
Abstract
The lysosome is a redox-active compartment containing low-mass iron and copper liberated by autophagic degradation of metalloproteins. The acidic milieu and high concentration of thiols within lysosomes will keep iron in a reduced (ferrous) state, which can react with endogenous or exogenous hydrogen peroxide. Consequent intralysosomal Fenton reactions may give rise to the formation of lipofuscin or “age pigment” that accumulates in long-lived postmitotic cells that cannot dilute it by division. Extensive accumulation of lipofuscin seems to hinder normal autophagy and may be an important factor behind aging and age-related pathologies. Enhanced oxidative stress causes lysosomal membrane permeabilization, with ensuing relocation to the cytosol of iron and lysosomal hydrolytic enzymes, with resulting apoptosis or necrosis. Lysosomal copper is normally not redox active because it will form non–redox-active complexes with various thiols. However, if cells are exposed to lysosomotropic chelators that do not bind all the copper coordinates, highly redox-active complexes may form, with ensuing extensive lysosomal Fenton-type reactions and loss of lysosomal stability. Because many malignancies seem to have increased amounts of copper-containing macromolecules that are turned over by autophagy, it is conceivable that lysosomotropic copper chelators may be used in the future in ROS-based anticancer therapies. Antioxid. Redox Signal. 13, 511–523.
Introduction
Apart from their long-recognized role in necrosis, lysosomes were recently found to be increasingly involved in several forms of programmed cell death, including typical apoptosis. Lysosomes exist in all kinds of plant and animal cells, except mature erythrocytes. Inside the lysosomal compartment, the degradation of endocytosed or autophagocytosed materials takes place in an acidic environment (pH about 4 to 5), which is maintained by ATP-dependent proton pumps present in the lysosomal membrane. Such pumps also are present in the plasma membrane, especially in tumor cells. In the latter, these pumps enable cell survival despite the high intracellular production of lactic acid that otherwise would result in intracellular acidosis (5, 91, 110).
After synthesis in the endoplasmic reticulum, lysosomal hydrolases are tagged with mannose-6-phosphate (MP) at the cis-Golgi area and then enclosed in transport vesicles (sometimes called primary lysosomes, although they have a neutral pH) in the trans-Golgi network (TGN), with the help of MP receptors. The vesicles containing the newly produced hydrolases are then transported to slightly acidic (pH about 6) late endosomes, which arise from early endosomes containing endocytosed material. Finally, the late endosomes mature to lysosomes that lack MP receptors, are rich in acid hydrolases, have a pH of 4 to 5, and contain material to be degraded.
The acidic lysosomal compartment contains a wide spectrum of hydrolytic enzymes, which play a major role in the intracellular degradation of proteins, polysaccharides, phospholipids, and other biomolecules. Lysosomal proteases (cathepsins) are apparently the most important group of these enzymes. Lysosomal cathepsins can be categorized as cysteine (cathepsins B, C, F, H, K, L, O, S, V, W, and X), aspartic (cathepsins D and E) and serine (cathepsin G) proteases (35, 58, 107). They have pH optima around 5, although several of them remain active at neutral pH for minutes (cathepsin L) to hours (cathepsin S) (31).
