for the development of renovascular hypertension and endothelial dysfunction (131).
2. ROS
In HeLa cells, the endogenously released ROS associated with activation of Rac further upregulates a signal transduction pathway associated with NF-κB (269). A pull-down assay has revealed that in retinal pigment epithelial cells, the small GTPase activated by H2O2 is Rac1 (110). The activation state of Rac1 and RhoA responds rapidly to changes in oxygen tension via PI3K and Nox-dependent signaling pathways. Their coordinated actions regulate the endothelial barrier function in the endothelial cells of conduit porcine pulmonary artery endothelial cells (298).
An in vivo study has shown that cardiomyocyte-specific Rac1, but not other Rac isoforms (Rac2 and Rac3), is critical to generating oxidative stress via interaction with gp91 phox and other cytosolic components of NAD(P)H oxidase (i.e., p47 phox ) that are linked to development of cardiac hypertrophy in an adult mice heart in response to vasoactive substances such as AII (240, 242, 274). The action of AII can be attenuated by statin treatment, resulting in downregulation of the Rac1-mediated NAD(P)H oxidase activation and reduction of ROS production in SMCs (288). Notably, ROS released from upregulation of NAD(P)H oxidase coupled with increased Rac1 activity often results in failure of the left ventricular myocardium of patients with ischemic cardiomyopathy or dilated cardiomyopathy, and oral statin treatment permits downregulation of Rac1 activity in the human heart (181). Intriguingly, a study using the Cos7 cell line shows that Cdc42 can serve as a competitor of Rac for cytochrome b 558 (61).
b. Other diseases and relevance to stem cells
Although most of the research into Rac GTPases and ROS is associated with cardiovascular diseases (vide supra), misregulation of Rac GTPase activity by ROS also is implicated in the pathogenesis of neurodegenerative diseases in PC12 cells and mice (15, 95).
Multiple isoforms of NAD(P)H oxidase—such as Nox1 and Nox2 as well as their regulatory subunits, including Rac GTPase—were expressed in hematopoietic stem or progenitor cells derived from bone marrow (219). The activation of Nox isoforms likely plays a role in regulation of self-renewal and differentiation in stem cells. For example, O2 •− derived from Nox2 plays an essential role in the mobilization, homing, and angiogenic capability of stem or progenitor cells that lead to the revascularization of ischemic tissue (279). Hence, it is evident that an appropriate regulation of the activity of Nox isomers and its subunits, including Rac, also is required to give stem cell properties such as mobilization and proliferation.
2. Downregulation of Rho protein by redox agents
Most research results suggest that a redox agent activates Rho proteins by an increase in Rho GNE. However, an opposite effect of RNS on regulation of the activity of RhoA proteins has been reported (256). In that report, iNOS was not only involved in the increased expression of RhoA but also in prevention of the upregulation of RhoA in diabetic rat hearts. Although indirect, an explanation has been proposed for the mechanism in the ROS-dependent downregulation of RhoA (207): An activation of Rac GTPase stimulates release of O2 •− from NAD(P)H oxidase, which in turn inhibits the low-molecular-weight protein tyrosine phosphatase (LMW-PTP). Because p190Rho-GTPase-activating protein (p190Rho-GAP) is a substrate of LMW-PTP, inactivation of LMW-PTP results in accumulation of the active phosphorylated form of p190Rho-GAP. This activated p190Rho-GAP stimulates the hydrolysis of bound GTP to produce inactive GDP-bound RhoA. This downregulated RhoA contributes to the spread of integrin-dependent cells.
C. Diseases associated with dysregulation of redox signaling of the CGNKXD motif-containing GTPases
The redox-dependent pathophysiological function of Dexras1 has been investigated. However, despite investigation of the relevance to some specific diseases of the dysregulation of Ran associated with oxidative stress, these studies were performed before identification of the redox-sensitive CGNKXD motif in these GTPases. No reports have been made that directly allege pathophysiological relevance(s) that is linked to the misregulation of redox signaling associated with the CGNKXD motif of Ran.
