
Abstract
The hypoxia-inducible factor-2α (HIF-2α) contributes to the vascular response to hypoxia. Hypoxia inhibits prolyl hydroxylation of the N-terminal transactivation domain (N-TAD), thus preventing binding of the von Hippel–Lindau protein (pVHL) and proteasomal degradation; additionally, hypoxia inhibits asparagyl hydroxylation of the C-TAD, thus diminishing cofactor recruitment. Reactive oxygen species (ROS) derived from NADPH oxidases (NOXs) have been shown to control vascular functions and to promote vascular remodeling. However, whether HIF-2α, ROS, and NOXs are linked under such nonhypoxic conditions is unclear. We found that activation of NOX4 by thrombin or H2O2 increased HIF-2α protein because of decreased pVHL binding in pulmonary artery smooth muscle cells (PASMCs). Thrombin, H2O2, and NOX4 overexpression increased HIF-2α N-TAD and C-TAD activity, which was prevented by ascorbate treatment or mutation of the hydroxylation sites in the TADs. HIF-2α also mediated induction of plasminogen activator inhibitor-1 and the proliferative response to thrombin, H2O2, or NOX4 overexpression. Thus, ROS derived from NOX4 in response to thrombin stabilize HIF-2α by preventing hydroxylation of the N- and C-TAD, thus allowing formation of transcriptionally active HIF-2α, which promotes PASMC proliferation. Together, these findings present the first evidence that HIF-2α is critically involved in the ROS-regulated vascular remodeling processes. Antioxid. Redox Signal. 13, 425–436.
Introduction
The heterodimeric HIFs belong to the PAS [PER-ARNT (arylhydrocarbon receptor nuclear translocator)-SIM] family of basic helix–loop–helix (bHLH) transcription factors and are composed of an oxygen-sensitive α-subunit and a constitutively expressed β-subunit, also known as ARNT (44). To date, three HIFs (HIF-1, −2, and −3) have been identified. HIF-1 and HIF-2 have been clearly established in the adaptive response to hypoxia, whereas the role of HIF-3 in the hypoxic regulation of target-gene expression is not yet completely clear (44).
HIF-1 was the first HIF family member to be characterized. HIF-1α contains four distinct domains, including a bHLH-domain for DNA binding and dimerization, a PAS domain for dimerization and target gene specificity, and two transactivation domains located in the C-terminal portion of the protein (N-TAD and C-TAD). An oxygen-dependent degradation domain (ODD) required for degradation of the protein by the ubiquitin–proteasome pathway, overlaps with the N-TAD (41). HIF-1 has been considered an important regulator of the cellular response to hypoxia because it is ubiquitously expressed and promotes the expression of many hypoxia-inducible genes.
HIF-2α, also termed HIF-like factor (HLF) (14), HIF-related factor (15), or endothelial PAS protein-1 (EPAS1) (45), was the second HIF family member to be identified that is structurally similar to HIF-1α containing bHLH, PAS, and ODD motifs, with high amino acid sequence homology to HIF-1α. Initially, HIF-2α was considered to be specifically expressed in endothelial cells and highly vascularized tissues (14, 45), although more recently, it also was found in several other cell types, including cardiomyocytes, fibroblasts, and hepatocytes, as well as in cancer cells (35, 40).
Under normoxia, HIF-α subunits are targeted for proteasomal degradation by the von Hippel–Lindau (pVHL) tumor suppressor, which is the substrate recognition component of an E3 ubiquitin ligase complex. The interaction between pVHL and HIF-α requires the hydroxylation of specific proline residues located in the ODD, which are targets of a family of O2-dependent prolyl-4-hydroxylase domain (PHD)-containing enzymes that require, in addition to oxygen, Fe(II), α-ketoglutarate, and the reducing agent ascorbate as cofactors. Once the prolines are hydroxylated, they are recognized by pVHL, which initiates the ubiquitinylation process, and ubiquitinylated HIF-α is thereafter degraded by the 26S proteasome (25, 26).
When O2 availability is limited, the PHDs loose their ability to hydroxylate the proline residues, which prevents binding of pVHL and ubiquitinylation of HIF-α proteins. This results in HIF-α stabilization and translocation to the nucleus, where the PHDs heterodimerize with ARNT and bind to hypoxia-responsive elements (HREs) located within regulatory regions of HIF target genes. Once stabilized, the HIF-α/ARNT heterodimer activates transcription by recruiting the transcriptional activators p300 and CBP. The interaction between HIF and p300/CBP also is regulated in an oxygen-dependent manner by factor-inhibiting HIF-1 (FIH-1), an α-ketoglutarate- and Fe(II)-dependent asparagine hydroxylase. FIH hydroxylates asparagine residues located within the HIF-α C-TAD and prevents p300/CBP binding (32). Thus, full activation of HIF transcriptional activity requires both HIF-α stabilization and C-TAD activation (44).
Through the last decade, an increasing awareness has existed that HIF-1α is also responsive to different stimuli, including growth factors and prothrombotic factors, inflammatory cytokines, as well as reactive oxygen species (ROS) under nonhypoxic conditions (2, 20, 28) and may thus play a key role in the pathogenesis of various disorders, also independent of hypoxia. Regulation of HIF-1α levels under nonhypoxic conditions not only occurs posttranscriptionally, but also occurs at the level of transcription involving NF-κB (5).
In contrast to that with HIF-1α, however, only limited information is available regarding the role of HIF-2α under stress conditions independent of hypoxia. Thrombin has been recognized as an important stimulus of ROS production by NADPH oxidases in smooth muscle cells, which can lead to the induction of various genes, including chemokines, coagulation factors, and vascular growth factors (6, 19, 21, 22). Several homologues of the catalytic NOX subunit of NADPH oxidases have been identified in smooth muscle cells, including NOX1, NOX4, and NOX5 (3, 12, 27, 33). Among them, NOX4 appears to be of specific importance in the pulmonary vasculature (12, 37). However, the role of HIF-2α in the response of smooth muscle cells to ROS is not well understood. We thus investigated whether thrombin, H2O2, and NOX4 can control the levels of HIF-2α in pulmonary artery smooth muscle cells.
Materials and Methods
Reagents
All biochemicals and enzymes were of analytic grade and were purchased from commercial suppliers.
Cell culture
Human pulmonary artery smooth muscle cells (PASMCs) were from Lonza (Wuppertal, Germany) and cultured in the medium provided as recommended. PASMCs (passages 3 to 11) were serum deprived for 24 h before stimulation. A7r5 rat smooth muscle cells (rSMCs, ATCC) were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco, Karlsruhe, Germany) with 10% fetal calf serum. Cells were serum starved for 16 h before experiments. Human renal clear-cell carcinoma cells (RCC4) and RCC4 cells reconstituted with pVHL were kindly provided by Dr. M. Wiesener, Erlangen, Germany. Cells were grown in DMEM medium supplemented with 10% fetal calf serum, 2 mM glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin (Sigma, Taufkirchen, Germany).