Lysosomes fuse with autophagosomes/endosomes to form “hybrid” organelles containing material to be degraded, originating both from the outside and the inside of the cell. After complete degradation of the enclosed material, lysosomes turn into “resting” organelles, which, in the electron microscope, look homogeneous and moderately electron dense. They are then ready for new rounds of fusion (69). The active fusion and fission of the lysosomal compartment (69) allows lytic enzymes and other lysosomal contents to be distributed between different lysosomes. From a physiological point of view, the lysosomal compartment can be looked on as a “super organelle,” built of vacuoles that constantly fuse and divide, that receives enzymes from the TGN and substrates from either the outside or the inside of the cell. After substrate degradation inside individual lysosomes, the products diffuse or are actively transported to the cytosol for reutilization (Fig. 1 depicts the functions of the lysosomal compartment in a schematic form).
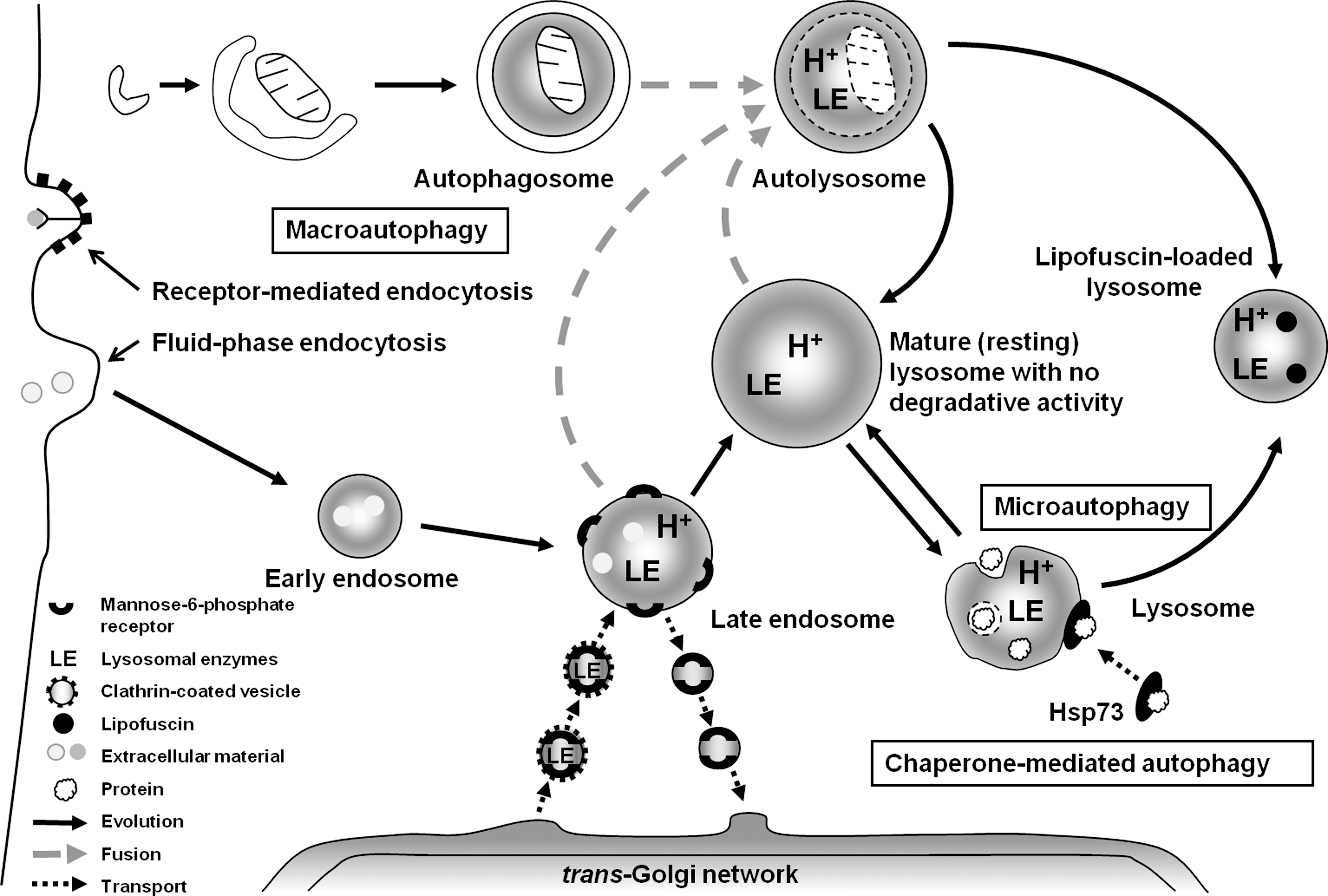
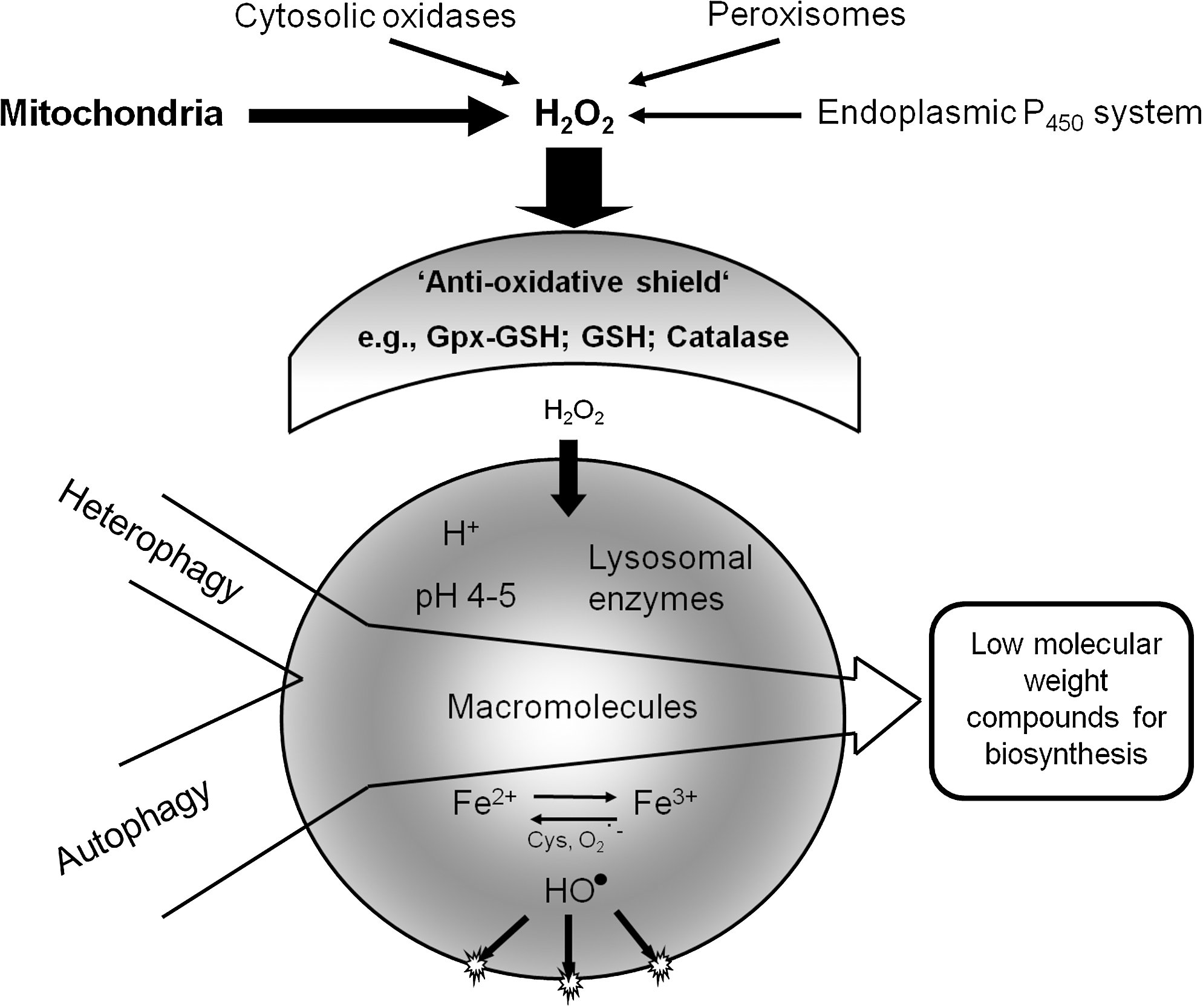
Because of the degradation of ferruginous materials, such as mitochondria, ferritin, and a variety of other Fe-containing macromolecules, the lysosomal compartment is rich in “loose” redox-active iron, making lysosomes sensitive to oxidative stress through intralysosomal Fenton-type reactions. The generation of hydroxyl radicals and ferryl and perferryl species may give rise to peroxidation of material under degradation, resulting in lipofuscin (see later section on lipofuscin) or, if substantial, to lysosomal membrane permeabilization (LMP) (Fig. 2). Lysosomal destabilization, with relocalization to the cytosol of potent hydrolytic enzymes and low-mass iron, is able to induce either apoptosis or necrosis. As is schematically shown in Fig. 3, crosstalk between lysosomes and mitochondria is an important process in apoptosis, and either of these organelles may induce the process. Obviously, a concerted and balanced action of released lysosomal cathepsins and cytosolic caspases is required for typical apoptosis, whereas massive release of cathepsins into the cytoplasm will give rise to necrosis (62).
After receptor-mediated endocytosis, the initially plasma membrane–bound receptors are often, but not always, returned to the plasma membrane, whereas the ligands are degraded within the lysosomal compartment. One exception to this “rule of ligand degradation” is transferrin, which is returned to the plasma membrane together with its receptor, whereas the iron, which is bound to transferrin, is released into the late endosomes because of their slightly acidic environment (pH about 6) and transported to the cytosol by transport proteins such as Nramp [reviewed in (62)].
The processing and presentation of antigens in immunocompetent cells is dependent on a form of endocytotic–exocytotic activity, whereas autophagic degradation is vital not only for the normal turnover of cellular constituents, but also for the elimination of damaged structures and cytosolic microorganisms that have invaded the cell. Some cell types are able to exocytose lysosomal contents or even intact lysosomes (secretory lysosomes) [reviewed in (69)]. It has been recognized that tumor cells often secrete lysosomal proteases, which, in combination with the acidic interstitial milieu, enhances the activity of lysosomal proteolytic enzymes, which, in turn, promote tumor cell infiltration and metastasis. Because many iron-containing macromolecules are degraded intralysosomally, low-mass iron is released inside the lysosomal compartment. Because the lysosomes also contain reducing agents [for example, glutathione, ascorbic acid, and, above all, the amino acid cysteine (estimated concentration, 2 mM)], some or most low-mass iron will exist as Fe(II), with the capacity to generate highly reactive radicals if exposed to hydrogen peroxide [reviewed in (62)]. As a result, lysosomes are very sensitive to oxidative stress, and their membranes are easily peroxidized and permeabilized by radicals formed secondary to the Fenton-type reactions taking place in the lysosomes. The rupture of lysosomes with relocation of lytic enzymes results in apoptosis or necrosis, depending on the magnitude of this relocation [reviewed in (15)]. Keeping the concentration of redox-active iron in lysosomes as low as possible is consequently important for the survival of cells under oxidative stress (62).