1. Upregulation of Ran and Dexras1 by redox agents
A body of physiological study results on the cellular redox stress associated with Ran has been reported (92, 148, 170, 197, 224, 307, 310). A H2O2-induced oxidative stress-induced perturbation of the Ran nucleo-cytoplasmic concentration gradient has been reported (148, 310). In HeLa cells, the oxidant H2O2 inhibits classical nuclear import, including perturbation of the Ran nucleo-cytoplasmic gradient (148). The Ran gradient in HeLa cells dissipates in response to a stress-induced depletion of GTP-bound Ran, diminishing the efficiency of Ran nuclear import (148, 310). The study also showed that the H2O2-mediated oxidative stress induces a relocation of the nucleoporin (Nup153) as well as the nuclear carrier importin-β. The result is to reduce docking of the importin-α/β/cargo complex at the nuclear envelope. Moreover, the H2O2-induced oxidative stress causes Ran, importin-β, and Nup153 to undergo a caspase/proteasome-dependent (but ubiquitin-independent) proteolysis (148). The nuclear accumulation of importin-α appears to be triggered by a collapse in the Ran gradient, causing suppression of the nuclear export of importin-α (197).
Dexras1, which also possesses the CGNKXD motif, is mainly involved in redox regulation of neural cells (173, 252). However, unlike with Dexras1, no in vivo or cell studies have been reported on redox regulation of the activity of the Rhes protein.
a. Cancers
Aberrant regulation of Ran-mediated cellular signaling events results in unchecked cell proliferation (7, 45). Hence, there are grounds for speculation that misregulation of Ran redox signaling may be linked to the formation of malignant tumors.
1. RNS
The pathophysiological effect of misregulation of RNS-mediated Ran signaling has not been investigated.
2. ROS
Although it is unclear whether the effect of ROS on Ran is direct or indirect, one study showed that Ran/TC4 was upregulated by H2O2 in nonmalignant breast epithelial cells but not in malignant cells (307). Further, a proteomic approach has identified in plants a thioredoxin-dependent activation of the Ran/TC4 protein (170).
b. Neuronal diseases
Studies suggest that dysregulation of the redox role of Dexras1 may be linked to neuronal disorders (63, 173).
1. RNS
Dexras1 forms a ternary complex with the NO-producing nNOS as well as the carboxy-terminal PDZ ligand of nNOS in rat brains (63). Formation of this complex enhances the ability of nNOS to activate Dexras1 in rat brains. In a separate study, colocalization of Dexras1 with carboxy-terminal PDZ ligand of nNOS and nNOS also was observed in the neurons and glial cells of rats (173). In vitro and cell studies also show that the Dexras1 Cys11 site can be S-nitrosated (120). A subsequent study using HEK293T and PC12 cells has shown that stimulation of the NMDA receptors activates nNOS, leading to S-nitrosation at the Dexras1 Cys11 site and activation of Dexras1, which is linked to NMDA-mediated neurotoxicity (37).
2. ROS
The pathophysiological relevance of misregulation of Dexras1 associated with ROS has not been investigated.
2. Nonredox response of Ran under certain conditions
As discussed in Section III, these redox-sensitive GTPases such as Ras and Rho proteins sometimes can be downregulated by RNS and/or ROS or they can be insensitive to one or the other or to both. A recent study shows that a mild oxidative stress (conditions that do not induce death in the majority of cells as defined by the author) induced by diethyl maleate has, without exception, little effect on the nucleocytoplasmic concentration gradient of Ran (149).
IV. Concluding Remarks and Future Research
This review article attempts to link physiological and pathophysiological relevance to aspects of the molecular mechanisms of the redox regulation of small GTPases. However, many provocative questions remain unanswered about this topic.
A. Investigation of the redox regulations of Rho, Rab, and Ran GTPases
Past investigation of the redox role and function of small GTPases has mainly focused on Ras while largely ignoring Rho, Rab, and Ran GTPases. For example, although the Wnt signaling pathway coupled with Rho GTPase plays a key role in embryogenesis and tumorigenesis, the direct role of redox signaling along the Wnt signaling pathway via targeting the Rho protein has not been investigated. Also, although redox regulation of Ran GTPases was recently discovered, its physiological and/or pathophysiological function has not yet been explored. Investigation of the action of a redox agent on Rho, Rab, and Ran GTPases promises to yield a better understanding of their presumed role(s) in human diseases, including cancer formation, cardiovascular disorders, and even embryogenesis.
