
Abstract
Pulmonary vascular remodeling associated with pulmonary hypertension is characterized by media thickening, disordered proliferation, and in situ thrombosis. The p21-activated kinase-1 (PAK-1) can control growth, migration, and prothrombotic activity, and the hypoxia-inducible transcription factor HIF-1α was associated with pulmonary vascular remodeling. Here we studied whether PAK-1 and HIF-1α are linked in pulmonary vascular remodeling. PAK-1 was expressed in the media of remodeled pulmonary vessels from patients with pulmonary vasculopathy and was upregulated, together with its upstream regulator Rac1 and HIF-1α in lung tissue from lambs with pulmonary vascular remodeling. PAK-1 and Rac1 were activated by thrombin involving calcium, thus resulting in enhanced generation of reactive oxygen species (ROS) in human pulmonary artery smooth muscle cells (PASMCs). Activation of PAK-1 stimulated HIF activity and HIF-1α expression involving ROS and NF-κB, enhanced the expression of the HIF-1 target gene plasminogen activator inhibitor-1, and stimulated PASMC proliferation. Importantly, HIF-1 itself bound to the Rac1 promoter and enhanced Rac1 and PAK-1 transcription. Thus, PAK-1 and its activator Rac1 are novel HIF-1 targets that may constitute a positive-feedback loop for induction of HIF-1α by thrombin and ROS, thus explaining elevated levels of PAK-1, Rac1, and HIF-1α in remodeled pulmonary vessels. Antioxid. Redox Signal. 13, 399–412.
Introduction
The p21-activated kinase-1 (PAK-1) has been implicated in signaling cascades regulating cytoskeletal remodeling, migration, proliferation, and survival (2, 14, 26), and we previously showed that PAK-1 plays an important role in promoting a prothrombotic state by upregulating tissue factor expression in response to thrombin (16). PAK-1 belongs to a highly conserved serine/threonine kinase family with six members in eukaryotes. Although several Rho GTPase-independent activation mechanisms have been reported (2, 14, 26), PAK-1 becomes activated primarily by GTP-bound Rac1, which is a member of the RhoGTPase family of proteins. In its inactive state, PAK-1 exists as a dimer in a trans-autoinhibitory conformation in which the autoinhibitory domain of one PAK-1 molecule blocks the catalytic domain of the other. Binding of Rac-GTP induces a series of conformational changes, which start with the disruption of the dimer and end with the release of the kinase domain in a stable catalytically active conformation. Furthermore, phosphorylation of threonine 423 (T423) within the activation loop is critical for PAK-1 activation.
In addition to PAK-1 and thrombin, the hypoxia-inducible transcription factor HIF-1 has been suggested to play an important role in pulmonary vascular remodeling and pulmonary hypertension in response to hypoxia (48). It is composed of two subunits: the constitutively expressed HIF-1β and the inducible HIF-1α, of which the latter is regulated by the cellular O2 concentration (38). The predominant oxygen-dependent regulation of HIF-1α occurs posttranslationally. Under normoxic conditions, HIF-1α becomes hydroxylated within the oxygen-dependent degradation domain overlapping the N-terminal transactivation domain. This allows physical interaction with the tumor-suppressor von Hippel–Lindau protein (pVHL), which promotes HIF-1α ubiquitinylation and degradation by the 26S proteasome (37). With hypoxia, hydroxylase activity is diminished, and nonhydroxylated HIF-1α escapes the proteasomal degradation, accumulates, and dimerizes with HIF-1β to bind to specific hypoxia-responsive elements (HREs) in the promoters or enhancers of target genes (34). In addition to hypoxia, HIF-1α also can be induced and activated under normoxic conditions in response to a variety of stimuli, including thrombin and oxidative stress, which can involve a transcriptional mechanism (3, 6, 18). Thus, this transcription factor may contribute to physiologic and pathophysiologic processes also independently of hypoxia.
Because PAK-1 was shown to control growth, migration, and prothrombotic activity, and HIF-1α was suggested to be involved in pulmonary vascular remodeling, we hypothesized that PAK-1 and HIF-1α are linked in pulmonary vascular remodeling. Thus, it was the aim of the present study to investigate the signaling pathways linking PAK-1, HIF-1α, and vascular remodeling processes in vivo and in thrombin-stimulated pulmonary artery smooth muscle cells, the main cell type involved in pulmonary vascular remodeling.
Materials and Methods
Reagents
All chemicals were of analytic grade and were purchased from Sigma (Taufkirchen, Germany) if not otherwise stated.
Cell culture
Human pulmonary artery smooth muscle cells (PASMCs) were obtained from Lonza (Cologne, Germany) and cultured in the medium provided, as recommended. PASMCs (passages 3 to 12) were serum deprived for 24 h before stimulation. A7r5 rat smooth muscle cells (rSMCs) and HepG2 cells (ATCC) were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco, Karlsruhe, Germany) with 10% fetal calf serum. Cells were serum starved for 16 h before experiments.
Animal experiments
As a model for pulmonary vascular remodeling, we used an ovine model of congenital heart disease with increased pulmonary blood flow, as described previously (32). Pregnant ewes (135–143 days' gestation; term, 145 days) underwent in utero surgery to anastomose an 8.0-mm Gore-tex vascular graft (W. L. Gore, Milpitas, CA) between the ascending aorta and main pulmonary artery of the fetus. The incisions in the uterus and the abdomen were closed, and the sheep were allowed to deliver normally. Three weeks after spontaneous delivery, lambs were anesthetized with propofol (0.2 ml, IV), diazepam (0.12 mg/kg/h, IV), and ketamine hydrochloride (0.18 mg/kg/h, IV), intubated, mechanically ventilated, and a midsternotomy incision was performed. Oxygen saturation was evaluated in the aorta, right ventricle, right atrium, and distal pulmonary artery, and hemodynamic measurements were performed to confirm graft potency, increased pulmonary blood flow, and mean pulmonary arterial pressure. Biopsies of peripheral lung tissue were harvested from randomly selected lobes. At the end of the protocol, all lambs were killed with a lethal injection of 7.45% KCl. All protocols and procedures were approved by the local legislation on protection of animals (Regierung von Oberbayern, Munich, Germany).
Immunohistochemistry
Archival lung tissue from patients with pulmonary vascular disease associated with pulmonary hypertension was formalin fixed under vacuum and paraffin embedded. Immunohistochemistry was performed by using antibodies against PAK-1 (Abcam, Cambridge, United Kingdom) and α-actin (Dako, Glostrup, Denmark), as previously described (1). Counterstaining was performed by using Hemalum, and specificity of the antibody staining was tested by omitting the primary antibody.
Plasmids
The expression vectors encoding myc-tagged PAKT423E or PAKR295, RacT17N or RacG12V, pcDNA3.1HIF-1α (HIF-1α), the luciferase vectors pGL3-EPO-HRE-luc and NFkB-luc, and the luciferase constructs containing the human wildtype or mutated PAI-1 promoter, PAI-1-796 or PAI-1-796m, respectively, or the human wildtype or mutated HIF-1α promoter, HIF-1α-538 or HIF1α-538m, respectively, have been described (3, 8, 16, 17, 43). Short-hairpin RNA targeting HIF-1α or control shRNA (siCtr) were previously described (3). The luciferase construct containing the genomic 5′ HIF-1α sequence from bp −538 up to +284 (HIF-1α-538) was kindly provided by Dr. C. Michiels (Namur, Belgium) (30).
Transfections and luciferase assays
PASMCs were cultured for 24 h to a density of 70%, and transfections were as described (10). Luciferase activity assays were performed in rSMCs, which were transfected by using Superfect reagent (Qiagen, Hilden, Germany), according to the manufacturer's instructions (10).