Plasmids and transfections
The GAL4, GAL4-HIF-2α-NTAD, GAL4-HIF-2α-NTAD-P530A, GAL4-HIF-2α-CTAD, GAL4-HIF-2α-CTAD-N851A, GAL4-HIF-1α-TADN, GAL4-HIF-1α-TADC, and p5GE1B-Luc constructs were previously described (16, 34). Short-hairpin RNA (shRNA) against HIF-2α or an unspecific random sequence (siCtr) was created by using the siSTRIKE U6 Hairpin Cloning System (Promega, Mannheim, Germany). All constructs were confirmed by DNA sequencing. The vectors coding for NOX4 (pcDNA3-NOX4) or for shRNA against NOX4 were previously described (12). Cells were plated to a density of 70% and cultured for 24 h. Transfections were performed as described previously (2). Because PASMCs do not efficiently express luciferase constructs, A7r5 cells were used for reporter gene assays and transfected with calcium phosphate, as described (12). Transfection efficiency was around 40%, as confirmed with fluorescence microscopy. A Renilla luciferase expression vector was co-transfected to adjust for variations in transfection efficiencies.
Western blot analysis
Western blot analysis was performed as described (10). In brief, 10 μg of isolated proteins was separated by 8% SDS-PAGE and transferred to nitrocellulose membranes. After blocking for 1 h in TBS containing 5% nondry milk (TBS), membranes were incubated overnight at 4°C with a polyclonal antibody raised against a NOX4 peptide (NH3- CSYGTRFEYNKESFS). The antibody for NOX4 was diluted at 1:500 in TBS with 5% milk. For HIF-2α, 50 μg of protein was used. Membranes were blocked in TBS containing 5% nondry milk and 0.3% Tween20 (TBS-T), and a HIF-2α antibody (Chemicon, Schwalbach, Germany) diluted at 1:500 in TBS-T was used. After incubation with a horseradish peroxidase–conjugated secondary antibody for 1 h, proteins were visualized with luminol-enhanced chemiluminescence. Loading of equal amounts of proteins was confirmed by reprobing the membranes with an actin antibody (Santa Cruz, Heidelberg, Germany). Blots were scanned and analyzed by using GelDoc software (Bio-Rad, Munich, Germany).
GST pull-down assays
Rat smooth muscle cells were pretreated for 30 min with ascorbate (100 μM) or left untreated, and stimulated with thrombin (3 U/ml), H2O2 (50 μM), or were exposed to hypoxia (1% O2) for 1 h. For the pVHL pull-down assay, [35S]VHL protein was synthesized from pCMV-HA-VHL as a template by using 35S-methionine and the TNT coupled reticulocyte lysate system (Promega). Cells were homogenized at 4°C, and cell extracts were prepared as previously described (34). The cell extracts (300 μg protein/ml) were then incubated at 37°C for 30 min in 40 mM Tris-HCI (pH 7.5), 0.5 mM dithiothreitol, 50 μM ammonium ferrous sulfate, 1 mM ascorbate, 2 mg/ml bovine serum albumin, 0.4 mg/ml catalase, 1 mM 2-oxoglutarate, and either 20 μg GST protein or GST-ODD proteins. The reaction products were then incubated at 4°C in 200 μl buffer (50 mM Tris-HCl, pH 8, 120 mM NaCl, 0.5% NP-40) supplemented with glutathione-sepharose beads and 50.000 dpm [35S]-labeled protein. After 2 h, beads were washed 3 times with cold buffer (20 mM Tris-HCI, pH 8, 100 mM NaCl, 1 mM EDTA, 0.5% NP-40). The bound proteins were eluted in 10 mM reduced glutathione and analyzed with SDS-PAGE, followed by autoradiography on a phosphorimager screen.
Determination of reactive oxygen species
The generation of ROS was determined by using the fluoroprobe 5- (and 6-) chloromethyl-2',7'-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCFDA; Molecular Probes, Karlsruhe, Germany), as described (12).
Determination of cellular iron content
Iron levels were determined as described (17). In brief, 2 × 106 cells were lysed in acetate buffer (pH 4.6) by sonification. Lysates were incubated with 100 μM Na-bathophenanthrolinsulfonate for 1 h at 37°C to determine Fe2+ levels or with thioglycol acid and 100 μM Na-bathophenanthrolinsulfonate to determine total Fe levels. Absorbances were detected at 550 nm. Fe3+ was calculated as total Fe minus Fe2+.
Determination of intracellular ascorbate concentrations
Ascorbate levels were measured as previously described (1, 38).by determining the conversion from ascorbate to dehydroascorbate. In brief, A7R5 cells were grown to confluence and serum-starved overnight with medium supplemented with ascorbate (250 μM). One hour before stimulation, medium was replaced by fresh medium with ascorbate. After stimulation, cells were trypsinized, washed, counted, and lysed with ultrasonification. The pH was adjusted to 5.5, and the supernatant was treated with ascorbate oxidase for 30 min before the optical density was read at 346 nm.
Proliferation assays
DNA synthesis was assessed with 5-bromo-2′deoxyuridine (BrdU) labeling (Roche, Mannheim, Germany), as described previously (10).
Statistical analysis
Values are presented as mean ± standard deviation (SD). Results were compared with ANOVA for repeated measurements followed by the Student-Newman-Keuls t test. A value of p < 0.05 was considered statistically significant.