B. Exploration of the role of Ras-SNO
Although this review presents various potential mechanisms for the formation of Ras-SNO, neither the mechanism of Ras-SNO formation nor the biological role of Ras-SNO has been established. In particular, the structural role and function of the NO moiety of the Ras-SNO in cell signaling events remains unclear. A second obvious question associated with Ras-SNO formation is whether Ras-SNO is really a chemical end product. A third question that is related to this second question also can be asked: What is the potential mechanism of denitrosation and/or degradation of Ras-SNO? Identification of any role or function, if such a role or function exists, of the NO moiety of Ras-SNO in the Ras-mediated cellular signaling cascade(s) also is essential to understanding the redox regulation of Ras GTPases and its physiological and pathophysiological relevance to the action of Ras. Exploration of the cellular mechanism and/or process of denitrosation and/or degradation of Ras-SNO is crucial to a better understanding of the cellular redox signaling events associated with small GTPases containing the NKCD motif.
C. Clarification of the redox regulation mechanisms proposed for Ras, Rho, and Ran GTPases
This perspective is closely linked to the potential exploration of the structural and biological roles of Ras-SNO (vide supra). Although potential mechanisms relevant to the redox action of RNS and ROS on small GTPases have been proposed, vigorous examination and validation of these proposals will be essential. For example, the proposed radical intermediates, such as a small GTPase thiyl radical (e.g., Ras Cys118-S•), G•+-DP, G•-DP, and/or the transient aromatic cluster•+, in the radical-based mechanisms for Ras, Rho, and Ran GTPases remain elusive. Identification and/or characterization of these putative radical intermediates is essential to prove or disprove the proposed radical-based molecular mechanisms for redox-sensitive small GTPases.
A sequence analysis shows that ERas and HRas have the NKCD motif in common. ERas also possesses an electron-mediating residue, Phe66, which is equivalent to HRas Phe28 that has been shown to play a key role in the redox response of HRas (Section II). Hence, it is possible that ERas is redox sensitive, but no investigation has been made of this possible redox property associated with RNS or ROS. Investigation of the redox regulation of ERas would be particularly important to understanding regulation of human ERas activity under oxidative stress. The results of such an investigation may provide insight into the mechanisms of tumors induced by expression of ERas.
It is difficult to define the role of tyrosine (e.g., Ras Tyr141 or Rac1 Tyr40) in radical-mediated nucleotide dissociation from small GTPases, yet any possible role of Ras tyrosine associated with the redox regulation of Ras GTPases is open for future research.
D. Application of Marcus theory to cellular redox regulation mechanisms
Further efforts to incorporate biophysical insight into the cellular consequences associated with physiological and pathophysiological relevance also are essential. Although Marcus theory may be applicable to elucidating the redox regulation of certain proteins, its application as an explanation of electron transfer in cell signaling proteins has not been developed as rapidly as expected. This is partly because of the extreme complexity of the protein systems in which electron transfers play a role. This complexity increases the difficulty of measuring rates of electron transfer and also adds to the difficulty of identifying specific electron donors and acceptors. Although these features involve vigorous and rigorous research, understanding the mechanistic elements of the redox regulation of small GTPases will in turn provide the basis for a better understanding of the nature of redox-sensitive GTPase signaling. This understanding, in turn, will aid development of methods to alleviate or cure disorders associated with dysregulation of these small GTPases.
Footnotes
Acknowledgments
The author is deeply grateful to many anonymous reviewers for their criticism of, comments on, and suggestions for this article. Their help and guidance was essential to whatever merit it may claim. The author also thanks J. Stanford Fisher and Michael Wey for their proofreading of this article.
Abbreviations Used
Reviewing Editors: Sharon L. Campbell, Paola Chiarugi, Chunming Dong, Thomas Kietzmann, Bokyung Kim, Harry Lander, and Hugo Monteiro