Northern blot analysis, RT-PCR, and real time-PCR
Total RNA from pulmonary artery smooth muscle cells was isolated by using the RNeasy Mini Kit (Qiagen), according to the manufacturer's instructions (3). Then 10 μg RNA was subjected to Northern blot analysis. Hybridizations were performed with digoxigenin-labeled antisense RNA probes for PAI-1 or HIF-1α at 65°C (3, 25). For PCR analysis, first-strand cDNA was synthesized from 1 μg RNA by using reverse transcriptase (Invitrogen, Karlsruhe, Germany). PCR analysis was performed by using Phusion High-Fidelity DNA Polymerase (New England Biolabs, Frankfurt, Germany) with the following primers to detect Rac1, 5′-CCC TAT CCT ATC CGC AAA CA-3′ as forward and 5′-CAG CAG GCA TTT TCT CTT CC-3′ as reverse primer (366-bp PCR product); PAK-1, 5′-CGT GGC TAC ATC TCC CAT TT-3′ as forward and 5′-GGA TCG CCC ACA CTC ACT AT-3′ as reverse primer (150 bp PCR product); HIF-1α, 5′-GAA GAC ATC GCG GGG AC-3′ as forward and 5′-TGG CTG CAT CTC GAG ACT TT-3′ as reverse primer (105-bp PCR product); β-actin, 5′-CCA ACC GCG AGA AGA TGA-3′ as forward and 5′-CCA GAG GCG TAC AGG GAT AG-3′ as reverse primer (97-bp PCR product). The PCRs for Rac1, PAK-1, and β-actin were run for 28 cycles in the following cycle profile: 98°C for 15 s, 59°C for 15 s, and 72°C for 10 s. The PCR for HIF-1α was run for 30 cycles in the following cycle profile: 98°C for 10 s, 63°C for 10 s, and 72°C for 10 s. PCR products were separated on a 2% agarose gel, stained with ethidium bromide, and visualized by using GelDoc software (BioRad, Munich, Germany). PCR products were verified by sequence analysis. Real time-PCR analysis was performed by using the PerfeCTa SYBR Green FastMix (VWR) in a Rotorgene 6000 (LTF, Wasserburg, Germany) with the following primers to quantify Rac1 mRNA levels: 5′-AGG AAG AGA AAA TGC CTG-3′ as forward and 5′-AGC AAA GCG TAC AAA GGT-3′ as reverse primer: To detect PAK-1 and β-actin, the primers described were used. Quantification was performed by using ΔCT calculation.
Chromatin immunoprecipitation
Confluent HepG2 cells were serum starved for 16 h and exposed to thrombin (3 U/ml) for 3 h. Cells were fixed with formaldehyde, lysed, and sonicated to obtain DNA fragments in a size from 500 to 1,000 base pairs. Chromatin was then precipitated with an HIF-1α antibody (Novus, Herford, Germany) overnight at 4°C, and real-time PCR was performed with primers for the Rac1 promoter (forward: 5′-CGT ATG GTG TAT GAT TTT ATT CTT CC-3′, reverse: 5′-GGA GCC ATG TTC CTT GTC AC-3′), the PAK-1 promoter (forward: 5′-CAG TTG GGA GGG AAA ACA AA-3′, reverse: 5′-GCA AAC CAA CTG CTC CTT CT-3′) and as positive control for the PAI-1 promoter (forward, 5′-GCT CTT TCC TGG AGG TGG TC-3′; and reverse: 5′-GGA GCA CAG AGA GAG TCT GGA-3′) by using a Rotorgene 6000 (Corbett). As negative control to analyze unspecific binding and precipitation, real time-PCR using primers amplifying a region of the third intron of β-actin (gene ID: 60) without a putative HRE (5′-ACGTG-3′) was performed (forward: 5′-AAC ACT GGC TCG TGT GAC AA-3′; reverse: 5′-AAA GTG CAA AGA ACA CGG CT-3′). As background control, Chip with isotype IgG antibody (Santa Cruz, Heidelberg, Germany) was performed. Statistical analysis was performed by using a standard curve of the input, and increased HIF-1α binding to chromatin is represented after background subtraction as the relative amount of the input.
Western blot analysis
Western blot analysis was performed by using antibodies against HIF-1α, Rac1 (BD Transduction, Heidelberg, Germany), PAI-1 (American Diagnostica, Pfungstadt, Germany), phospho-PAK-1 (Thr423)/PAK-2 (Thr402), and PAK-1 (Cell Signaling, Frankfurt, Germany), c-myc (Santa Cruz), or actin (Sigma). Goat anti-mouse or anti-rabbit immunoglobulin G (Calbiochem) was used as secondary antibody. Blots were scanned and analyzed by using GelDoc software (BioRad).
Measurement of ROS production
The generation of ROS in PASMCs was measured by using the fluoroprobe 5-(and -6)chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCFDA, Molecular Probes, Karslruhe, Germany), as described (9).
Rac1 activity assay
Rac1 activity was evaluated with an affinity precipitation assay by using the PAK-1 PBD conjugated glutathione agarose beads, according to the manufacturer's instructions (Upstate, Hamburg, Germany), as described previously (10).
Proliferation assay
Proliferative activity of pulmonary artery smooth muscle cells was evaluated with 5-bromo-2′-deoxyuridine (BrdU) labeling (Roche Diagnostics, Mannheim, Germany), according to the manufacturer's instructions, as described (11).
Statistical analysis
Values are presented as mean ± standard deviation (SD). Results were compared with ANOVA for repeated measurements followed by the Student–Newman–Keuls t test. A value of p < 0.05 was considered statistically significant.
Results
The p21-activated kinase-1 is involved in pulmonary vascular remodeling
Enhanced proliferation of pulmonary artery smooth muscle cells (PASMCs) leads to media thickening of pulmonary vessels as a hallmark of pulmonary vascular remodeling. To determine whether PAK-1, as a central regulator of migratory and proliferative events, may be associated with pulmonary vascular remodeling, we evaluated PAK-1 in lung biopsies from patients with pulmonary vascular remodeling. Interestingly, PAK-1 was found primarily in the media of remodeled pulmonary vessels colocalizing with α-actin as a marker of smooth muscle cells (Fig. 1A), suggesting that PAK-1 may be important during the course of media hypertrophy of pulmonary vessels. To test this assumption further, we determined PAK-1 in lung tissues derived from a lamb model of pulmonary vascular remodeling. On intrauterine application of an aortopulmonary shunt, the lambs displayed pulmonary vascular remodeling 3 weeks after birth, compared wih their control nonshunted siblings. Western blot analyses of lung-tissue samples from shunt lambs demonstrated enhanced levels of PAK-1 protein compared with those of their control siblings. Similarly, the levels of Rac1, the upstream activator of PAK-1, as well as of HIF-1α, were enhanced in shunt lambs compared with control lambs (Fig. 1B). These data led us hypothesize that Rac1, PAK-1, and HIF-1α may be directly linked in pulmonary vascular remodeling.