Results
HIF-2α protein is increased in response to thrombin and H2O2
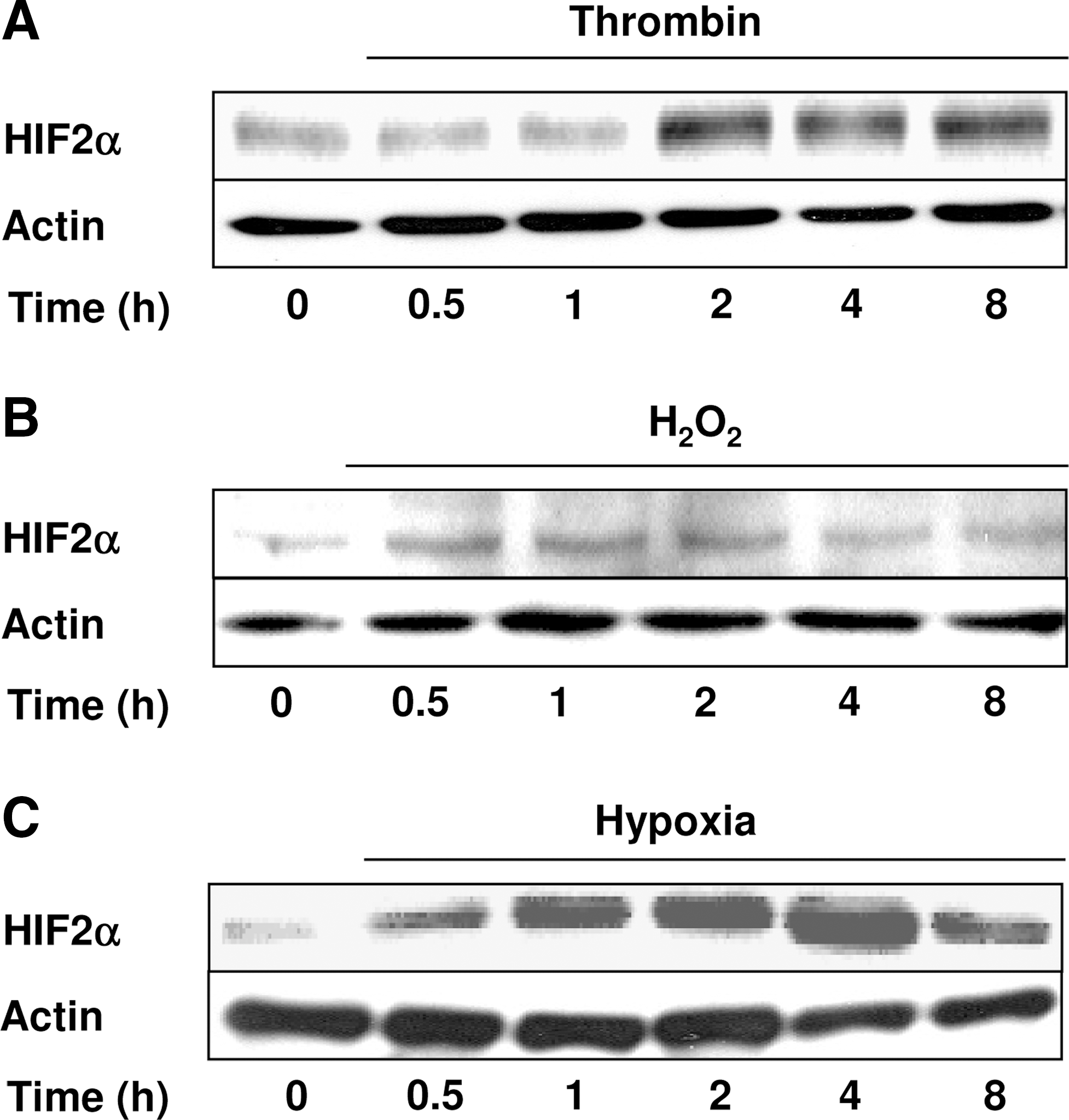
To investigate whether HIF-2α can be regulated independent of hypoxia, pulmonary artery smooth muscle cells (PASMCs) were exposed to thrombin or H2O2 for various time periods, and Western blot analysis was performed with an antibody against HIF-2α (Fig. 1A and B). Thrombin and H2O2 increased HIF-2α protein levels with slightly varying kinetics. The thrombin-dependent increase in HIF-2α first became visible after 2 h of exposure and then remained constant for up to 8 h. The H2O2-mediated HIF-2α increase was transient and started after 0.5 h, reaching its maximum at 2 h, which was followed by a decline up to 8 h. In comparison, exposure to hypoxia increased HIF-2α levels after 0.5 h, similar to the situation with H2O2, reaching a maximum at 4 h of exposure (Fig. 1C).
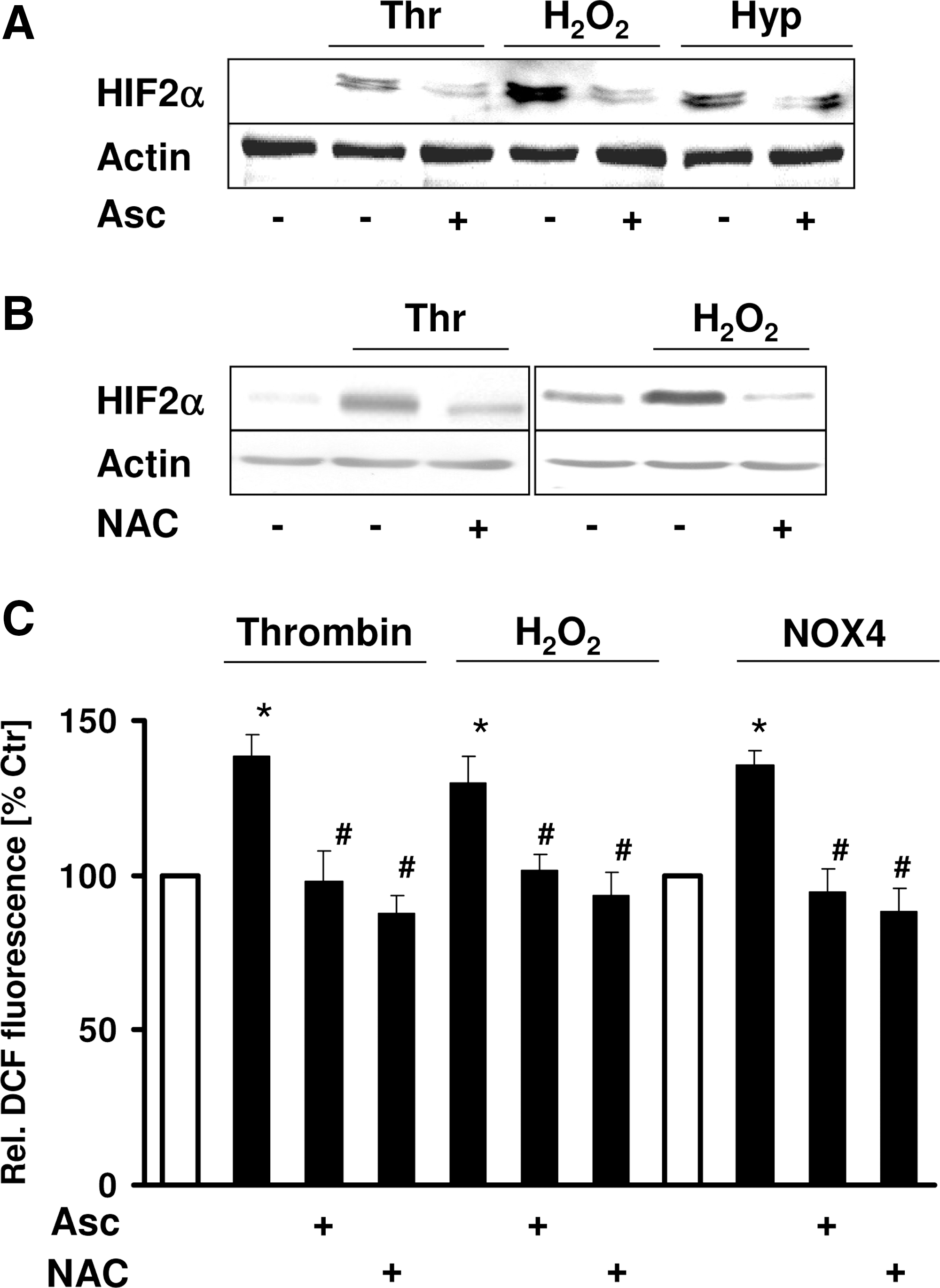
Because the reducing agent ascorbate has been shown to be required for prolyl hydroxylase activity, we investigated whether ascorbate treatment would affect HIF-2α protein levels under nonhypoxic conditions. The upregulation of HIF-2α protein levels by thrombin or H2O2 was completely prevented by ascorbate, and the hypoxia-induced HIF-2α levels were reduced by about 50% (Fig. 2A). Because ascorbate also has been suggested to act as an antioxidant, we then treated PASMCs with N-acetylcysteine (NAC), a thiol also acting as antioxidant. Similar to the situation with ascorbate, NAC diminished HIF-2α upregulation by thrombin and H2O2 (Fig. 2B). Ascorbate and NAC were able to decrease ROS levels in the presence of thrombin or H2O2 (Fig. 2C). Together, these results indicate that thrombin and H2O2 induce HIF-2α and that ROS are required for the ability of thrombin and H2O2 to induce HIF-2α in PASMCs.