Apart from the occurrence of redox-active iron inside lysosomes, low-mass iron exists also in a weakly chelated labile iron pool (LIP) (38), or transient iron pool (44). The nature of these low-molecular-weight chelators is not known but, among others, complexes with citrate, phosphate, polypeptides, and nucleotides have been suggested (49, 55, 56, 83). Methodologically, the LIP is defined as iron that can be bound by iron chelators, especially calcein. Whether the iron in the LIP is redox active or not is presently unknown. However, some reports suggest that redox-active iron in the LIP may exist, and that some of it is associated with the endoplasmic reticulum, where it is involved in the signaling of hypoxia-regulated genes (68). This iron may be relocated from lysosomes, from which it is released into the cytoplasm to be taken up by iron-binding proteins, such as ferritin.
The Main Functions of the Lysosomal Compartment
Autophagy
Autophagy is an unceasing biologic renewal mechanism providing for lysosomal degradation of the cell's own constituents. It represents one of the main pathways for the turnover and reuse of worn-out long-lived proteins and organelles and is a perfectly normal process. Interestingly, the multicatalytic proteinase complexes, proteasomes, which also play an important role in the turnover of macromolecules, are themselves degraded by autophagy (28). The implication of this is that hampered autophagy might result in defective proteasomes, because they, in common with mitochondria and other organelles, are then not properly renewed. The mechanisms involved in the formation of the phagophore, the inclusion of materials to be degraded, and the fusion of autophagosomes and lysosomes were recently worked out as a result of the discovery in yeast of a large family of phylogenetically well-preserved autophagy-related genes (ATGs) (53, 92, 97, 114).
To date, three different mechanisms of autophagy have been described in mammalian cells: macroautophagy (also known as just autophagy), microautophagy, and chaperone-mediated autophagy (CMA). Macroautophagy, which, in at least a subset of cases, is a nonselective process (90), involves the sequestration within a double membrane–enclosed vacuole (the phagophore) of portions of the cytosol, including aggresomes, dysfunctional mitochondria or proteasomes, as well as long-lived soluble proteins. The initially formed sequestration vacuole is devoid of lysosomal enzymes and is termed an autophagosome. In consecutive steps, it fuses with lysosomes and with other sequestration vacuoles, eventually resulting in the formation of an autophagolysosome (also called an autolysosome), within which the degradation of the cargo allows recycling of amino acids and other monomeric molecules (25, 62, 114) (see Fig. 1). Macroautophagy is the most universal type of autophagy, being involved in the degradation of practically any type of cellular material. It becomes activated under stress conditions, such as starvation, to generate ATP and essential building blocks by means of the nonspecific degradation of organelles and cytosolic macromolecules that are not critical to the survival of the cell (100).
Microautophagy is probably also involved in the turnover of lysosomes themselves, as suggested by the fact that fibroblasts exposed to the sequestration inhibitor 3-methyladenine accumulate large numbers of altered lysosomes containing a lipofuscin-like material (95). In support of this view, lysosomes with active hydrolytic enzymes have been found within autophagosomes (74). Microautophagy involves the invagination of portions of the lysosomal membrane into the lumen of the lysosome/vacuole, resulting in the internalization of cytosolic material. A variation of this process, known as “piecemeal microautophagy of the nucleus” or PMN, also was recently described in yeast, in which a small nonessential section of the nucleus is “pinched off” at nucleus–vacuole (NV) junctions [for a review, the reader is directed to (63)].
Chaperone-mediated autophagy (CMA) is a selective mechanism of lysosomal degradation, specific for soluble cytosolic proteins, which contain a targeting motif biochemically related to the penta-peptide KFERQ (for Lys-Phe-Glu-Arg-Gln). Unlike other forms of mammalian autophagy, CMA does not require vesicle formation or major changes in the lysosomal membrane to proceed; rather, substrate proteins directly cross the lysosomal membrane to reach the lumen, where they are rapidly degraded. This pathway requires the cooperation of cytosolic and lysosomal chaperones, including cytosolic hsc70 (cyt-hsc70), lysosomal hsc70 (lys-hsc70), lysosomal-associated membrane protein 2A (LAMP-2A), and co-chaperones including Hsp90, Hsp40, Hip, Hop, and Bag-1 (1). The upregulation of CMA is observed in response to nutritional starvation and mild stress induced by toxic compounds or oxidants (25, 114). The importance of CMA and LAMP-2A in particular is emphasized by recent observations that this protein decreases with advancing age and that upregulation of LAMP-2A in aged mice decreases age-associated decrements in hepatic function (119).
Subsequent to cellular damage, reparative autophagy follows, and malfunctioning structures are replaced. Such reparative autophagy is commonly seen, for example, after ionizing irradiation, virus infection, and hypoxic or oxidative stress (7, 50). Interestingly, in newborns, the postpartum period of starvation is bridged by a period of enhanced autophagy in the liver, explaining why certain mutations that hinder autophagy are lethal (113). Recent evidence suggests that regular day-long periods of starvation, or exposure to rapamycin (a TOR inhibitor), may, by stimulating autophagy, help to “keep cells clean” and be beneficial (26, 42).
Autophagy is the most efficient mitochondrial turnover mechanism, providing for the complete removal of irreversibly damaged mitochondria (mitophagy). It is believed that mitochondria are normally replaced every 2 to 4 weeks in rat brain, heart, liver, and kidneys (72), although recent studies showed that the turnover rate might be considerably higher (75). Mitophagy is particularly important for long-lived postmitotic cells, whose mitochondria may have pronounced oxidative damage. Under normal conditions, the biogenesis of mitochondria through mitochondrial fission is balanced by mitophagy, resulting in a relatively constant number of mitochondria within postmitotic cells. As a result of fission, oxidatively damaged mitochondrial biomolecules are diluted, as are also damaged components of dividing cells. The regulatory link between mitochondrial fission and mitophagy follows from a recent study showing that the pro-fission mitochondrial protein Fis1 induces mitochondrial fragmentation and the formation of autophagic vacuoles that contain mitochondria (37). Oxidant-induced injury to mitochondrial components seems to initiate asymmetric mitochondrial fission, generating daughter mitochondria with unequal membrane potentials (i.e., with different respiratory capacities and, probably, with different degrees of oxidative damage) (4, 109).