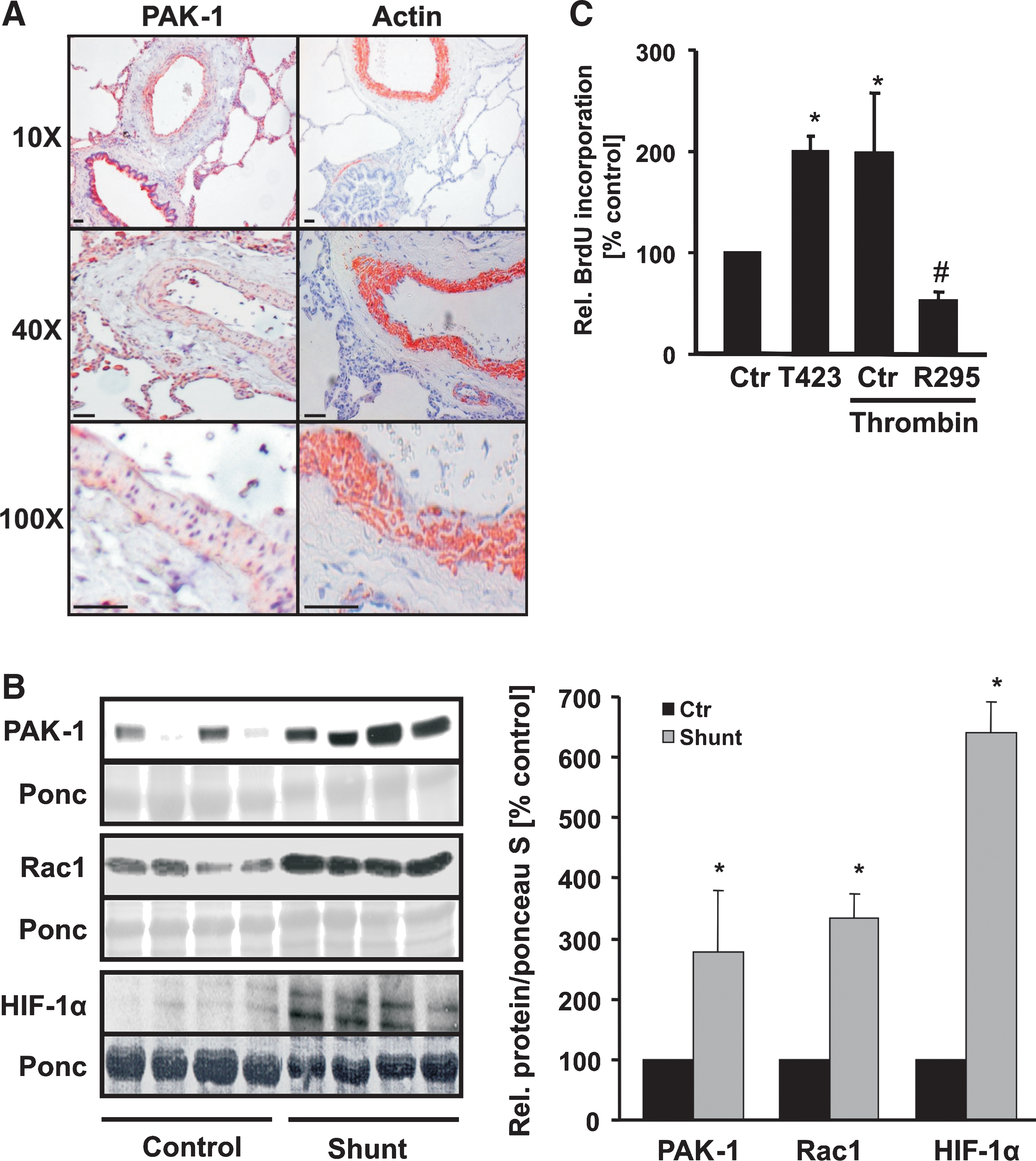
In addition to enhanced proliferative activity of pulmonary artery smooth muscle cells (PASMCs), a prothrombotic state and fibrin deposition are frequently observed in pulmonary vascular remodeling. We thus next determined whether PAK-1 is involved in the control of PASMC proliferation in response to thrombin. PASMCs were exposed to thrombin, and the proliferation of PASMCs was evaluated with BrdU incorporation (Fig. 1C). Thrombin-stimulated proliferation of PASMCs was completely abrogated in PASMCs expressing a dominant-negative PAK-1 variant (PAKR295). In contrast, overexpression of a constitutively active PAK-1 variant (PAKT423E) enhanced the proliferative activity of PASMC to a similar extent as thrombin, indicating a substantial role of PAK-1 in this response.
Thrombin activates and upregulates p21-activated kinase-1
To test whether PAK-1 is responsive to thrombin, we exposed PASMCs for increasing periods to thrombin. Western blot analysis revealed, consistent with previous reports that thrombin rapidly, but transiently increased phosphorylation of PAK at Thr 423 (16), starting at 30 s of exposure, which reached maximal levels at 1 min and returned to basal levels after 7 min (Fig. 2A). Because PAK-1 can be activated by direct interaction with the GTPase Rac1, we tested whether activation of PAK-1 by thrombin is dependent on Rac1. PASMCs were transfected with an active Rac1 mutant (RacG12V) or dominant-negative Rac1 (RacT17N). Whereas RacG12V increased PAK-1 phosphorylation, RacT17N reduced thrombin-stimulated phosphorylation of this kinase (Fig. 2B).

Thrombin has been previously described to increase intracellular calcium levels (36), and we therefore investigated whether calcium could also influence activation of PAK-1. To this end, PASMCs were pretreated with the intracellular calcium chelator BAPTA-AM (7.5 μM) for 30 min. In the presence of BAPTA-AM, thrombin-induced PAK-1 phosphorylation and activation of Rac1 were diminished (Fig. 2C and D), indicating that calcium release is upstream of PAK-1 activation by thrombin.
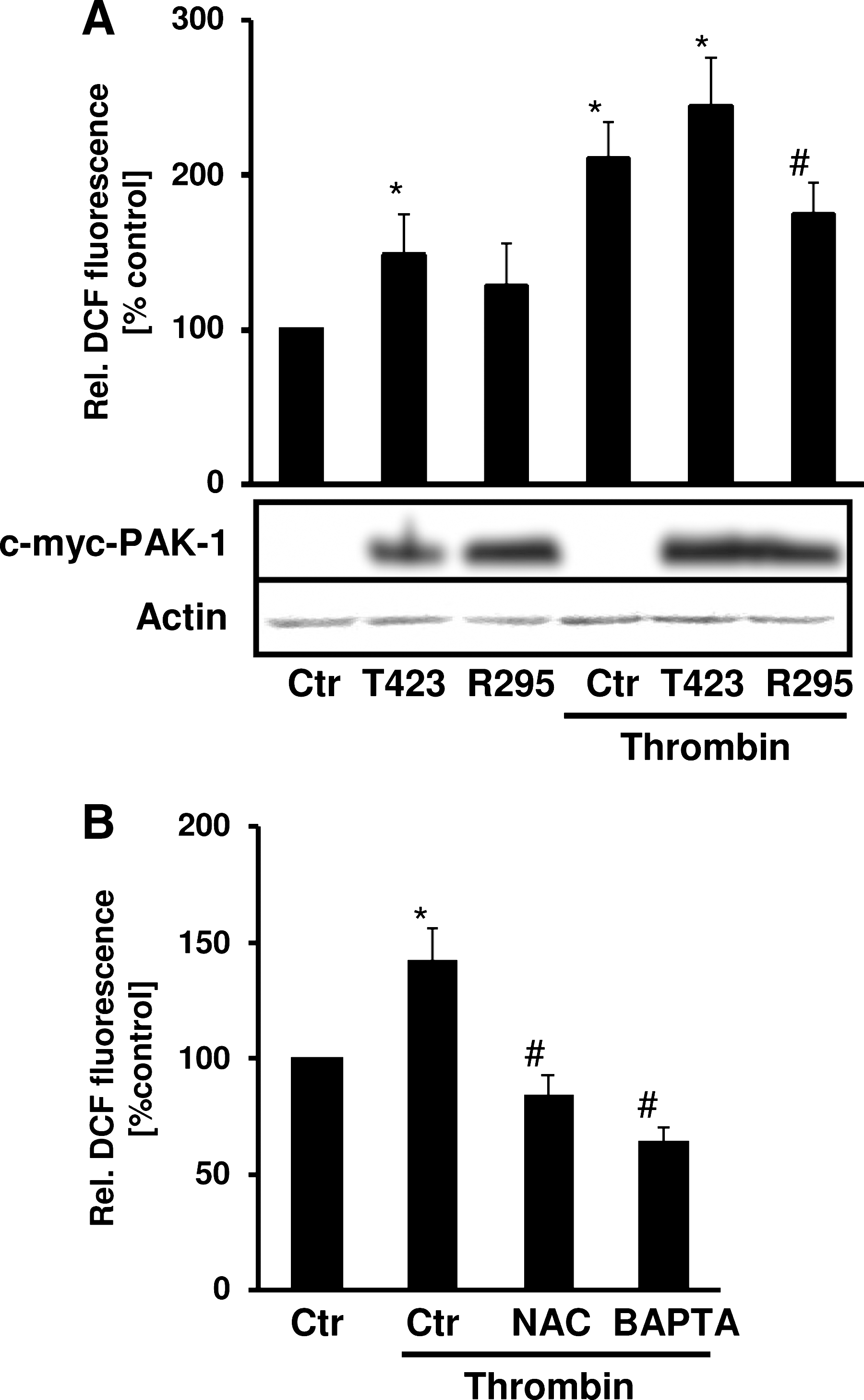
In addition, Rac1 has been shown to increase ROS production in response to thrombin (10). We thus hypothesized that PAK-1 may also be involved in the regulation of ROS production by thrombin. Expression of dominant-negative PAK-1 decreased thrombin-induced ROS production, whereas expression of active PAK-1 increased ROS production. This response was enhanced in the presence of thrombin, indicating that PAK-1 activation mediates ROS production (Fig. 3A). ROS levels in the presence of thrombin were decreased by addition not only of the antioxidant N-acetylcysteine (NAC) but also by BAPTA-AM (Fig. 3B).