Induction of HIF-2α by thrombin and H2O2 involves pVHL
Under normoxia, HIF-2α is hydroxylated by prolyl hydroxylases (PHDs) within an oxygen-dependent degradation domain (ODD) overlapping the N-terminal transactivation domain (N-TAD), thus allowing binding of the von Hippel–Lindau protein (pVHL) and proteasomal degradation. To test whether pVHL is important for nonhypoxic regulation of HIF-2α, we determined HIF-2α levels in a renal carcinoma cell line (RCC4) lacking pVHL. Western blot analysis revealed that HIF-2α was detectable under normoxic conditions. Exposure to thrombin or H2O2 had almost no effect on HIF-2α levels, similar to the situation with hypoxia (Fig. 3A). In contrast, in RCC4 cells reconstituted with pVHL, HIF-2α levels were barely detectable under normoxic conditions, as expected, but were substantially increased by hypoxia as well as by thrombin and H2O2 (Fig. 3A). These data suggest that thrombin and H2O2 increase HIF-2α by a pVHL-dependent mechanism.
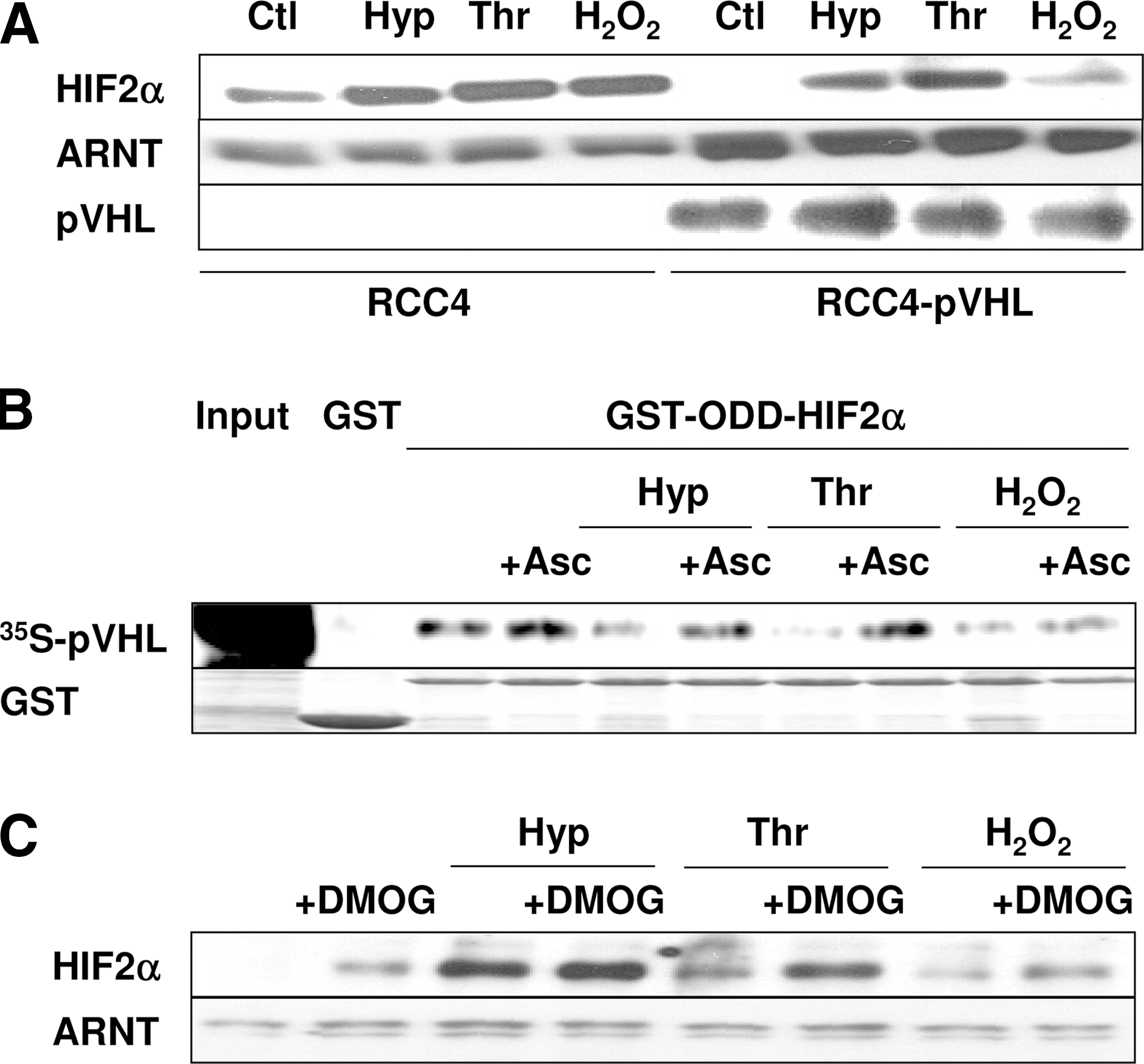
Next we tested whether smooth muscle cells treated with thrombin or H2O2 modulate the interaction between pVHL and HIF-2α-ODD by using a GST-pulldown assay. Lysates from normoxic control cells promoted binding of 35S-labeled recombinant pVHL to the HIF-2α-ODD, and this capacity was severely inhibited with lysates from hypoxic cells, as expected (Fig. 3B). Interestingly, pVHL binding also was attenuated when lysates were used from cells treated with thrombin or H2O2. However, when lysates were used from cells treated with ascorbate, binding of pVHL to the HIF-2α-ODD was restored, not only under hypoxia, but also in the presence of thrombin or H2O2 (Fig. 3B).
The interaction of pVHL with the HIF-ODD occurs after PHD-dependent hydroxylation of the HIF-ODD, and therefore we next tested whether treatment of cells with the PHD inhibitor dimethyloxaloylglycine (DMOG) affects the thrombin- and H2O2-dependent HIF-2α induction. We found that DMOG increased HIF-2α protein levels to a similar extent as thrombin or H2O2, whereas combined treatment further enhanced HIF-2α protein levels, suggesting that modulation of PHD activity is involved in HIF-2α upregulation by thrombin and H2O2. As expected, treatment with DMOG did not affect the levels of ARNT (Fig. 3C).
