
Abstract
Cytosolic ferritins sequester and store iron, consequently protecting cells against iron-mediated free radical damage. However, the mechanisms of iron exit from the ferritin cage and reutilization are largely unknown. In a previous study, we found that mitochondrial ferritin (MtFt) expression led to a decrease in cytosolic ferritin. Here we showed that treatment with inhibitors of lysosomal proteases largely blocked cytosolic ferritin loss in both MtFt-expressing and wild-type cells. Moreover, cytosolic ferritin in cells treated with inhibitors of lysosomal proteases was found to store more iron than did cytosolic ferritins in untreated cells. The prevention of cytosolic ferritin degradation in MtFt-expressing cells significantly blocked iron mobilization from the protein cage induced by MtFt expression. These studies also showed that blockage of cytosolic ferritin loss by leupeptin resulted in decreased cytosolic ferritin synthesis and prolonged cytosolic ferritin stability, potentially resulting in diminished iron availability. Lastly, we found that proteasomes were responsible for cytosolic ferritin degradation in cells pretreated with ferric ammonium citrate. Thus, the current studies suggest that cytosolic ferritin degradation precedes the release of iron in MtFt-expressing cells; that MtFt-induced cytosolic ferritin decrease is partially preventable by lysosomal protease inhibitors; and that both lysosomal and proteasomal pathways may be involved in cytosolic ferritin degradation. Antioxid. Redox Signal. 13, 999–1009.
Introduction
Although the mechanisms of iron-mediated post-transcriptional regulation of cytosolic ferritin synthesis and iron incorporation into the ferritin protein cage have been well characterized, iron release from cytosolic ferritin and iron recycling have been less studied (3, 30). Two models regarding iron exit and reutilization have been proposed. The first model is based on lysosome-mediated protein degradation and iron release. The second model postulates that ferritin pores at the junctions of ferritin subunits comprise iron channels that mediate iron release (30). In vitro studies support the concept that iron exits the ferritin shell via the hydrophilic channel on the 3-fold axes (30). These studies have shown that iron stored in the ferritin core can be mobilized by reductants in the presence of iron chelators (19). Further investigations have demonstrated accelerated iron release following mutagenesis of conserved amino acids in gated pores (21, 50). All of these results were obtained from studies using purified proteins. However, studies with mammalian cell cultures have shown that cytosolic ferritin iron is primarily released after proteolytic degradation of the protein (3). Evidence of lysosomal cytosolic ferritin degradation and subsequent iron release has been found in cells after different treatments. For example, inhibitors of lysosomal proteases, but not proteasomal inhibitors, block the degradation induced by iron chelators in different cell lines (22, 52). Lysosomal degradation of cytosolic ferritin has also been seen in cells treated with horse spleen ferritin-derived cationic ferritin (43), and has been seen in cells exposed to doxorubicin (25). Recent studies by Domenico et al. (9, 10) have shown that overexpression of the cellular iron exporter, ferroportin, caused cytosolic ferritin degradation and iron release and that different iron chelators induced different routes of cytosolic ferritin degradation. Interestingly, iron release is independent of cytosolic ferritin degradation, and iron can be mobilized in the absence of the degradation (9). Other studies have shown that oxidation-induced cytosolic ferritin turnover (33), and degradation of L-ferritin mutants linked to neuroferritinopathy (8), occur mainly via a proteasomal degradation pathway. Collectively, these studies suggest that multiple cytosolic ferritin degradation pathways may exist, and that different degradation mechanisms may play different roles under various physiological and pathological conditions. It is not clear, however, which degradation pathway is related to cytosolic ferritin degradation induced by mitochondrial ferritin (MtFt) or whether the protein shell degradation is a necessary step for iron release and reutilization.
The recent discovery of mitochondrial ferritin (MtFt) has contributed to our understanding of mitochondrial iron metabolism (7, 36, 37) and sideroblastic anemia (6, 7, 11, 28). Overexpression of MtFt results in a significant decrease in cytosolic ferritin levels, mobilizes iron exit from ferritin protein shells (37), and sensitizes tumor cells against oxidative stress (31). To evaluate the roles of different proteolysis pathways on cytosolic ferritin degradation and to characterize the mechanisms of iron exit from the ferritin cage, we examined cytosolic ferritin degradation in cells overexpressing MtFt in the presence of various proteolysis inhibitors. Our results show that lysosomal proteolysis was the primary pathway of cytosolic ferritin degradation in MtFt-expressing cells, and the blockage of cytosolic ferritin degradation also prevented iron release from the protein cage. Inhibitors of lysosomal proteases not only decreased new ferritin synthesis, but also altered iron redistribution between cytosolic ferritin and MtFt. Finally, in addition to lysosomes, proteasomes were also responsible for cytosolic ferritin degradation in iron overload cells.