The p21-activated kinase-1 activates HIF-1 and increases HIF-1α expression
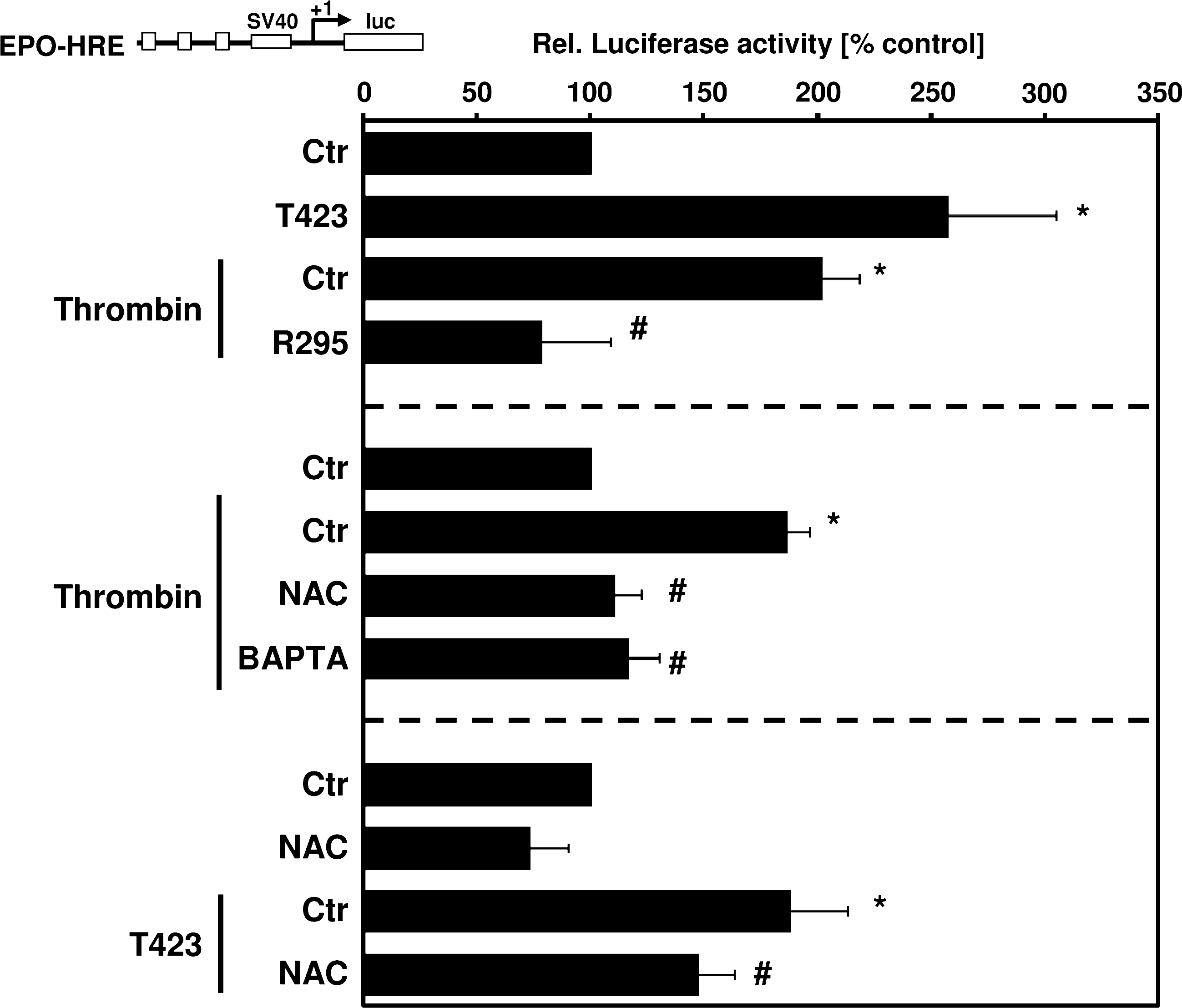
Next, we investigated whether PAK-1 is involved in activating HIF-1 in response to thrombin. First, we determined the involvement of PAK-1 in regulation of HIF-1 activity by using a luciferase reporter gene construct containing three hypoxia-responsive elements (HRE) known to bind HIF-1 in front of the SV40 promoter (pGl3-EPO-HRE-Luc). Thrombin and active PAKT423E increased HIF-1 activity (Fig. 4), whereas PAKT423E-stimulated HIF-1 activity was decreased in the presence of NAC, indicating that PAK-1–mediated ROS production is involved in the regulation of HIF-1 activity. Further, thrombin-stimulated luciferase activity was abrogated in the presence of dominant-negative PAKR295 and also was diminished by BAPTA-AM and NAC (Fig. 4), indicating involvement of calcium and ROS in this pathway.