Thrombin and H2O2 decrease the cellular levels of ascorbate and Fe(II)
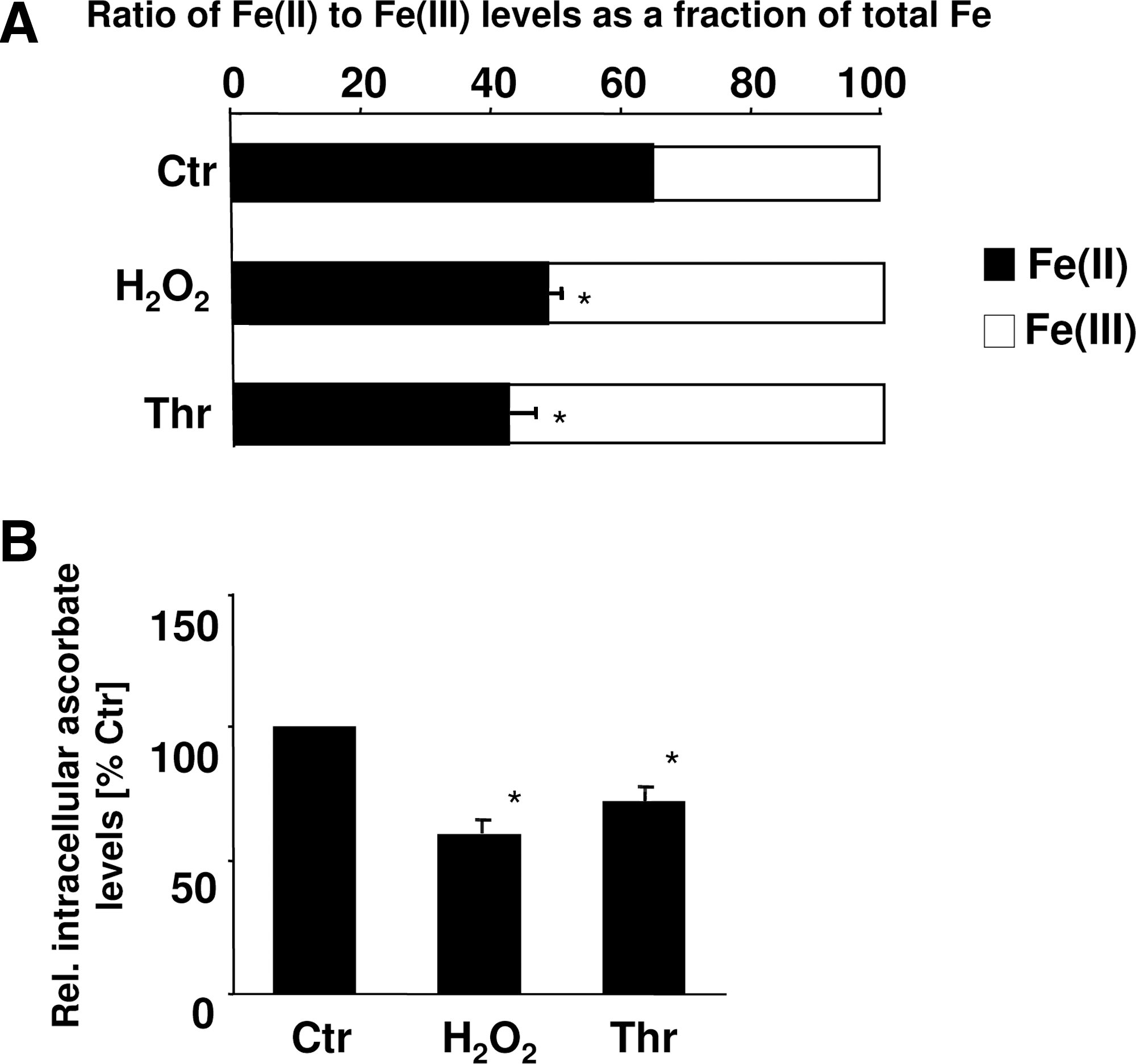
Because the action of PHDs requires Fe(II) and ascorbate, in addition to oxygen, which did not appear to be the limiting factor under our conditions, and our VHL-pulldowns indicated that ascorbate may influence pVHL binding, we were interested to test whether thrombin and H2O2 influence the cofactor availability for the HIF-PHDs. Interestingly, in PASMCs treated with thrombin or H2O2, the intracellular ascorbate levels as well as the Fe(II) levels were reduced (Fig. 4). These findings suggest that thrombin as well as H2O2 may decrease PHD activity by reducing the cellular levels of the cofactors Fe(II) and ascorbate, subsequently diminishing pVHL binding and proteasomal degradation.
Thrombin and H2O2 promote HIF-2α transactivation
Because pVHL interacts with HIF-2α at specific hydroxylated proline residues within the ODD, overlapping the N-TAD, we next investigated whether thrombin and H2O2 may affect HIF-2α protein stability through the N-TAD. To this end, smooth muscle cells were transfected with constructs encoding fusion proteins from the Gal4 DNA-binding domain and the HIF-2α N-TAD, along with a luciferase (Luc) construct containing 5 Gal4 binding sites in front of the adenovirus E1B promoter. We found that treatment of cells with thrombin or H2O2 increased Luc activity by about twofold, similar to the situation under hypoxia (Fig. 5). Interestingly, pretreatment of cells with ascorbate prevented stimulation of N-TAD–mediated Luc activities by thrombin, H2O2, or hypoxia. However, when the PHD target P530 was mutated to alanine, overall Luc activities increased, but the N-TAD became unresponsive to thrombin, H2O2, hypoxia, or ascorbate (Fig. 5), indicating that activation of the N-TAD by hypoxia, as well as by thrombin or H2O2, was dependent on prolyl hydroxylation.
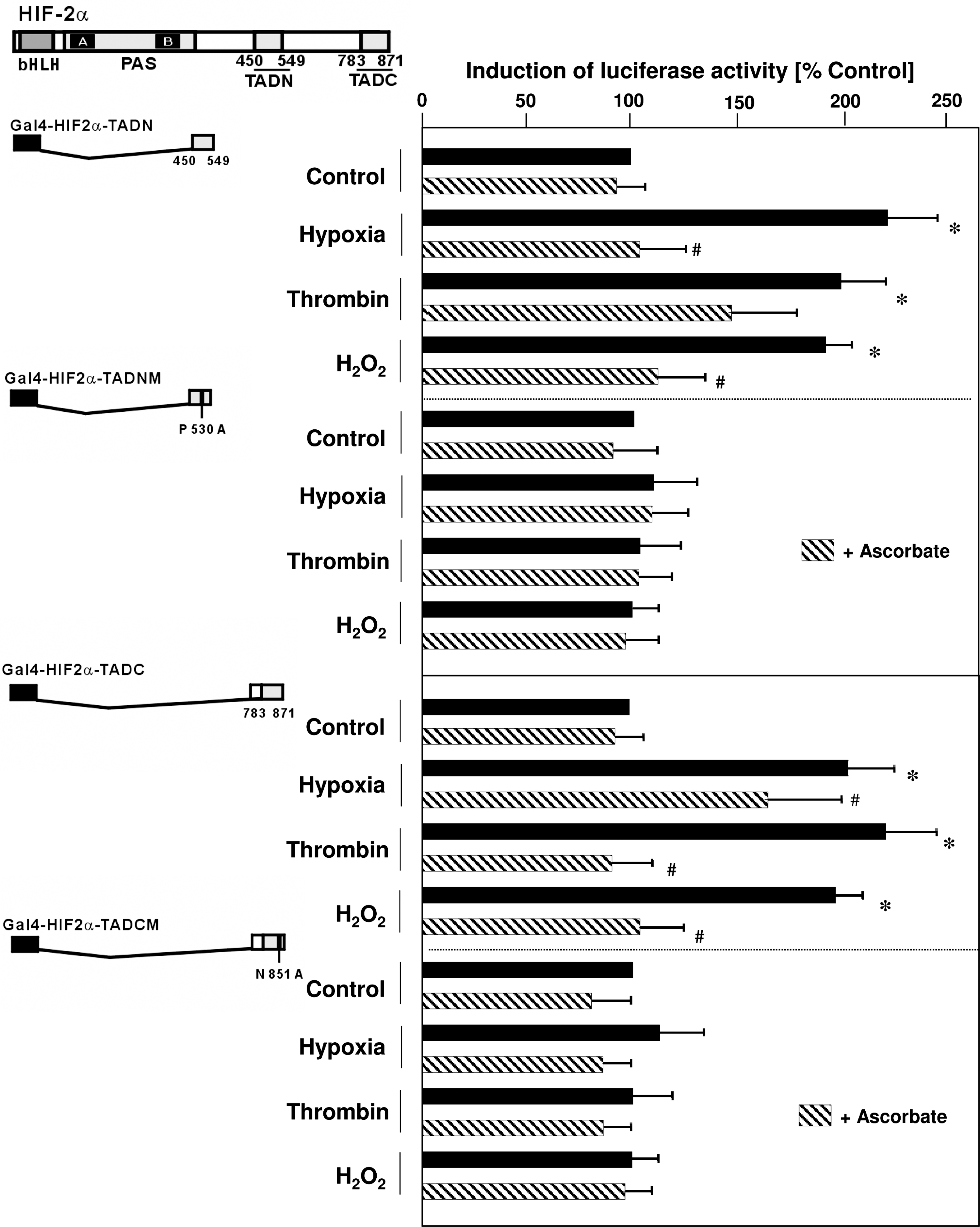
Because activation of the C-TAD is required for full activation of HIF-2α, we next investigated whether thrombin and H2O2 also act through the C-TAD. By using the same assay, we found that both stimuli increased C-TAD–mediated Luc activity, again similar to the situation with hypoxia, and this response was prevented by ascorbate (Fig. 5). Because hydroxylation of N851 by FIH is important for cofactor recruitment and full activity of the C-TAD, we used a construct in which this residue was mutated (Fig. 5). Neither thrombin, H2O2, hypoxia, nor ascorbate was able to modulate Luc activity of the mutated C-TAD, indicating that hydroxylation is important for regulating C-TAD activity under nonhypoxic conditions.