Questions yet unanswered are whether mitophagy is selective, and how mitochondria are targeted for degradation. Early studies performed on isolated hepatocytes exposed to amino acid starvation showed that macroautophagy may be a nonselective process (90), suggesting that the degradation of mitochondria occurs randomly. This is probably true for starving cells, in which autophagy activation is a rescue mechanism, using the cells' own constituents to generate energy and limited vital anabolism. All the functions of starving cells, perhaps with the exception of autophagy, are downregulated, with many mitochondria, including normal ones, becoming unnecessary.
Although the molecular mechanisms responsible for selective mitochondrial degradation remain to be further elucidated, extensive evidence indicates that oxidant-induced mitochondrial damage and related mitochondrial permeability transition with decreased inner membrane potential often initiate a sequence of events resulting in mitophagy [reviewed in (52, 108)]. Interestingly, it appears that superoxide (which may be generated in increased amounts by damaged mitochondria) is a trigger of autophagy (23).
Apoptosis and necrosis
An active role for lysosomes in the induction of cell death was suggested many years ago [reviewed in (29)]. Although apoptosis was not yet identified in those days, its characteristic morphology was described by morphologists. As pointed out in the introduction to this review, the role lysosomes play in apoptosis did not become recognized until recently. Major reasons for this considerable delay were the facts that (a) lysosomal membrane permeabilization may occur without any apparent ultrastructural alterations, creating an impression, still reflected in textbooks, that lysosomes are sturdy organelles that break only when cells are dead or almost dead; and that (b) many caspase inhibitors used experimentally to block apoptotic cell death also inhibit cysteine proteases, including lysosomal cathepsins (54).
The finding that some apoptogenic drugs that are also lysosomotropic detergents or aldehydes—such as MSDH (o-methylserine dodecylamide hydrochloride), sphingosine, Leu-Leu-OMe, and 3-aminopropanal—specifically destabilize lysosomes raised the possibility that lysosomal rupture might be an upstream event in some forms of apoptosis (10, 12, 24, 48). A strong argument for the idea that moderate lysosomal rupture causes apoptosis was the finding that the apoptogenic effects of these lysosomotropic drugs are prevented by an initial exposure to pH-raising lysosomotropic substances [for example, ammonia or chloroquine (80)]. The raised lysosomal pH prevents intralysosomal accumulation of the apoptogenic lysosomotropic agents (48, 67, 116). The resulting preservation of lysosomal stability and lack of apoptosis certainly linked lysosomal leak to apoptosis, although the causal molecular mechanisms were not explained by these experiments.
Oxidative stress during apoptosis, and lysosomal involvement in apoptosis due to such stress, is increasingly recognized (15, 24, 39, 79). The influence of such stress on cells depends on the dose. Depending on the extent of oxidant damage, moderate lysosomal rupture induces apoptosis, whereas pronounced lysosomal leakage results in necrosis without caspase activation (120).
The puzzling lack of apoptotic response after major lysosomal damage was difficult to explain. It was hypothesized to be a result of a violent degradation of procaspases, which, however, could not be verified. Very recently, Galaris and co-workers (2, 13) pointed out that a pronounced lysosomal breakup results in a major release to the cytosol of low-mass lysosomal iron that seems to prevent the activation of caspase-9 within the apoptosome (a complex of procaspase-9, cytochrome c, and ATP). This lack of activation was suggested to be an effect of iron binding to thiol groups in the active center of the procaspase (2, 13).
As pointed out earlier, the amount of redox-active lysosomal iron seems to be crucial for lysosomal stability during oxidative stress, probably explaining the variability in lysosomal sensitivity to such stress (77). The fact that cells with upregulated metallothioneins, heat-shock proteins, or apo-ferritin are relatively resistant to oxidative stress may, at least partially, be explained by autophagic transfer of such macromolecules into the lysosomal compartment, where they may temporarily bind redox-active iron before being degraded. In the event that autophagy of such proteins continues, as would be expected as a part of normal turnover, a steady state between lysosomal redox-active and non–redox-active iron would be established that is dependent on the cytosolic amounts of these proteins.
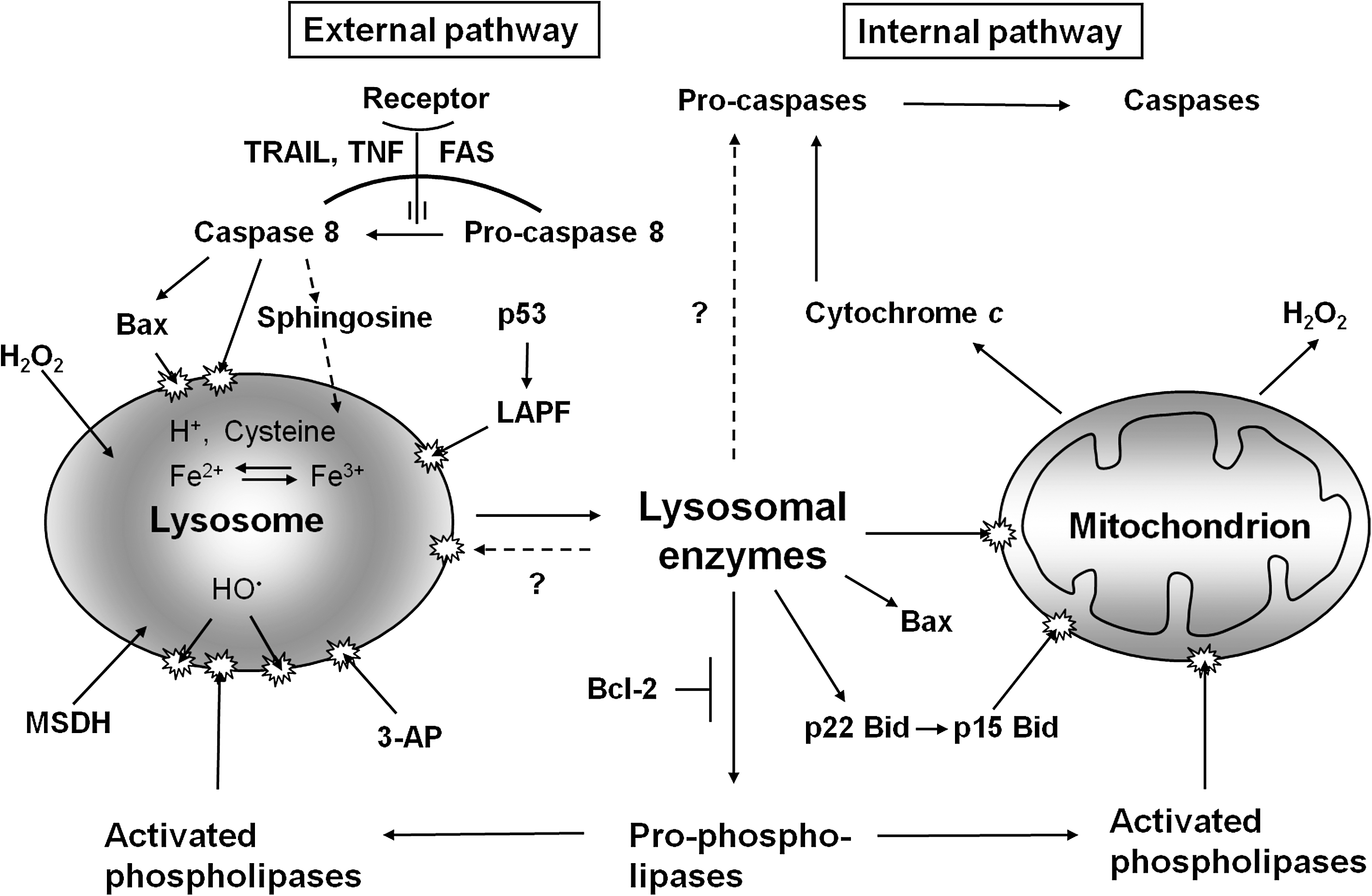
Lysosomal enzymes have been shown to reduce mitochondrial membrane potential (MMP) as well as to promote MPT (mitochondrial permeability transition), through proteolytic activation of phospholipases and/or the Bcl-2 family members Bid, Bax, and Bak. Calpain-like lysosomal cysteine proteases were found to induce cleavage of Bid (24), which, in its truncated form, relocates to mitochondria, resulting in Bax/Bak activation with the ensuing formation of pores in the outer membrane and the resulting escape of cytochrome c. Moreover, cathepsin D, but not cathepsin B, has been reported to modify/activate Bax to a pore-forming structure (18). Subsequent release occurs into the cytosol of proapoptotic mitochondrial factors such as cytochrome c, apoptosis-inducing factor (AIF), and Smac/DIABLO (9, 15, 24, 39, 43). Moreover, the direct activation of caspases-2 and - 8 by lysosomal cathepsins has been described for at least certain cell types (6, 112), although further studies are needed to confirm this.