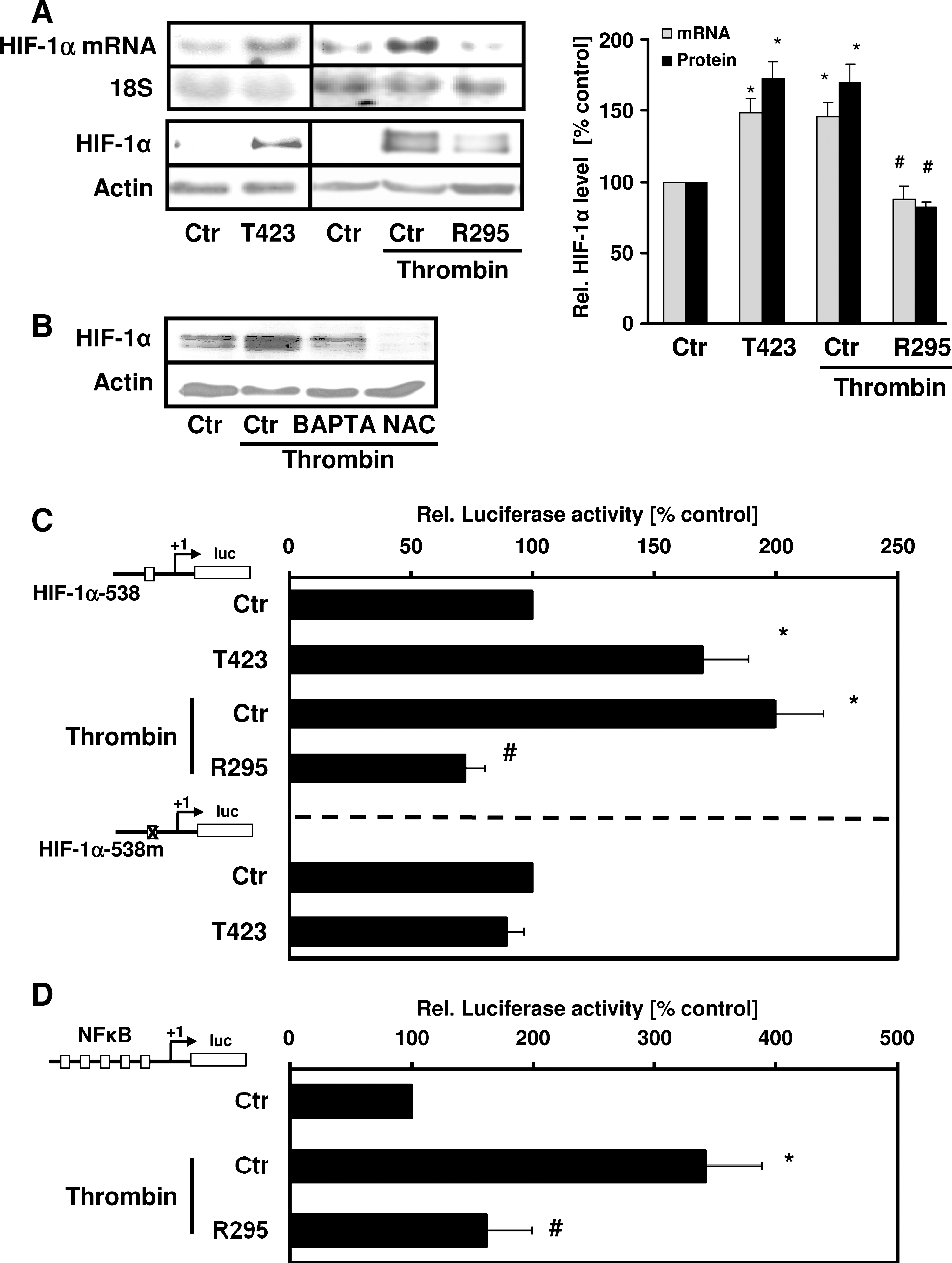
In the next step, we investigated whether PAK-1 is involved in the regulation of the HIF-1α subunit. Overexpression of active PAKT423E increased HIF-1α protein levels in PASMCs compared with cells transfected with control vector (Fig. 5A). In contrast, PAKR295 abrogated HIF-1α upregulation by thrombin, indicating that activation of PAK-1 results in increased HIF-1α levels. BAPTA-AM and NAC pretreatment decreased HIF-1α upregulation by thrombin (Fig. 5A). Because we have shown that thrombin can also increase HIF-1α at the transcriptional level, involving the activation of NF-κB (3), we tested whether PAK-1 would regulate HIF-1α mRNA levels. PASMC transfected with PAKT423E showed enhanced HIF-1α mRNA levels, whereas PAKR295 prevented thrombin-dependent upregulation of HIF-1α mRNA (Fig. 5B). In addition, active PAK-1 increased (similar to the situation with thrombin) HIF-1α promoter activity (Fig. 5C), whereas PAKR295 decreased HIF-1α promoter activity in response to thrombin. However, PAKT423E did not affect luciferase activity of a HIF-1α–promoter construct where an NF-κB binding site at −197/−188 bp was mutated, indicating that NF-κB mediates PAK-1–induced HIF-1α promoter activity. Thrombin-induced NF-κB activity was diminished in the presence of PAKR295 (Fig. 5D). Together, these data indicate that the generation or release (or both) of calcium by thrombin activates PAK-1 and ROS generation, leading to the activation of NF-κB and the subsequent transcriptional induction of HIF-1α.
The p21-activated kinase-1 regulates PAI-1 expression
Plasminogen-activator inhibitor-1 (PAI-1) is a serine protease inhibitor that targets the two plasminogen activators, tissue-type plasminogen activator (t-PA) and urokinase-type plasminogen activator (u-PA) (13), thereby maintaining normal hemostasis by limiting the activity of the fibrinolytic system. Furthermore, enhanced PAI-1 levels have been shown to promote fibrin deposition in several animal models and have been associated with tissue remodeling (39). Because PAI-1 is a known HIF-1α target (25), we determined the involvement of PAK-1 in the regulation of PAI-1. To this end, PASMCs were transfected with PAKT423E or PAKR295. Whereas PAKT423E enhanced PAI-1 protein levels and PAI-1 promoter activity (Fig. 6A and B), PAKR295 diminished thrombin-stimulated PAI-1 protein levels and promoter activity (Fig. 6A and B), confirming that this kinase regulates PAI-1 expression by thrombin.
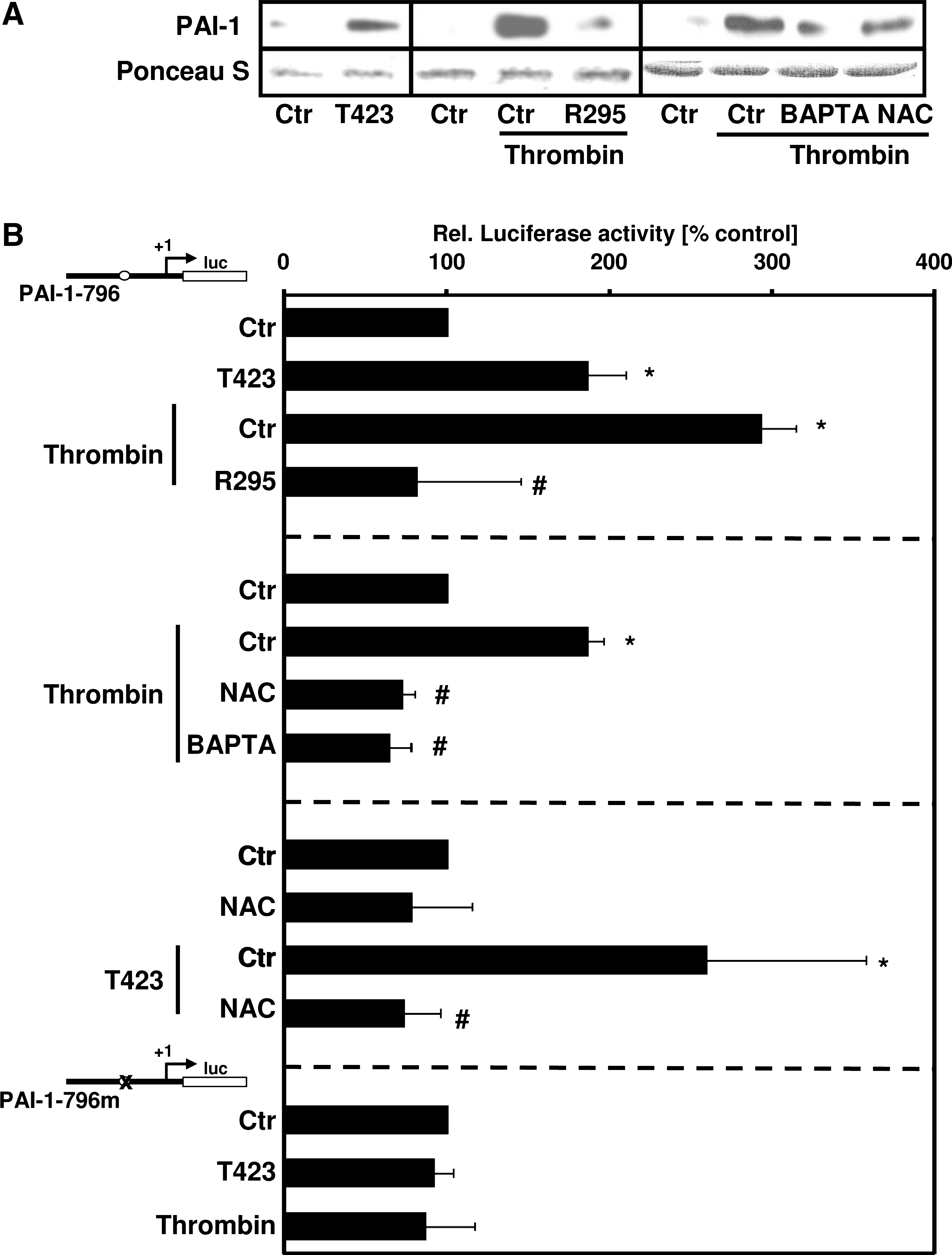
Similar to thrombin, PAKT423E was not able to increase luciferase activity of a PAI-1–promoter construct in which the HIF-1–binding site was mutated (Fig. 6B). In addition, thrombin-induced PAI-1 expression and promoter activity were diminished by NAC or BAPTA-AM (Fig. 6A and B), further indicating that calcium and ROS are involved not only in the regulation of HIF-1α but also in PAI-1 expression.