NOX4 is involved in stabilization of HIF-2α by thrombin and H2O2
Because the NOX4-containing NADPH oxidase is a major source of ROS in PASMCs, we investigated whether NOX4 is involved in stabilization of HIF-2α under normoxic conditions. First, we enhanced the levels of NOX4 by transfecting an expression vector coding for NOX4. Compared with control cells, HIF-2α levels were increased in NOX4-overexpressing PASMCs (Fig. 6A). Next, we depleted PASMCs from NOX4 by transfecting a vector coding for shNOX4. Compared with control cells, the upregulation of HIF-2α by thrombin or H2O2 was prevented (Fig. 6A). Further to investigate whether stabilization of HIF-2α by NOX4 overexpression is dependent on pVHL, we determined HIF-2α protein levels in RCC4 cells lacking pVHL or in pVHL-reconstituted RCC4 cells.

Overexpression of NOX4 did not affect HIF-2α protein levels in pVHL-deficient cells, but increased HIF-2α levels in pVHL-reconstituted RCC4 cells (Fig. 6B), indicating that NOX4 acts on HIF-2α in a pVHL-dependent manner.
We then investigated the effect of NOX4 overexpression on HIF-2α N-TAD and C-TAD activities in the mammalian two-hybrid assay. Compared with cells expressing an empty control vector, NOX4 overexpression increased the levels of the transfected N- and C-TAD constructs and enhanced N-TAD– and C-TAD–dependent Luc activities, whereas these latter responses were diminished in the presence of ascorbate (Fig. 6C). However, NOX4 overexpression did not affect Luc activity in the presence of mutated N-TAD (P530A) or C-TAD (N851A) (Fig. 6C), indicating that NOX4 is involved in the stabilization of HIF-2α by affecting hydroxylation of N-TAD and C-TAD.
HIF-2α contributes to proliferation and PAI-1 expression in response to thrombin and NOX4 overexpresssion
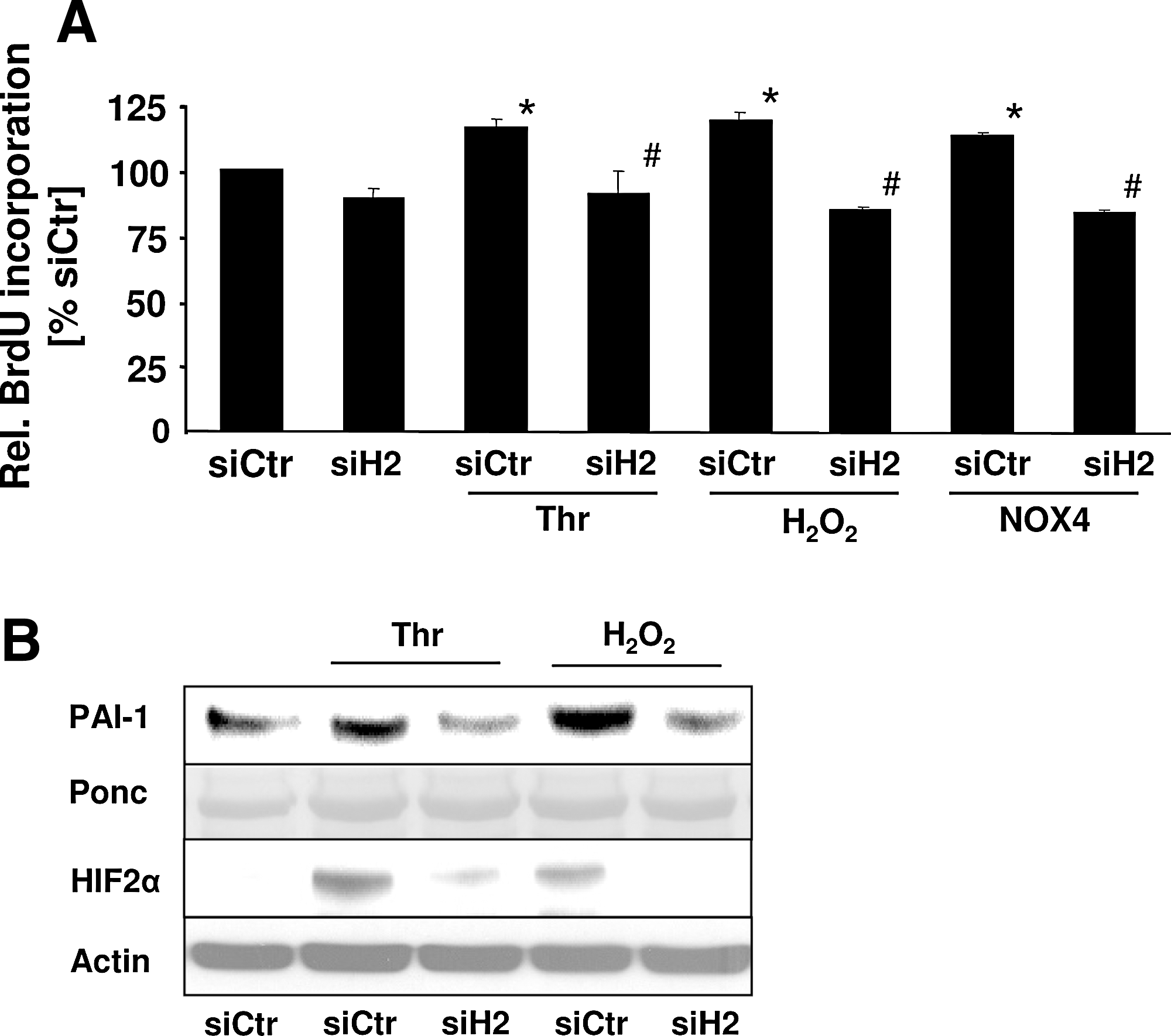
Finally, to determine the functional consequences of HIF-2α upregulation by thrombin, NOX4 overexpression, and H2O2, we studied the proliferative response of PASMCs by using BrdU incorporation in control cells and PASMCs depleted of HIF-2α. Both thrombin and H2O2 significantly increased the proliferation of PASMCs, and this response was similar in NOX4-overexpressing PASMCs (Fig. 7A). Depletion of HIF-2α did not affect basal proliferation, but abolished the proliferative response to thrombin, H2O2, or NOX4 overexpression. We previously described that PAI-1 contributes to thrombin-induced PASMC proliferation and that HIF-1α is involved in this process. Because the proliferation data also indicated an involvement of HIF-2α in PASMC proliferation, we aimed to determine whether HIF-2 is also involved in the regulation of PAI-1 in PASMCs. Exposure to both thrombin and H2O2 increased PAI-1 levels (Fig. 7B), whereas depletion of HIF-2α by shRNA decreased PAI-1 levels in stimulated PASMCs, indicating that thrombin and H2O2 can induce transcriptionally active HIF-2α. Together, these data indicate that upregulation of HIF-2α by thrombin and ROS is an important mechanism to promote the proliferation of PASMCs.