Materials and Methods
Materials
59FeCl3 was purchased from Perkin Elmer (Boston, MA). The iron chelator salicylaldehyde isonicotinoyl hydrazone (SIH) was synthesized as described by Ponka et al. (40). Anti-hemagglutinin (HA) antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-transferrin receptor (TfR) antibody was purchased from Zymed (South San Francisco, CA). Anti-ferritin antibodies were from Dako (Carpinteria, CA) and US Biological (Swampscott, MA). Anti-LC3 antibody was from Novus Biologicals (Littleton, CO). Leupeptin, E-64D, chymostatin, MG132, and lactacystin were purchased from Sigma (Oakville, Canada). All other reagents were purchased from Sigma unless otherwise specified.
Cell culture
B9 cells, which stably overexpress MtFt were generated from the TA-H1299 cell line previously reported (37), and were maintained in a medium consisting of DMEM supplemented with 10% (v/v) tetracycline (tet)-free fetal bovine serum (Clontech, Mississauga, Canada), 2 mM glutamine, 100 units/ml of penicillin, and 0.1 ng/ml of streptomycin. MtFt expression was repressed by inclusion of tetracycline (tet) in the medium (tet-on cells, defined as uninduced cells). MtFt expression was induced by removal of tet from the medium (tet-off cells, defined as induced cells).
Western blot analysis
Western blot analysis was carried out as described (37). The blots were hybridized with anti-HA rabbit polyclonal IgG (Y11) for HA-MtFt expression, anti-ferritin antibodies for cytosolic ferritin expression, anti-TfR antibody, or anti-β-actin antibody, respectively. Dilutions were 1:1000 for primary antibodies and 1:10,000 for anti-rabbit or anti-mouse IgG secondary antibodies. Peroxidase-coupled secondary antibodies were detected with Western Lighting (Amersham, Buckinghamshire, UK). The film was digitized and analyzed using NIH imaging software.
Iron uptake and release
Human apo-transferrin (Tf ) was labeled with 59Fe as described previously (42). Equal numbers of cells (3 × 106 cells) were seeded without or with tet and cultured for 48 h. The cells were then incubated with 1 μM 59Fe-Tf at 37°C for 48 h, followed by washing three times with ice-cold PBS. Radioactivity in the cell pellet and culture medium was measured in a gamma counter (Cobra II auto-gamma, Packard). For iron release experiments, the washed cells were resuspended in medium and returned to culture for another 24 h. Radioactivity in the cell culture medium was measured in a gamma counter.
Cytosolic ferritin ELISA
Cellular proteins were extracted with 150 mM NaCl, 10 mM EDTA, 40 mM Tris (pH7.4), 1% Triton X-100, and a protease inhibitor cocktail (Roche, Palo Alto, CA). Total protein concentrations were determined by the Bradford assay. Cytosolic ferritin levels from cell extracts were determined by cytosolic ferritin ELISA (Laguna Scientific, Laguna, CA) according to the manufacturer's instructions.
Cellular iron distribution
After labeling with 59Fe-Tf as described above, cells were harvested and lysed at 4°C with lysis buffer (0.14 M NaCl, 0.1 M HEPES, pH 7.4 and 1.5% Triton X-100), and lysates were analyzed by electrophoresis followed by autoradiography as described (44, 53). Briefly, following centrifugation at 10,000 g for 10 min at 4°C, equal amounts of lysate samples were loaded onto native gradient gels (3%–20 % polyacrylamide). After 2–3 h of electrophoresis, the gels were dried and exposed to X-ray film.
Metabolic labeling and immunoprecipitation
Cells were metabolically labeled with (100 μCi/ml) 35S-methionine and 35S-cysteine, followed by cytosolic ferritin immunoprecipitation as described (37).