The mechanisms described all work through activation of the internal (mitochondrial) pathway of apoptosis. However, indications also suggest that LMP is involved in apoptosis after the ligation of death receptors on the plasma membrane. Ligation of the FAS receptor was found to give rise to early LMP, although neither the mechanisms nor the possible relation to activation of caspase-8 was initially understood (16). Recently, however, it has been found that ligation of the receptors for tumor necrosis factor (TNF) and TRAIL, at least in certain types of cells, induces not only formation of active caspase-8 from its pro-form, but also the caspase-8–mediated activation of the proapoptotic protein Bax. This proapoptotic Bcl-2 family member, which was formerly believed to be involved only in MMP and MPT, was found also to participate in LMP, perhaps by forming pores similar to those of the mitochondria (111).
All these new findings suggest that lysosomal destabilization might play an integral part in apoptosis of both the internal and external pathways, and that the combined action of lysosomal hydrolases and caspases is necessary for apoptosis. Caspases may be needed for the degradation of a limited number of specialized proteins relevant to apoptosis, such as poly(ADP-ribose)polymerase and laminin, whereas lysosomal hydrolases may be needed for bulk degradation.
A form of apoptosis that is associated with pronounced autophagy is programmed cell death type II, or autophagic cell death that, for example, can be induced by serum starvation. It is often considered caspase independent, although whether this is completely correct remains to be clarified. An alternative interpretation of this phenomenon, at least in some cases, is that damaged cells try to heal themselves by reparative autophagy, which, in the event that it fails, is followed by apoptosis. Some indications exist of a mutually reinforcing link between autophagy and apoptosis, although mutual inhibition under certain conditions also was suggested (54, 70). Whether a completely caspase-free apoptosis-like cell death does exist still remains to be shown.
Surprisingly, we recently found (Kurz et al., unpublished data) that apoptosis induced by iron depletion after exposure to the lipophilic iron chelator, salicylaldehyde isonicotinoyl hydrazone (SIH), is preceded by lysosomal destabilization, indicating that lysosomal damage can be induced not only by iron-dependent oxidative stress but also by iron depletion. Any oxidative stress that might occur simultaneous with iron starvation due to general iron chelation should clearly not induce lysosomal labilization. Therefore, the destabilizing effect on lysosomes of iron starvation must be a consequence of other apoptogenic stimuli yet to be identified. The finding nevertheless gives additional support to the emerging view that LMP is a much more common phenomenon in apoptosis than is recognized.
Many forms of apoptosis are regulated by activation of the p53 gene. Exactly how the p53 protein triggers the apoptotic cascade is a topic of intense research (66). If lysosomal destabilization is a general phenomenon in apoptosis, it may be assumed that this phenomenon should be found early in the process of p53 activation. A mutated p53 protein exists in a thermo-labile form that is inactive at 37°C but structurally stable and active at 32°C (73). When cells that stably overexpress the thermolabile form of p53 are grown at 37°C, they remain normal, but when transferred to 32°C, they undergo apoptosis within 8 to 10 h. After transfer to the permissive temperature of 32°C, these cells start to show signs of early lysosomal destabilization within a few hours, long before any other apoptotic signs are found (117).
Even if lysosomes seem to be more deeply involved in apoptosis than is generally recognized, many questions remain to be answered. First among these is whether lysosomes are regularly destabilized early in forms of apoptosis that are not dependent on oxidative stress and, if so, how. Lysosomal rupture as a result of oxidative stress is well understood, but why are iron starvation, serum starvation (16), and p53 activation followed by lysosomal destabilization? The recently discovered protein LAPF (for lysosome-associated apoptosis-inducing protein containing PH and FYVE domains), a member of a widespread new family of Phafins (proteins containing the above domains), was shown to induce apoptosis by attaching itself to lysosomal membranes, causing lysosomal permeabilization and activation of the lysosomal–mitochondrial pathway (22). Perhaps this protein acts as a link between the p53 protein and lysosomal labilization? Figure 3 summarizes the role of lysosomes in apoptosis and emphasizes the crosstalk between lysosomes and mitochondria that seems to occur after lysosomal rupture.
Lipofuscin Formation and Its Influence on Autophagy
Influence of labile iron and ROS on lipofuscin formation
Lipofuscin (also known as age pigment or ceroid) is a nondegradable, yellowish brown, autofluorescent, polymeric compound that slowly accumulates within aging postmitotic cells at a rate that is inversely correlated with species longevity [reviewed in (17, 99)]. This interesting fact in and of itself suggests that lipofuscin accumulation may be hazardous to cells.
Long-lived postmitotic cells are very rarely replaced through division and differentiation of stem cells. In the case of the heart, one recent estimate is that cardiomyocytes renew at a rate of 1% annually in young adults and of less than 0.5% in old age (8). Therefore, biologic waste materials (such as irreversibly damaged mitochondria and aberrant proteins) accumulate and gradually replace normal structures, unless removed by autophagy, leading to functional decay and cell death (104). It should be noted that lipofuscin also accumulates in cultured cells undergoing replicative senescence (associated with a progressive decline in the cellular proliferation rate) and in cells whose proliferation is inhibited by pronounced density-dependent inhibition of growth [reviewed in (17)]. This suggests that lipofuscin can potentially form in various cell types, but only long-lived, nondividing cells accumulate significant amounts of the pigment.