Rac1 and p21-activated kinase-1 are upregulated by thrombin in an HIF-1α–dependent pathway
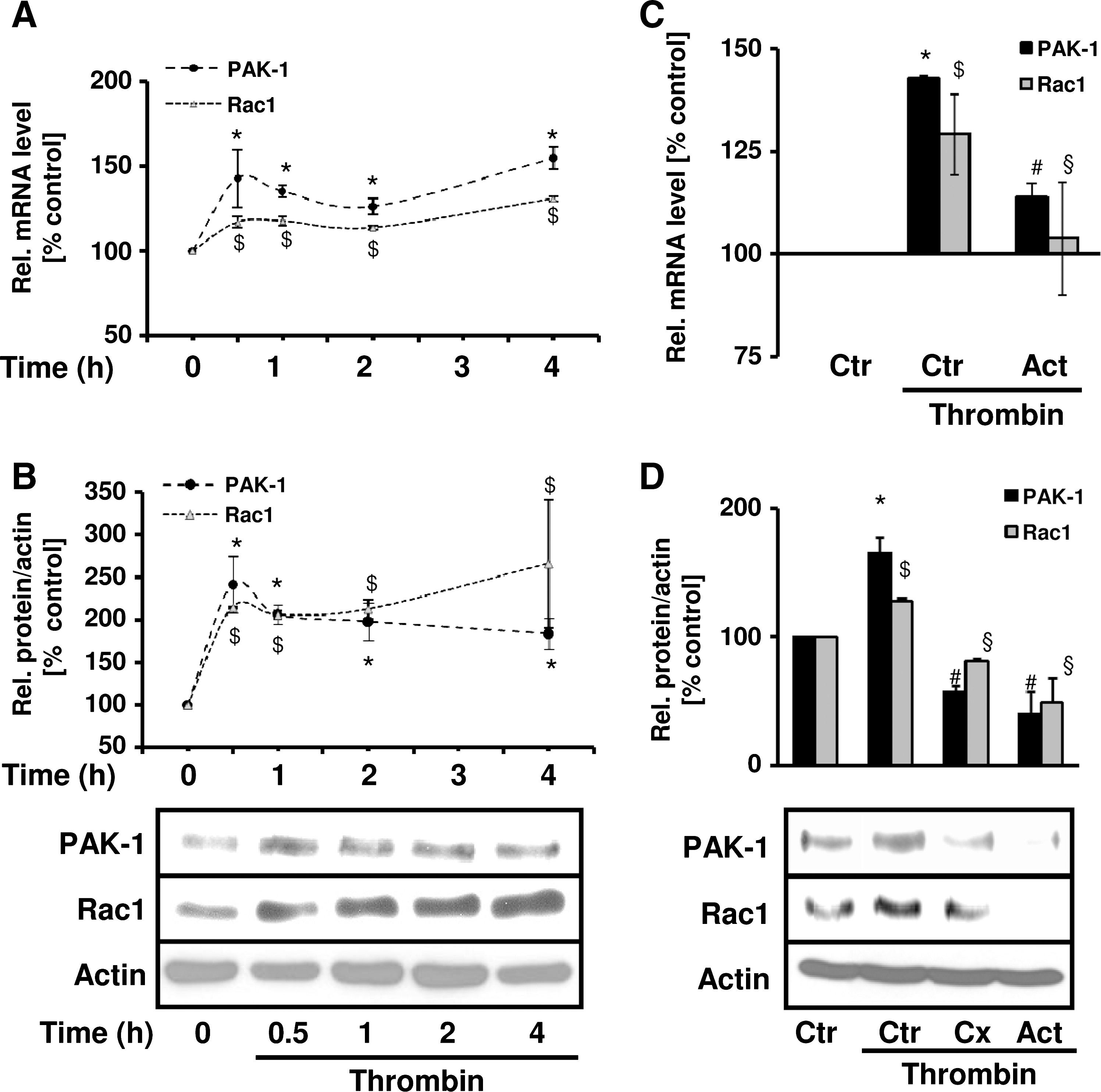
Because our in vivo experiments demonstrated enhanced levels not only of HIF-1α, but also of PAK-1 and Rac1 in pulmonary vascular remodeling, we investigated possible regulatory mechanisms underlying this observation. To this end, we exposed PASMCs to thrombin and determined Rac1 and PAK-1 expression. Interestingly, thrombin enhanced not only Rac1 and PAK-1 protein levels in a time-dependent manner, but also Rac1 and PAK-1 mRNA levels (Fig. 7A and B). To test the involvement of a transcriptional mechanism in this response, PASMCs were pretreated with actinomycin D. This treatment diminished thrombin-induced Rac1 and PAK-1 mRNA and protein levels (Fig. 7C and D). Treatment with cycloheximide prevented the upregulation of Rac1 and PAK-1 protein levels by thrombin (Fig. 7D), suggesting also the involvement of de novo protein synthesis.