Cytosolic ferritin and LC3 immunofluorescence and cellular localization
For immunofluorescence confocal microscopy, cells were washed once in PBS, fixed for 20 min in 100% methanol, permeabilized in PBS, 0.5% Triton-X-100 for 90 sec and washed in PBS. After blocking in buffer (5% FBS, PBS) for 1 h, anti-ferritin antibodies diluted in blocking buffer were added, and the cells were incubated at 37°C for 1 h. After washing three times in PBS, the cells were incubated at 37°C for 40 min with anti-rabbit antibody labeled with TRITC. Cells were then labeled with anti-LC3 antibody and FITC labeled secondary antibody using the procedure described above. Cytosolic ferritin and LC3 were examined by immunofluorescence colocalization using a Zeiss confocal microscope with laser lines at 488 nm (LC3-FITC) and 543 nm (cytosolic ferritin-TRITC). Expression of cytosolic ferritin is shown as red immunofluorescence and LC3 is shown as green immunofluorescence. Colocalization of cytosolic ferritin and LC3 was demonstrated by merged images (yellow fluorescence).
Flow cytometric measurement of TfR
Direct immunofluorescent labeling of TfR was carried out using PE-labeled mouse anti-TfR antibody according to the manufacturer's protocol (Invitrogen, Carlsbad, CA) using 2.5 μl TfR antibody per 0.5 × 106 cells for 90 min.
Statistical analysis
All experiments were performed in triplicate unless otherwise indicated. Results are representative of three independent experiments. Error bars represent standard deviations. A probability level of 95% (p < 0.05) was considered significant.
Results
Inhibitors of lysosomal proteases prevent cytosolic ferritin loss in both uninduced and induced B9 cells
Both lysosomes and proteasomes have been shown to be involved in cytosolic ferritin degradation. Expression of MtFt leads to a decrease in steady state levels of cytosolic ferritin and to a decrease in newly synthesized cytosolic ferritin (37). To investigate the roles of MtFt on cytosolic ferritin degradation, both uninduced and induced B9 cells were treated with various proteolysis inhibitors, followed by measurement of cytosolic ferritin levels. Consistent with previous observations (37), MtFt expression led to a significant