It is now generally accepted that the aging of long-lived postmitotic cells is at least partly induced by endogenously formed ROS affecting various cellular structures, but mainly mitochondria and lysosomes (3, 41, 57, 61, 64, 94, 103). Lipofuscin formation is one of the most important manifestations of ROS-induced oxidation that occurs within the lysosomal compartment (14).
Although rapid and effective, lysosomal (autophagic) degradation is not completely perfect. Even under normal conditions, some iron-catalyzed peroxidation occurs intralysosomally, resulting in oxidative modification of the autophagocytosed material, making it resistant to the hydrolytic activity of lysosomal enzymes. If cells do not divide, this material progressively accumulates in the form of lipofuscin inclusions. Lysosomes receive a wide variety of autophagocytosed subcellular structures, most importantly mitochondria, which are rich in lipidaceous membrane components and iron-containing proteins, such as cytochrome c. That lipofuscin to a large extent originates from mitochondrial components is indicated by the presence of the ATP-synthase subunit c in age pigment or ceroid granules (34). Thus mitochondria are not only the main generators of ROS, triggering lipofuscinogenesis, but are also a major source of the macromolecules from which lipofuscin forms.
Lipofuscinogenesis has pronounced similarities to the formation of advanced glycation end products (AGEs), which are involved in the aging of connective tissue components leading to wrinkling of the skin, cataract, and the stiffening of blood vessels. These effects are thought to be a consequence of alterations in extracellular matrix proteins, which undergo polymerization and lose their elasticity because of glycosylation, with accompanying Maillard reactions (30). The difference between formation of AGEs and lipofuscin is that no sugars are involved in the formation of the latter, because the linking aldehydes are produced by degradation of oxidized lipids. Basically, lipofuscin may be regarded as a nondegradable plastic-like polymer that slowly matures by intramolecular reorganization. It has been found that lipofuscin contains oxidized proteins in which tyrosine residues have been converted to DOPA (3,4-dihydroxy-
Oxidized proteins are usually thought to be degraded by the proteasome system secondary to ubiquitination, but recent studies have shown that such proteins may also be degraded through autophagy. Studies using a cell line with a thermolabile ubiquitin-conjugating enzyme showed that oxidized proteins were still being degraded at a normal rate, even in the absence of functioning ubiquitin-conjugating enzymes (93). Although it is not clear to what extent other pathways for ubiquitin conjugation could have been active, the latter finding suggests that the proteasomal pathway is not the only one involved in the degradation of oxidized proteins (45).
By using an approach in which oxidatively modified proteins were generated in vitro by allowing cells to incorporate DOPA into proteins, it was demonstrated that mildly modified proteins were efficiently degraded by proteasomes, because this process could be inhibited by the specific proteasome inhibitor, lactacystin (87). By increasing the amount of DOPA incorporated into proteins, it was, however, possible to generate proteins that were heavily modified and eventually to generate lysosomal autofluorescent lipofuscin aggregates in cells (33). Moreover, by using inhibitors of the proteasome and lysosomal proteases, it was found that the degradation of the more highly modified aggregate-prone DOPA-containing proteins began to switch from the proteasomal to the lysosomal pathway (87). This “cooperation between proteasomes and lysosomes” implies that extensively modified proteins are no longer substrates for proteasomes and, therefore, must be redirected to the endosomal–lysosomal pathway. It also was shown that soluble and ubiquitinated misfolded proteins localize to proteasome-rich perinuclear sites, whereas terminally aggregated proteins are sequestered in autophagic vacuoles (47).
Further evidence for crosstalk between the proteasomes and lysosomes was reported for human retinal pigment epithelial (RPE) cells, in which an accumulation of perinuclear ubiquitin-, Hsp70-, and LAMP2-positive aggregates occurred in response to MG-132 (a proteasome inhibitor), with the aggregates being removed after cessation of inhibition by a mechanism thought to involve autophagy (88). In an earlier study, increased levels of lipofuscin-like autofluorescence were found in cultured SH-SY5Y neuroblastoma cells exposed to the proteasome inhibitor MG-115 (96). It is to be noted that the accumulation of lipofuscin-like material and other protein aggregates, considered an effect of proteasome inhibition, may depend also on the inhibition of lysosomal functions. MG-115 and MG-132, aldehydic peptide proteasome inhibitors, are known to inhibit not only proteasomes but also lysosomal cathepsins (19, 71). Even stronger support for proteasomal–lysosomal crosstalk was provided by a study using a highly specific proteasome inhibitor, MG-262. When exposed to this substance, cultured human fibroblasts showed increased lysosomal lipofuscin accumulation (especially pronounced under hyperoxic conditions), suggesting that modified proteins that were not degraded by proteasomes underwent autophagic degradation, contributing to lipofuscinogenesis (105).