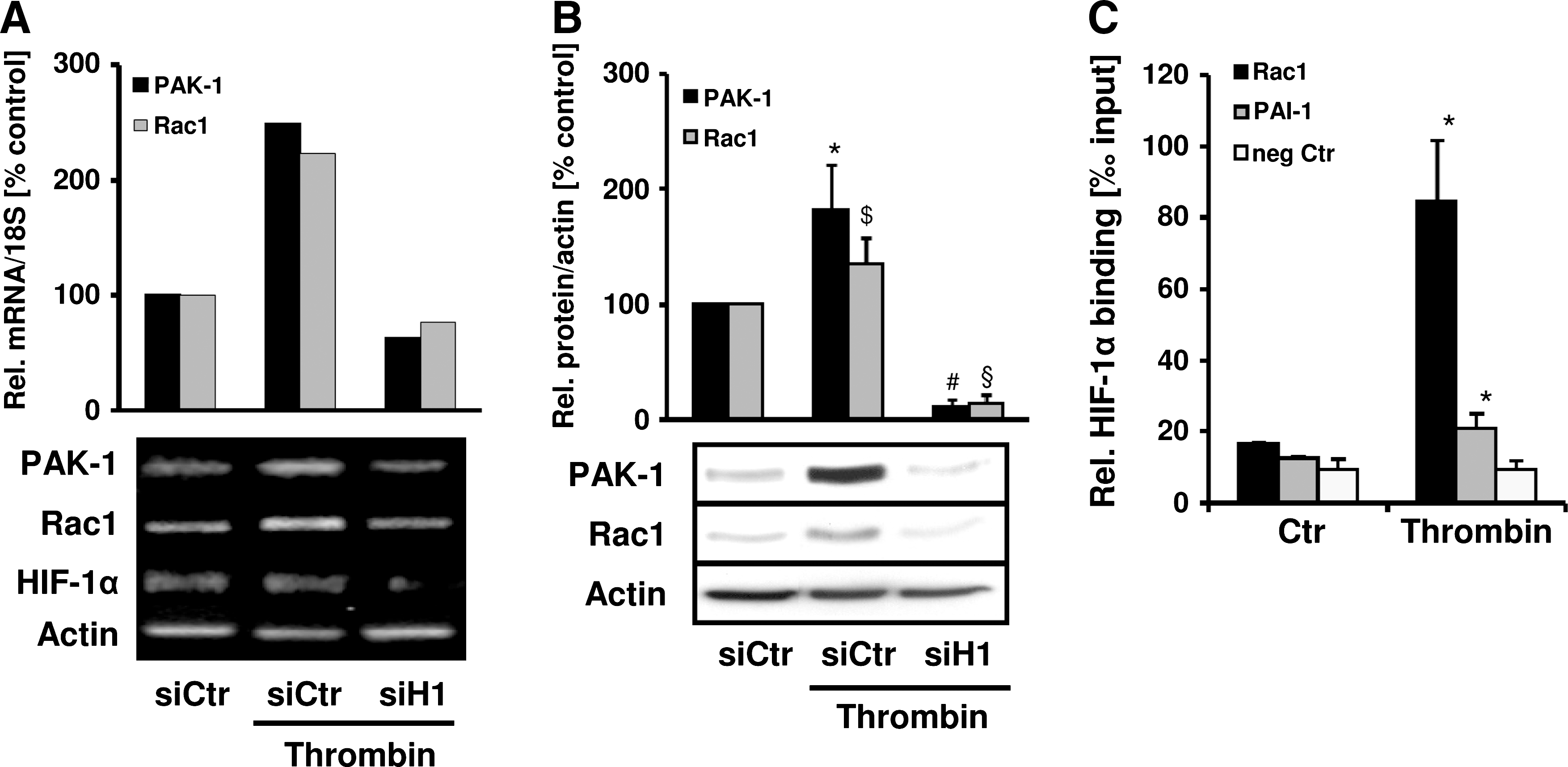
In the next step, we tested the hypothesis that HIF-1 itself could be involved in the regulation of Rac1 and PAK-1 expression by thrombin. To this end, PASMCs were depleted of HIF-1α by using specific shRNA. Compared with control cells, thrombin-induced Rac1 and PAK-1 mRNA and protein levels were diminished in HIF-1α–deficient PASMCs (Fig. 8A and B). To investigate whether a direct link might exist between HIF-1 and Rac1 and PAK-1 gene expression, we first performed bioinformatic analysis by using the Matinspector software and revealed several potential HREs in the promoters of Rac1 and PAK-1. In the next step, we determined whether HIF-1α could bind to these promoters by performing chromatin immunoprecipitation assays with primers amplifying a region containing two predicted HREs in the Rac1 promoter and a region containing one putative HRE in the PAK-1 promoter. Under control conditions, minimal binding of HIF-1α to the Rac1 promoter was observed, which was similar to HIF-1α binding to the PAI-1 promoter, which served as a positive control (Fig. 8C). In thrombin-stimulated cells, a substantial increase of HIF-1α binding to the Rac1 promoter was observed; it even exceeded the increase of HIF-1α binding to the PAI-1 promoter. In contrast, HIF-1α binding to the β-actin gene, which served as a negative control, was not enhanced. However, chromatin immunoprecipitation using primers amplifying the PAK-1 promoter fragment did not provide conclusive evidence that HIF-1α binding to this DNA sequence is enhanced in response to thrombin (data not shown), suggesting that Rac1 is a direct target gene of HIF-1α, whereas PAK-1 may be regulated by HIF-1α by a more indirect mechanism.
Discussion
In this study, we demonstrate that PAK-1 is a central component in pulmonary vascular remodeling by mediating a positive-feedback loop promoting HIF-1α–dependent target gene expression in response to thrombin. This is based on the findings that (a) PAK-1 was enhanced in patients and animal models with pulmonary vascular remodeling and promoted the proliferation of PASMCs; (b) thrombin activated PAK-1 in a calcium- and Rac1-dependent manner; (c) active PAK-1 enhanced HIF-1 activity involving transcriptional upregulation of HIF-1α through ROS and NF-κB; and (d) HIF-1α itself mediated transcriptional upregulation of Rac1 and PAK-1 in response to thrombin.
Thrombin activates p21-activated kinase-1: involvement in ROS production
Although fibrin deposition and in situ thrombosis frequently accompany structural changes of the pulmonary vessel wall in pulmonary hypertension, the exact contribution of hemostatic and prothrombotic factors to pulmonary vascular remodeling processes is not well understood. In smooth muscle cells, PAK-1 not only is a regulator of cytoskeletal organization but also mediates various signaling cascades. In PASMCs, PAK-1 controls procoagulant activity by upregulating tissue factor, which activates the extrinsic coagulation cascade, leading to the formation of thrombin (16). PAK-1 and its activator Rac1 have been shown to be central modulators of a prothrombotic cycle (21). In line with this study, we showed here that thrombin can phosphorylate PAK-1 in PASMCs in a calcium- and Rac1-dependent manner. Rac1 can bind to PAK-1, thereby allowing autophosphorylation of PAK-1 and kinase activation, although Rac1-independent activation of PAK-1 also has been described (26). Calcium was previously shown to activate PAK-1 in response to angiotensin-II (47), whereas thrombin was shown to modulate Ca2+ signaling through binding to protease-activated receptors (PARs) expressed on smooth muscle cells (36).
We previously showed that PAR-1 is critically involved in activation of Rac1 and ROS production in PASMCs (7). Furthermore, we demonstrated here that PAK-1 also contributes to ROS production in PASMCs, because active PAK-1 enhanced ROS production, whereas inactive PAK-1 prevented thrombin-induced ROS production. Similarly, transfection of an inactive PAK-1 mutant was shown to decrease ROS levels in endothelial cells (46). In neutrophils, PAK-1 activity was required for efficient superoxide generation by activation of the NADPH oxidase due to regulatory phosphorylation of the oxidase components p47 phox and possibly p67 phox , and direct association with p22 phox of the flavocytochrome b558 (29). Because we previously showed that not only Rac1 but also p47 phox , as well as p22 phox , are involved in thrombin-stimulated ROS production in smooth muscle cells (4, 10, 18), PAK-1 may, as in the situation in neutrophils, enhance ROS production by acting on cofactor recruitment of NADPH oxidases.
The p21-activated kinase-1 regulates HIF-1α transcription and protein levels
PAK-1 has been suggested to be involved in the activation and repression of various genes, although the exact mechanisms have not been elucidated in detail. Here we showed that activation of PAK-1 by thrombin by calcium and Rac1 is critically involved in the activity and upregulation of HIF-1α in a redox-dependent manner. ROS have been identified as important signaling molecules in the vasculature that mediate activity and expression of HIF-1α, also under nonhypoxic conditions (19), including stimulation with thrombin (3, 7, 18). Modulation of calcium levels has been shown to regulate HIF-1 activity and HIF-1α expression either positively or negatively in different cellular systems. BAPTA-AM increased HIF-1α levels by inhibiting prolyl hydroxylase activity and caused stabilization of HIF-1α in HepG2/cells (28), whereas conversely, BAPTA-AM decreased HIF-1α levels in PC12 cells potentially by affecting HIF-1α translation (23, 41). Furthermore, Rac1 was previously shown to increase HIF-1 activity and to enhance HIF-1α levels in thrombin-stimulated PASMCs (7). Consistently, RacT17N prevented HIF-1 activation by carbachol in HEK293 cells overexpressing a muscarinic acetylcholine receptor (22). In contrast, under hypoxic conditions, contradictory results were observed with regard to the effects of Rac1 mutants on HIF activity (17, 22). Similarly, in endothelial cells, active and inactive Rac1, as well as inactive PAK, decreased HIF-1 activity under hypoxic conditions without effects under normoxia (46). Although the reasons for these inconsistent results of the calcium–Rac1–PAK-1 cascade on the HIF-1 pathway are not resolved, one may speculate that additional regulatory elements contribute to Rac1, and possibly PAK-1, signaling under conditions of impaired oxygen availability or other stress environments. Given the importance of this signaling cascade in many critical cellular functions, cell-type–specific factors may be essentially involved in directing this signaling cascade to the exact cellular function needed at that instant.