decrease in cytosolic ferritin levels (Figs. 1Ai and Aii, lanes 1 and 3 vs. lanes 2 and 4). The inhibitor of lysosomal proteases, leupeptin, partially blocked cytosolic ferritin protein loss in both uninduced and induced cells. As a positive control, treatment with 10 μM ferric ammonium citrate (FAC) led to significantly increased cytosolic ferritin levels, most likely due to stimulation of cytosolic ferritin synthesis (Fig. 1A, lanes 5 and 6).
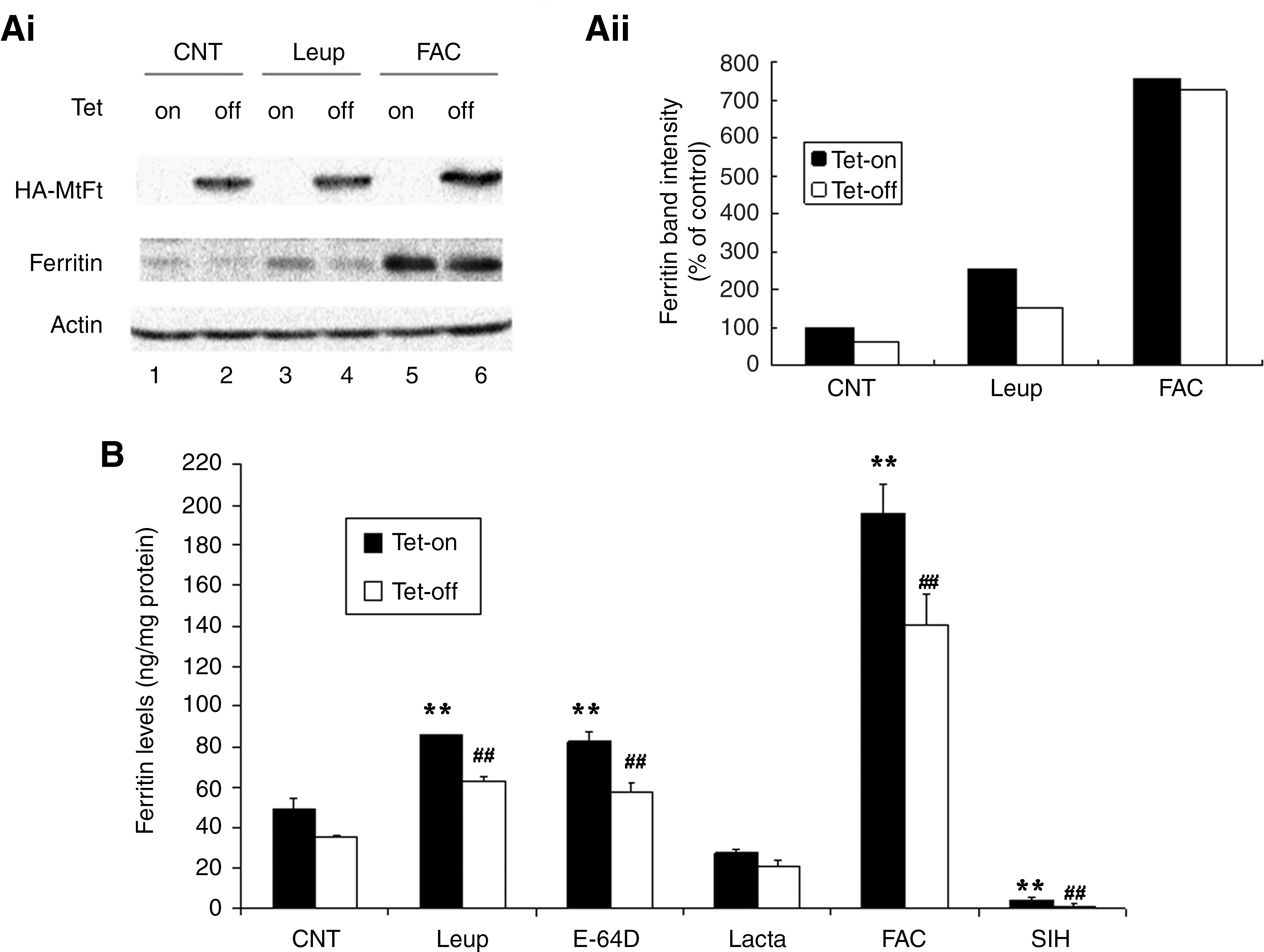
To further investigate the roles of lysosomes and proteasomes on cytosolic ferritin degradation in both uninduced and induced cells, the protein levels were measured by ELISA following treatment with lysosomal and proteasomal inhibitors. As shown in Figure 1B, consistent with results in Figure 1A, MtFt expression led to decreased cytosolic ferritin levels. The basal levels of cytosolic ferritin in B9 cells were about 49 ng/mg in uninduced cells and 36 ng/mg in induced cells. Treatment with inhibitors of lysosomal proteases leupeptin and E-64D significantly prevented the loss of cytosolic ferritins in both uninduced cells and in induced cells. However, treatment with the specific proteasomal inhibitor lactacystin did not effectively prevent cytosolic ferritin degradation, but rather led to decreased cytosolic ferritin levels in both uninduced and induced cells, most likely due to toxic effects of lactacystin on cells. Inhibition of proteasome activities by lactacystin has been reported to induce programmed cell death (34). MG 132 was not used because it is nonspecific and affects both lysosomal and proteasomal degradation pathways. MG 132 primarily targets the chymotrypsin-like activity of proteasomes; however, it also inhibits the activities of lysosomal cysteine proteasomes and the calpains (26). As a positive control, FAC treatment led to dramatically increased cytosolic ferritin levels compared to control cells. The negative control, SIH, a hydrophobic iron chelator reduced cytosolic ferritin levels to an almost undetectable level compared to untreated B9 cells. These results indicate that lysosomes may be responsible for cytosolic ferritin degradation in both MtFt-expressing and wild-type cells.