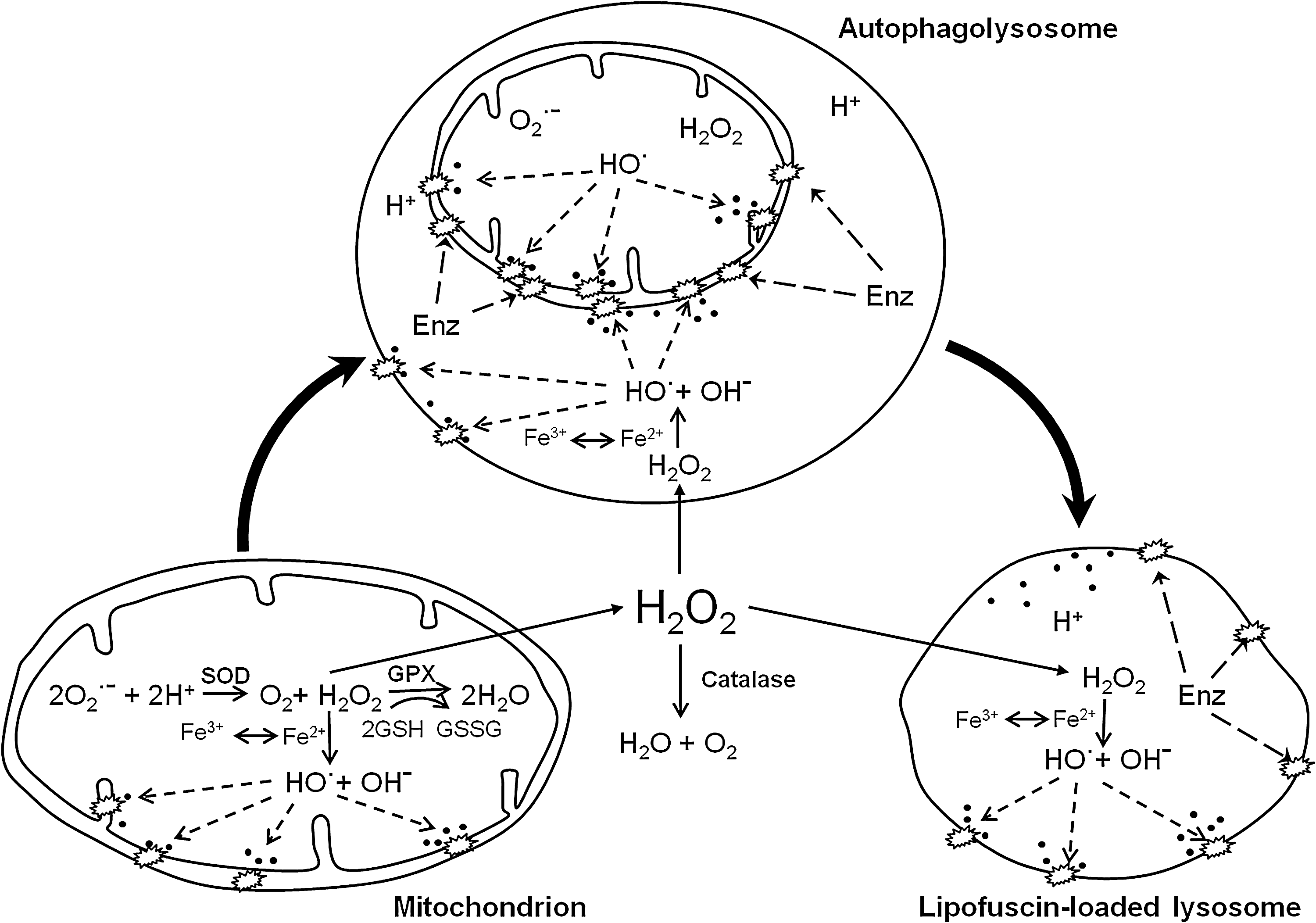
In professional scavengers, such as RPE cells and macrophages (foam cells) in atheroma, a large portion of lipofuscin (or ceroid) originates from endocytosed material (65, 78). Depending on the nature of the autophagocytosed/endocytosed material, the composition of lipofuscin varies among different types of postmitotic cells, and no chemical formula can be given for this complex substance that seems mainly to be composed of crosslinked protein and lipid residues. It should be pointed out, however, that all forms of lipofuscin contain considerable amounts of redox-active iron, which, as was mentioned before, sensitizes lipofuscin-loaded cells to oxidative stress [reviewed in (62)]. Figure 4 shows the basic mechanisms behind lipofuscinogenesis.
Consequences of lipofuscin accumulation
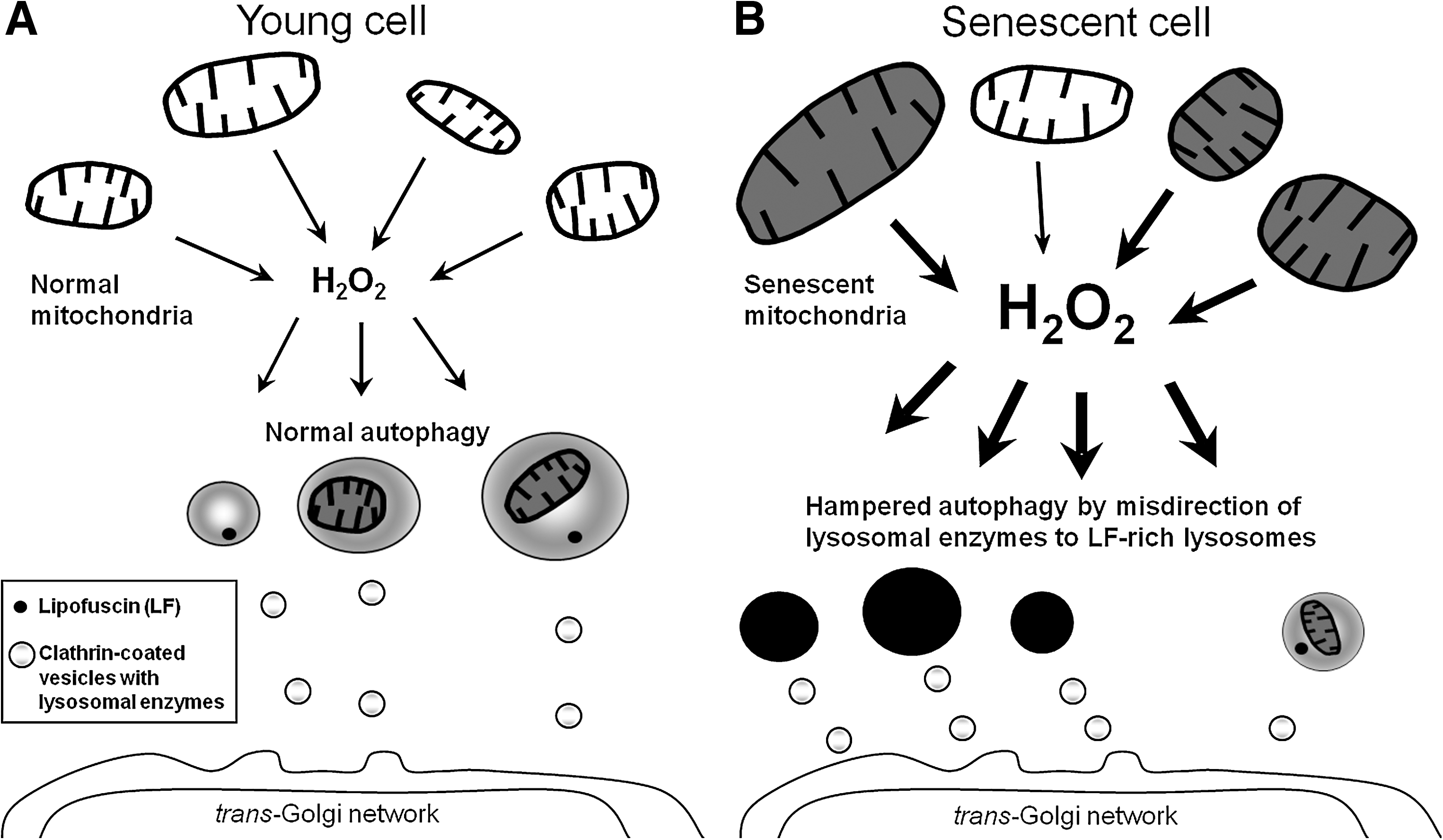
The accumulation of lipofuscin within the lysosomal compartment apparently compromises autophagic degradative capacity, prolonging the half-lives of long-lived proteins and organelles and creating a situation in which cells are forced to exercise their functions with less than perfect tools (Fig. 5). Consistent with this theory, the capacity for autophagic degradation is diminished in aged lipofuscin-loaded cells (27, 98, 102), which may lead to some serious consequences. For example, delayed degradation of mitochondria would result in accumulation of dysfunctional mitochondria and increased damage by mitochondrial ROS generation, further contributing to lipofuscinogenesis and perhaps inducing apoptotic cell death by LMP.