Our data in thrombin-stimulated PASMCs clearly show that PAK-1 not only increases HIF-1 activation, but also induces HIF-1α expression, involving a transcriptional mechanism. This latter response was mediated by binding of NF-κB to the HIF-1α promoter, because inactive PAK-1 decreased thrombin-stimulated activation of NF-κB, and mutation of an NF-κB binding site in the HIF-1α promoter prevented PAK-1–dependent HIF-1α promoter activation. PAK-1 was previously shown to activate NF-κB (26). Consistently, we recently showed that HIF-1α can be upregulated by a transcriptional mechanism in response to H2O2, thrombin, and Rac1, and this response required the transcription factor NF-κB (3, 7, 33).
The p21-activated kinase-1 regulates PAI-1 expression
PAI-1 is a known target gene of the transcription factor HIF-1 in response to hypoxia, but also to thrombin or oxidative stress (3, 7, 18, 25). Here we demonstrated that PAK-1 is critically involved in HIF-1–dependent PAI-1 expression in response to thrombin, involving calcium, Rac1, and ROS. Plasminogen activator inhibitor-1 (PAI-1) is known to inhibit fibrinolysis, to enhance fibrin deposition, and to promote a prothrombotic state (13). Interestingly, and in line with this study, we and others have shown that thrombin itself is able to enhance PAI-1 mRNA and protein levels in pulmonary artery and other smooth muscle cells in an Rac1- and ROS-dependent manner (3, 7, 17, 18, 31). The present study shows that PAK-1 increases PAI-1 expression in PASMCs, further indicating that PAK-1 is an important mediator of a prothrombotic state. Because elevated levels of PAI-1 have been associated with vascular remodeling processes (27, 39), our findings in PASMCs, the major cell type involved in pulmonary vascular remodeling, suggest an important contribution of PAK-1 in linking enhanced hemostatic activity and deteriorated fibrin clearance, as is observed in these conditions.
The p21-activated kinase-1 regulates proliferation and is associated with pulmonary vascular remodeling
PAI-1 was recently shown to promote proliferation of PASMCs in response to thrombin (7), and this study provides clear evidence that PAK-1 essentially regulates proliferation of these cells. PAK-1 is well known as a regulator of cytoskeletal remodeling and cell motility (2, 26) and can also promote transformation and tumor cell proliferation (26). However, to our knowledge, our study is the first to show that active PAK-1 is able to stimulate proliferation of PASMCs, which is a prerequisite for media hypertrophy, a hallmark of pulmonary vascular remodeling. We show here that PAK-1 is highly expressed in remodeled pulmonary vessels in the hypertrophic media of patients with pulmonary vasculopathy, and we previously showed that tissue factor, the activator of thrombin, also is present in the media of patients with pulmonary vascular remodeling (1), further supporting the assumption that PAK-1 contributes to pulmonary vascular remodeling by promoting a prothrombotic state and enhanced proliferation. We showed in this study that PAK-1, together with its activator Rac1 and its target HIF-1α, is upregulated in lungs of lambs with pulmonary vascular remodeling due to an intrauterine aortopulmonary shunt. Whereas this is the first report describing elevated levels of PAK-1 in pulmonary vascular remodeling in vivo, enhanced levels of Rac1 have been described in lung tissue of transgenic rats overexpressing mouse renin in extrarenal tissues, which showed pulmonary hypertension and vascular remodeling (5), as well as in lambs with in utero aorta-to-pulmonary artery vascular graft, which developed pulmonary hypertension 4 weeks after birth (20).
A role for HIF-1α in pulmonary vascular remodeling processes has been supported by an experimental model of hypoxia-induced pulmonary hypertension in which muscularization of small pulmonary arteries in response to chronic hypoxia was delayed in heterozygous HIF-1α+/– mice compared with wild-type mice (48). Moreover, HIF-1α has been found in remodeled vessels in tissue sections from patients with different forms of pulmonary hypertension (44), indicating that HIF-1α may indeed play a role in promoting pulmonary vascular remodeling, also independently of hypoxia.
Rac1 and the p21-activated kinase-1 are regulated by HIF-1α
Our findings that Rac1 and also PAK-1 are novel target genes of HIF-1α and are upregulated by thrombin further support the involvement of this signaling cascade in vascular remodeling processes, also independently of hypoxia. As indicated earlier, expressional control of Rac1 has been described, whereas data regarding expressional control of PAK-1 are scarce. Our data now indicate that Rac1 and PAK-1 are transcriptionally regulated by HIF-1α. Several putative binding sites for HIF transcription factors were predicted in the proximal Rac1 promoter, and chromatin immunoprecipitation confirmed that HIF-1α is binding to the Rac1 gene. Interestingly, similar to HIF-1α, Rac1 has been associated with vascular remodeling and vascular development early in embryonic development. Whereas the complete knockout of Rac1 was shown to affect gastrulation of all three germ layers (40), a tissue-specific knockout in endothelial cells showed defective development of major vessels and complete lack of small branched vessels in endothelial Rac1-deficient embryos and their yolk sacs, resulting in embryonic lethality in midgestation around E9.5 (42). Similarly, HIF-1α–knockout mice showed greatly disturbed vascularization of the embryonic yolk sac with complete lack of vascular organization and subsequent lethality around midgestation (35). Thus, similar to HIF-1α, Rac1 appears to be essential for the proper formation of the vascular network. Our findings that Rac1 is regulated by HIF-1α may explain observations implicating enhanced levels of these proteins in several cardiovascular disease models, including our findings that Rac1 and HIF-1α are upregulated in pulmonary vascular remodeling.
In addition, our data indicate that PAK-1 expression is regulated by HIF-1α. However, despite the presence of a putative HRE in the far distal promoter of PAK-1, we could not provide conclusive evidence that HIF-1α is directly binding to the PAK-1 gene in a regulated manner, under the conditions applied in this study. Nevertheless, our data that PAK-1 expression is diminished in HIF-1α–deficient cells and does not respond to thrombin clearly show that HIF-1α is involved in the regulation of this kinase. Although to our knowledge, this is the first report demonstrating increased levels of PAK-1 in vascular cells and in in vivo models of vascular remodeling, enhanced expression of PAK-1 has been, similar to HIF-1α, reported in several tumors (12). A role for activated PAK-1 has been implicated in the control of vascular proliferation (14, 15). Our study now suggests that PAK-1 may also play a major role in the pathogenesis of pulmonary vascular remodeling.
Taken together, our findings indicate a mechanism whereby thrombin may activate Rac1 in a calcium-dependent manner, which then activates PAK-1 to increase ROS levels, resulting in the activation of NF-κB (Fig. 9). NF-κB binds to the HIF-1α promoter, leading to increased HIF-1α levels and HIF-1 transcriptional activity. Under these conditions, HIF-1 target genes, such as PAI-1, are upregulated, which may deteriorate fibrin clearance and promote PASMC proliferation and thus pulmonary vascular remodeling. Importantly, activated HIF also increases the levels of Rac1 and PAK-1, which can stimulate proliferation of PASMCs and promote media hypertrophy and pulmonary vascular remodeling. In addition, as part of a positive feedback mechanism, enhanced levels of ROS may evolve from the increased abundance of Rac1 and PAK-1, further activating the HIF-1 pathway and pulmonary vascular remodeling. Thus, this novel pathway may play an important role in linking a prothrombotic state to advanced pulmonary vascular remodeling, as observed in pulmonary hypertension.

Footnotes
Acknowledgments
We thank Florian Riess for help with chromatin immunoprecipitation. This work was supported by Fondation Leducq (J.H., J.F., S.B., S.F., T.K., and A.G.), DFG grant GO709/4-4 (A.G.), Metoxia (HEALTH-F2-2009-222741) under the 7th research framework program of the European Union (A.G.), and Fonds der Chemischen Industrie (T.K.).
Author Disclosure Statement
No competing financial interests exist.