Cytosolic ferritin colocalizes with LC3, a marker for autophagosomes
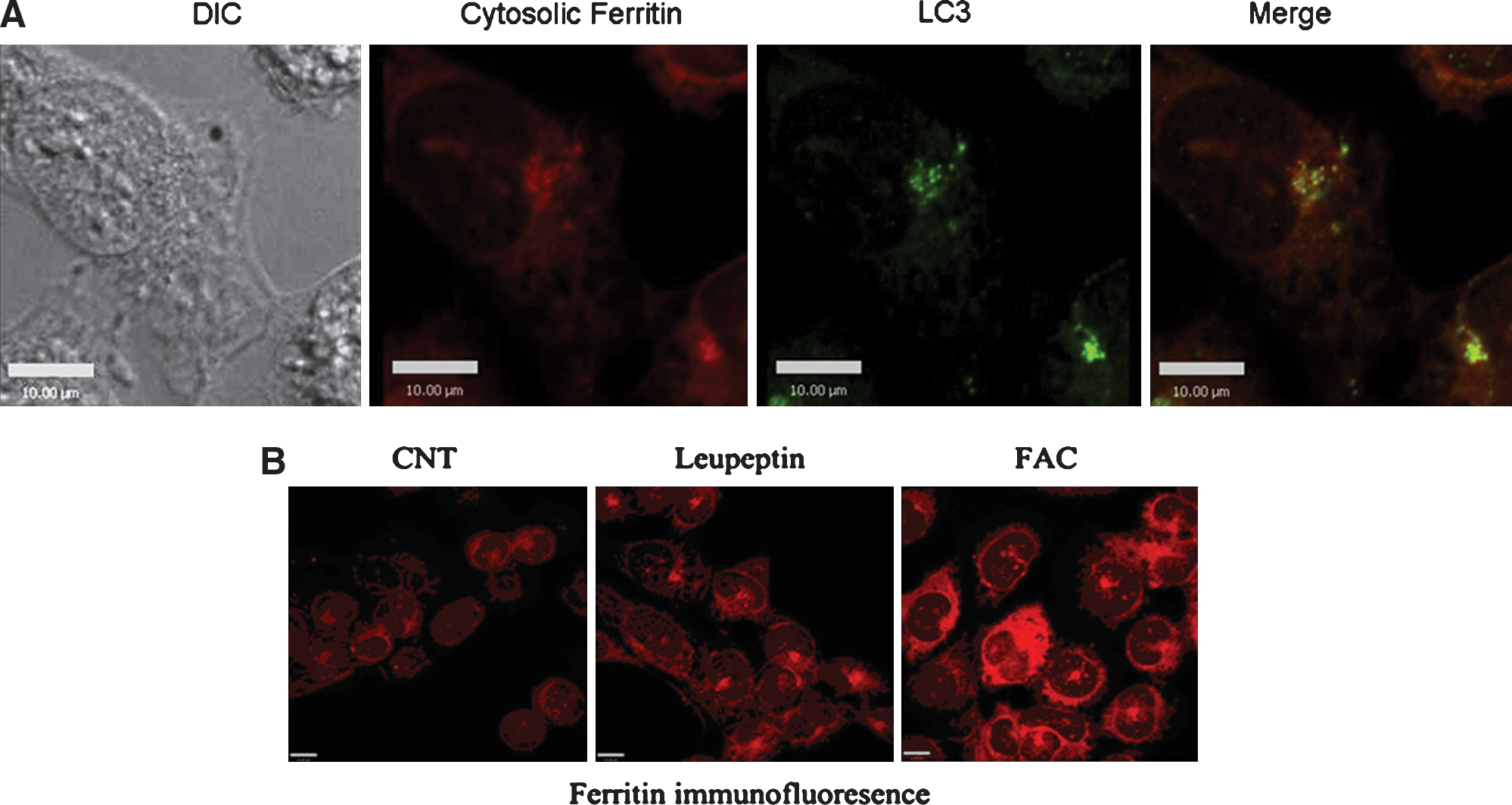
Lysosomes play an important role in cellular metabolism, such as in turnover of many cellular constitutes and in the processing of nutrients and other substances from external environments. One of the routes by which intracellular proteins gain access to organelles involves autophagy. To study the possibility that cytosolic ferritin is removed from the cytosol by autophagy, we studied possible colocalization with LC3, a marker for autophagosomes. Autophagy begins with the formation of double-membrane-bounded autophagosomes. These membrane associated structures fuse with lysosomes to form autophagolysosomes. LC3 is the only known marker that specifically localizes to autophagosomes and autophagolysosomal membranes (46). The pattern of cytosolic ferritins intracellular expression in uninduced B9 cells was studied by confocal microscopy using anti-ferritin antibodies and tetramethylrhodamine isothiocyanate (TRITC) labeled secondary antibody. Cytosolic ferritins exhibited strong diffuse cellular immunofluorescence staining consistent with a cytosolic distribution as described by Corsi et al. (7). The distribution of autolysosomes was investigated using a monoclonal antibody against LC3. LC3 showed punctuate staining in cytosol in addition to the diffuse pattern. In autolysosomes, cytosolic ferritin immunofluorescence demonstration also revealed a punctuate pattern which overlapped significantly with LC3 immunofluorescence staining (merged images, yellow fluorescence) (Fig. 2A). Leupeptin treatment enhanced cytosolic ferritin immunofluorescence, further supporting the notion that cytosolic ferritin degradation is via the lysosomal pathway. The control treatment with FAC also enhanced cytosolic ferritin immunofluorescence as a consequence of increased the protein synthesis (Fig. 2B).