Discussion
In this study, we demonstrated that thrombin and oxidative stress due to activation of NOX4 stabilize HIF-2α. This response is mediated by limited hydroxylation of the HIF-2α ODD and C-TAD and decreased interaction of HIF-2α with pVHL and has consequences for the proliferative response of PASMCs because (a) thrombin, H2O2, and NOX4 overexpression increased HIF-2α protein levels similar to those in hypoxia, and this response was dependent on ascorbate and enhanced by the PHD inhibitor DMOG; (b) these stimuli prevented pVHL binding to the HIF-2α-ODD and increased transactivation of the HIF-2α N-TAD and C-TAD in a hydroxylation-dependent manner; and (c) these stimuli increased cell proliferation that was dependent on HIF-2α.
HIF-2α is stabilized by thrombin and ROS through a pathway involving NOX4
Although HIF-2α is initially confined to endothelial cells, a variety of studies now indicate that HIF-2α is expressed not only in highly vascularized tissues, but also in a variety of other organs and cell types (40). Because of the structural similarity between HIF-1α and HIF-2α, debate increases about the actual importance of these two transcription factors, suggesting differences regarding target gene specificity, sensitivity to oxygen concentration, and possibly cell-type–specific context factors. However, despite the increasing awareness of HIF-1α as responsive to a variety of nonhypoxic stress stimuli in a redox-sensitive manner (20), only limited data are available regarding the importance of HIF-2α in response to nonhypoxic stress conditions (35).
In this study, we provided clear evidence that HIF-2α protein levels are increased in response to the coagulation factor thrombin and oxidative stress generated by H2O2 application or NOX4 overexpression in PASMCs. Thrombin-induced ROS production has been suggested to play an important role in several vascular disorders associated with media hypertrophy and remodeling of the vascular wall, including pulmonary and systemic hypertension and atherosclerosis (23).
We previously identified NADPH oxidases as important sources of ROS in response to thrombin in smooth muscle cells (19, 23) and found NOX4-based NADPH oxidases to be critically involved in ROS signaling in PASMCs (5, 12). Here we showed that NOX4 acts as an important source of ROS, mediating upregulation of HIF-2α by thrombin in PASMCs. Whereas the role of HIF-2α in smooth muscle function and in the response to thrombin is not well understood, HIF-1α has been shown to be activated by redox-sensitive signaling cascades involving NOX4 as well as the NADPH oxidase subunits p22 phox and Rac1 in thrombin-activated smooth muscle cells (5, 10, 19). Furthermore, evidence has been provided that H2O2 is important for controlling HIF-1α levels because, in addition to exogenous application (5), overexpression of CuZnSOD increased HIF-1α levels, whereas expression of catalase, which metabolizes H2O2, decreased thrombin-induced HIF-1α levels in PASMCs (2). Subsequently, NADPH oxidases also were implicated in the regulation of HIF-1α under different conditions in endothelial cells, fibroblasts, and several nonvascular cells, including tumor cells (20). However, this is the first study clearly demonstrating a role for thrombin, H2O2, and NOX4 in the regulation of HIF-2α in vascular cells.
Thrombin, H2O2, and NOX4 regulate HIF-2α in a pVHL-dependent manner
Our study shows for the first time that HIF-2α is stabilized by thrombin and H2O2 because of limited hydroxylation, thus leading to decreased pVHL binding and protein accumulation, similar to the situation with hypoxia.
Under normoxia, HIF-α proteins are hydroxylated by PHDs at an oxygen-dependent degradation domain (ODD) overlapping the N-TAD, thus allowing binding of pVHL and subsequent ubiquitinylation and proteasomal degradation. PHD activity is dependent on O2, Fe(II), and α-ketoglutarate. Thus, under hypoxia, the activity of PHDs is decreased. Our study suggests that also in the presence of thrombin and H2O2, hydroxylation of HIF-2α and subsequent pVHL binding are decreased, thus allowing protein accumulation. This is based on the findings that in pVHL capture assays, cells stimulated with thrombin or H2O2 clearly showed decreased binding of HIF-2α to pVHL to a comparable extent as that with hypoxia, and that both stimuli increased N-TAD–mediated Luc activity in a mammalian two-hybrid assay. Because the proline residues targeted by PHDs are located in the ODD overlapping the N-TAD, and mutation of these residues abolished activation of the N-TAD by thrombin, H2O2, and hypoxia, these findings further support the assumption that, in the presence of enhanced ROS levels, proline hydroxylation of HIF-2α is diminished, thus preventing pVHL binding.
Subsequently, treatment with ascorbate not only restored pVHL binding to HIF-2α, but also diminished N-TAD activity induced by thrombin and H2O2, or by hypoxia, and decreased HIF-2α protein accumulation by these stimuli. Because ascorbate was able to reduce ROS levels in response to thrombin or in NOX4-overexpressing cells, confirming previous studies (19), this suggests that ROS are involved in controlling hydroxylation and pVHL binding to HIF-2α. In support, by using an in vitro hydroxylation assay with the HIF-1α ODD as a template, we showed that H2O2 was able to decrease hydroxylation, and this effect could be reversed by ascorbate (39).