Recently, an elegant study on Caenorhabditis elegans added new evidence for the hypothesis that lipofuscin accumulation is causally related to aging and to the deterioration of postmitotic cells. Fortunately, these tiny nematodes are transparent, which permits lipofuscin to be measured directly by spectrofluorometry in vivo. It was found that mutant nematodes that live either for longer or shorter periods than wild-type animals accumulate lipofuscin at a slower or quicker pace, respectively. It was also found that calorie-restricted worms lived longer and accumulated lipofuscin more slowly than did animals fed ad libitum. Finally, when wild-type siblings that aged differently, as evaluated by changes in their motility, were compared, it was found that the still mobile and youthful ones at day 11 of their life spans contained only 25% of the lipofuscin that was found in severely motility-impaired siblings of the same age. This implies that lipofuscin accumulation reflects biologic rather than chronologic age (36).
Besides accumulating the intralysosomal “waste” material lipofuscin, aging postmitotic cells also form extralysosomal “garbage,” such as damaged dysfunctional mitochondria and indigestible protein aggregates (aggresomes) that for some reason are not efficiently autophagocytosed or degraded by the proteasome pathway [reviewed in (100, 101)]. Aged mitochondria are enlarged and show considerably reduced fusion and fission activity. Their autophagy may be prevented by their size, because the autophagy of large structures is apparently energy consuming, and autophagosomes seem to have an upper volume limit (76, 104).
Lysosomes and Ionizing Irradiation
Exposure of cells to ionizing radiation not only destabilizes lysosomes, because of the resulting formation of superoxide and hydrogen peroxide that interact with lysosomal redox-active iron, as described earlier, but also triggers reparative autophagy of damaged intracellular material (82). Coincident with the increased autophagy, the lysosomal apparatus increases in volume. This enhanced autophagy has major consequences, inasmuch as still viable cells exposed to a second dose of radiation appear to be more radiosensitive. This, in turn, may be due to an increase in lysosomal “loose” iron, which results from enhanced intralysosomal digestion of ferruginous material, perhaps most important, mitochondria (Fig. 6).

The damage caused by ionizing radiation appears to be due, in part, to reactions involving intralysosomal iron. Support for this derives from experiments in which pretreatment of target cells with a high-molecular-weight derivative of the iron chelator, desferrioxamine, both stabilizes lysosomes and partially protects cells against radiation-induced killing (82). Desferrioxamine does not penetrate the plasma membrane, is taken up by endocytosis, remains in the lysosomal apparatus, and suppresses oxidant-induced LMP.
Lysosomes and Metal Chelators
Water- and lipid-soluble iron chelators, such as 1-propyl-2-methyl-3-hydroxypyrid-4-one (CP22) and salicylaldehyde isonicotinoyl hydrazone (SIH), also are protective against oxidative stress and, thereby, are radioprotective. These chelators can be washed away (unlike desferrioxamine, which cannot leave the lysosomes and ultimately kills cells by iron depletion) and will cause no further interference with iron metabolism (60, 115). Overall, these observations suggest that cells with more reactive iron should be more sensitive to ionizing radiation and that chelators that suppress the redox activity of iron are radioprotective. Although firm data are presently lacking, it may be that types of tumors known to be highly radiosensitive may also be relatively rich in reactive iron or, alternatively, have decreased antioxidant protection. Further to this point, it has been found that radiation-resistant tumors often contain high levels of metal-binding proteins (such as ferritin, metallothioneins, and heat-shock proteins), all of which will act to reduce the amounts of intralysosomal redox-active iron after autophagy (59).
Up to this point, we have emphasized the importance of intralysosomal iron in sensitization to oxidants, radiation, and other insults. However, iron is not the only transition metal present in lysosomes; copper also is present because copper-containing biomolecules, such as SOD and mitochondrial complex IV, are also degraded by autophagy. In contrast to iron, copper is not in redox-active form in the lysosomal compartment because this metal forms stable non-redox–active complexes with thiol-containing compounds, such as cysteine and GSH.
Recently, a number of tridentate chelators were developed and tried as cytostatics, the rationale being that they would induce iron starvation specifically in malignant cells (84). Some of these have very low LD50 values and, interestingly, are also active when applied in the form of iron complexes, creating some doubt about their activity being exclusively a result of iron chelation. The structural formulas and pK a values of several of these metal chelators indicate that they are lysosomotropic and will undergo “proton trapping” in an acidic compartment. If these tridentate chelators bind copper as well as iron, the tridentate copper chelates should be very redox-active at a 1:1 chelator/copper ratio (iron is bound at a 2:1 chelator/iron ratio) and be able to react with a variety of reducing agents, including the abundant intralysosomal cysteine. Copper is a more potent catalyst of Fenton-type reactions than is iron. Consequently, the small amounts of hydrogen peroxide that normally reach the lysosomal compartment by diffusion might be enough to induce substantial production of hydroxyl radicals in lysosomes, especially in malignant cells that may be already under increased oxidant stress.
Interestingly, many malignancies are significantly richer in copper than are normal cells [reviewed in (20, 40)], and we may thus assume that tumor lysosomes should be rich in low-mass copper. Neocuproine and diethyl dithiocarbamate are examples of such lysosomotropic copper chelators that may promote the redox activity of copper by binding fewer than all coordinates leading to LMP. The well-known tendency of tumor cells to generate excess acid may further promote the accumulation of chelators susceptible to proton trapping. We expect to see, in the near future, the initiation of clinical trials on selected copper chelators, because they have already proven effective on malignant cells in culture (20, 21, 40).
Summary and Conclusions
The lysosomal compartment constitutes a redox-active partition of the cell. It contains low-mass iron and copper liberated as a consequence of autophagic degradation of metalloproteins. Its acidic interior and high concentrations of thiols, such as cysteine and GSH, allow iron to exist in partly redox-active form. The reduced metal can react with hydrogen peroxide (most of it incidentally generated by mitochondria), leading to intralyosomal Fenton reactions and resulting in the formation of lipofuscin. This “age pigment” accumulates in long-lived postmitotic cells that cannot dilute it by division. Extensive accumulation of lipofuscin seems to hinder normal autophagy and may be an important factor behind aging and age-related pathologies. Enhanced oxidative stress causes lysosomal membrane permeabilization, with ensuing relocation to the cytosol of iron and lysosomal hydrolytic enzymes that in turn may induce apoptosis or necrosis. Lysosomal copper is probably not redox active, because it will complex in a non–redox-active form by various thiols. However, if cells are exposed to lysosomotropic copper chelators that do not bind all coordinates of copper, a highly redox-active complex could be formed that may produce extensive lysosomal Fenton-type reactions, with ensuing loss of lysosomal stability. Because many malignancies seem to have increased amounts of copper-containing macromolecules that are necessarily turned over by autophagy, it is conceivable that lysosomotropic copper chelators may in the future be used in ROS-based anticancer therapies.