Treatment with leupeptin and E-64D increases cytosolic ferritin stability and decreases synthesis
In Figure 1 we showed that leupeptin and E-64D effectively blocked cytosolic ferritin degradation. Cytosolic ferritin levels and LIP iron content are part of a regulatory feedback loop based on the IRP–IRE system. Cytosolic ferritin levels are post-transcriptionally regulated by iron availability, and in turn, LIP iron levels are regulated by cytosolic ferritin, which stores excess iron. We hypothesized that if iron release from the ferritin protein shell were dependent on ferritin shell degradation, leupeptin treatment, which has been shown to prevent cytosolic ferritin protein degradation, would lead to increased iron storage in cytosolic ferritins, and a consequent reduction of iron in LIP and a reduction in newly synthesized cytosolic ferritins. To test this hypothesis, we measured the effect of leupeptin treatment on cytosolic ferritin protein stability and synthesis using metabolic labeling with 35S-methionine and 35S-cysteine.
To measure the effect of leupeptin treatment on the synthesis of cytosolic ferritins, B9 cells were induced to express MtFt for 24 h, followed by incubation with 10 μM leupeptin for an additional 16 h in the absence of tet. The cells were then labeled with 35S-methionine and 35S-cysteine for 1 h, and the radiolabeled cytosolic ferritin was analyzed by immunoprecipitation with anti-ferritin antibodies. As shown in Figures 3Ai and 3Aii, there was a significant decrease in cytosolic ferritin synthesis (especially for H-ferritin) in leupeptin treated cells compared to control cells in both uninduced (lane 1 vs. 3) and induced cells (lane 2 vs. 4). In addition to confirm that MtFt expression led to decreased cytosolic ferritin synthesis (lane 1 vs. 2, lane 3 vs. 4), while FAC treatment resulted in increased cytosolic ferritin synthesis (lane 5 vs. 1, and lane 6 vs. 2), Figure 3A also showed that expression of MtFt (tet-off ) leads to a switch towards a less H-chain rich cytosolic ferritin species marked by a changed H:L chain ratio. This observation may indicate that H-chain ferritin, compared to L-chain, is less stable for iron deficiency-induced ferritin degradation. These results showed that leupeptin treatment resulted in a decrease in cytosolic ferritin synthesis in both uninduced and induced cells. Therefore, in the absence of cytosolic ferritin synthesis, the increase in cytosolic ferritin levels previously observed by Western blotting and ELISA in leupeptin-treated cells (Figs. 1A and 1B) must be a consequence of an increase in the protein stability.