Furthermore, ascorbate itself has been found to be important for ensuring the activity of prolyl hydroxylases, probably by helping to keep iron in a reduced state (30). Of note, in the presence of thrombin and H2O2, the levels of ascorbate and Fe(II) were reduced, thus at least partially explaining our findings that pVHL binding to HIF-2α is diminished, even with sufficient concentrations of oxygen. Because thrombin can increase ROS levels, one may speculate that ROS-dependent oxidation of Fe(II) to Fe(III) may prevent full PHD activity, thus allowing the accumulation of HIF-2α. Previously, angiotensin-II was described to affect the availability of ascorbate and Fe(II), thus leading to enhanced HIF-1α levels in smooth muscle cells (38). We showed that treatment with DMOG, an inhibitor of PHDs, further enhanced HIF-2α levels in the presence of thrombin, H2O2, and hypoxia. In support, reduced levels of Fe(II) due to increased levels of ROS were implicated in decreasing hydroxylation and pVHL binding and thus accumulation of HIF-1α in jun-/- cells (17).
Our findings now clearly demonstrate that NOX4 is important in regulating HIF-2α stability, because overexpression of NOX4 increased not only HIF-2α protein levels, but also the activity of the N-TAD, similar to the situation with H2O2, thrombin, and hypoxia. Regulation of HIF-2α by NOX4 overexpression was absent in RCC4 renal carcinoma cells deficient in pVHL, mimicking the situation with hypoxia, but was restored on reintroduction of pVHL, indicating that NOX4 controls HIF-2α through a pVHL-dependent mechanism. In contrast, in 786-O renal carcinoma cells, which are deficient in pVHL and HIF-1α, knockdown of NOX4 by siRNA decreased HIF-2α mRNA and protein levels independent of pVHL (36). It was suggested that NADPH oxidases regulate HIF-2α by a translational mechanism (4). Although we cannot rule out that ROS derived from NADPH oxidases regulate HIF-2α by a translational mechanism, or transcriptionally, as has been reported for HIF-1α (5), our data in RCC4 cells lacking pVHL indicate that, under our experimental conditions, pVHL is important in controlling HIF-2α in response to thrombin, H2O2, or NOX4.
Our data also provide evidence that thrombin, H2O2, and NOX4 control asparagines hydroxylation of the HIF-2α C-TAD, because C-TAD–mediated transactivation was upregulated by these stimuli, similar to the situation with hypoxia, but was diminished in the presence of ascorbate. Importantly, the responsiveness of the C-TAD to these stimuli was abrogated on mutation of the asparagine residue, which is targeted by the asparagine hydroxylase FIH. Decreased hydroxylation of the C-TAD is important for recruitment of cofactors and thus for transcriptional activity. Thus, our study is the first showing that thrombin and ROS derived from NOX4 can affect HIF-2α through hydroxylation of the C-TAD. Because FIH also belongs to the family of α-ketoglutarate dioxygenases and has the same cofactor requirements, although slightly different kinetics than PHDs (31), activity of this enzyme may be similarly affected by ROS derived from thrombin and NOX4 because of limitation of the availability of ascorbate and Fe(II). Interestingly, FIH has been described to have a higher K m for ascorbate than PHDs, although the physiological consequences of this observation are not clear (31).
In addition to our report, demonstrating redox-sensitive regulation of the HIF-2α C-TAD through FIH-dependent hydroxylation, a previous report demonstrated that the HIF-2α C-TAD can also be directly regulated through a redox-sensitive mechanism because of the presence of a redox-sensitive cysteine in the C-TAD of HIF-2α (13). Subsequent experiments must show whether NOX4-derived ROS can affect HIF-2α also through this mechanism.
HIF-2α mediates proliferation and PAI-1 induction in response to thrombin, H2O2 and NOX4 overexpression
Because the functional importance of HIF-2α in smooth muscle cells has not been well characterized, we tested the involvement of HIF-2α in the control of PASMC proliferation. Whereas depletion of HIF-2α did not affect basal proliferative activity, it reduced the proliferative response to thrombin, H2O2, and NOX4 overexpression, indicating that HIF-2 also is transcriptionally active under nonhypoxic conditions, and that it is acting downstream of NOX4 and ROS to promote PASMC proliferation.
We further showed that depletion of HIF-2α reduced the levels of PAI-1 in response to thrombin and H2O2, indicating that HIF-2 is transcriptionally active, also independent of hypoxia. The involvement of HIF-2 in the control of PAI-1 levels appears to be in contrast to our previous studies, in which we reported that PAI-1 contributes to thrombin-induced PASMC proliferation through induction of HIF-1α in PASMCs (10), and that HIF-1α mediates the response to hypoxia in liver cells (18, 29). However, in line with the findings of the present study, an involvement of HIF-2 in the regulation of PAI-1 was reported previously in RCC4 and Hep3B cells (8, 24), indicating that PAI-1 may be targeted by both HIF-1 and HIF-2, depending on the cell type and the stimulus. It was shown that the N-terminal HIF-1α and HIF-2α domains are involved in specificity effects and that interaction with cofactors either present or absent in the cell mediates target-gene specificity (24). In addition, because the PAI-1 promoter contains three elements that could bind HIF-1 with different affinity (11), it is possible that those elements binding HIF-1 with less affinity may bind HIF-2 with higher affinity. However, this must be determined, and currently the exact molecular mechanisms and circumstances under which PAI-1 can be switched on through HIF-1 or HIF-2 remain open. The present study shows that both HIF transcription factors can regulate this prothrombotic and pro-proliferative factor. In contrast, depletion of HIF-2α had no effect on PDGF-induced PASMC proliferation, whereas depletion of HIF-1α inhibited PDGF-stimulated proliferation of vascular smooth muscle cells (42). Thus, one may speculate that cell type–, but also stimulus-specific pathways exist and may preferentially activate HIF-1 or HIF-2. However, our present study, together with our previous findings, clearly indicates that both, HIF-1α and HIF-2α can be upregulated independent of hypoxia by redox-sensitive pathways, and can promote PASMC proliferation in response to thrombin, but also, as is shown here, to H2O2 and NOX4.
Taken together, our data show that HIF-2α is upregulated under nonhypoxic conditions in PASMCs in response to enhanced levels of ROS, stimulated by thrombin, H2O2, or NOX4 overexpression. This response was mediated by decreased hydroxylation of HIF-2α at the ODD, which was accompanied by diminished availability of the PHD cofactors ascorbate and Fe(II), resulting in decreased pVHL binding. Subsequently, HIF-2α C-TAD activity was increased under nonhypoxic conditions, thus promoting transcriptional activation of target genes and, subsequently, enhanced proliferation of PASMCs. Because a prothrombotic state and enhanced ROS levels together with increased proliferation of PASMCs have been associated with pulmonary vascular remodeling in pulmonary hypertension, and HIF-2α has been suggested to play a prominent role in lung physiology and the development of hypoxia-induced pulmonary hypertension (7, 9), ROS-dependent stabilization of HIF-2α may contribute to remodeling of the pulmonary and possibly also the systemic vasculature, also independent of hypoxia.
Footnotes
Acknowledgments
This work was supported by DFG grant GO709/4-4 (A.G.), Metoxia (HEALTH-F2-2009-222741) under the 7th research framework program of the European Union (A.G.), Fondation Leducq (J.H., T.K., and A.G.), and Fonds der Chemischen Industrie (T.K.).
Author Disclosure Statement
No competing financial interests exist.