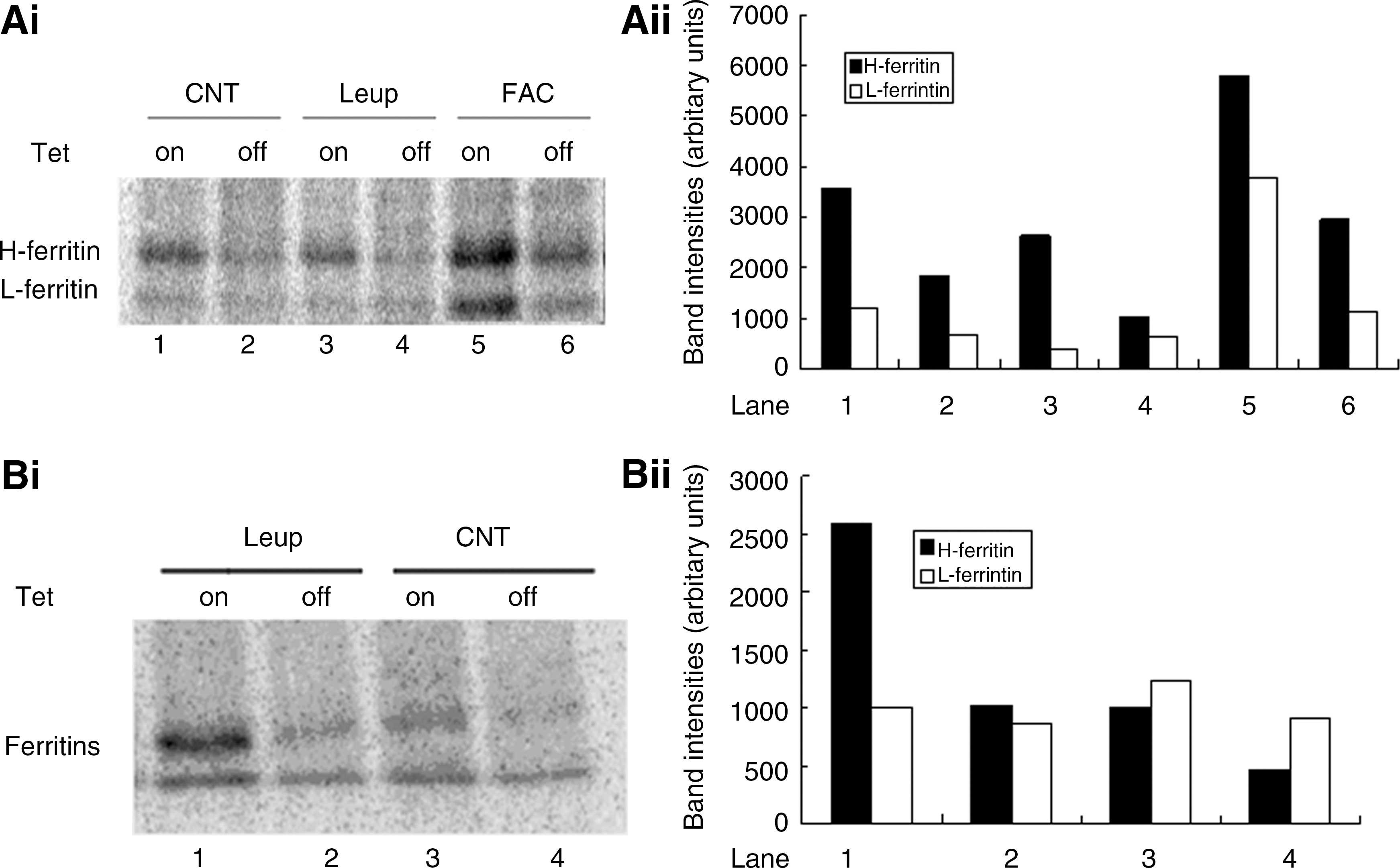
To investigate the effect of leupeptin treatment on the stability of cytosolic ferritins, B9 cells were induced to express MtFt for 24 h, followed by 35S-methionine and 35S-cysteine labeling for an additional 24 h. The cells were then treated with leupeptin for 16 h, and cytosolic ferritin was analyzed by immunoprecipitation with anti-ferritin antibodies. As can be seen in Figures 3Bi and 3Bii, the 35S-labeled cytosolic ferritin band from leupeptin-treated cells was more intense than from control cells (especially in uninduced cells, lane 1 vs. 3). MtFt expression decreased the stability of cytosolic ferritins (lane 2 vs. 1 and lane 4 vs. 3) and counteracted the stabilizing effect of leupeptin on the cytosolic ferritin band. Together, these cytosolic ferritin synthesis and stability experiments show that leupeptin treatment resulted in a decrease in cytosolic ferritin synthesis and in an increase in cytosolic ferritin stability. Therefore, the increase in steady state levels of cytosolic ferritins observed in Figures 1A and 1B is not due to increased protein synthesis, but must be a consequence of increased protein stability in cells treated with leupeptin. Furthermore, these results also support the hypothesis that iron release from the cytosolic ferritin protein shell is dependent on ferritin shell degradation, because leupeptin treatment, which has been shown to prevent cytosolic ferritin protein degradation, led to an increase in the amount of iron stored in cytosolic ferritin, and very likely to a reduced amount of iron in LIP and, consequently, to reduced cytosolic ferritin synthesis.
Inhibitors of lysosomal proteases prevent iron release from cytosolic ferritin
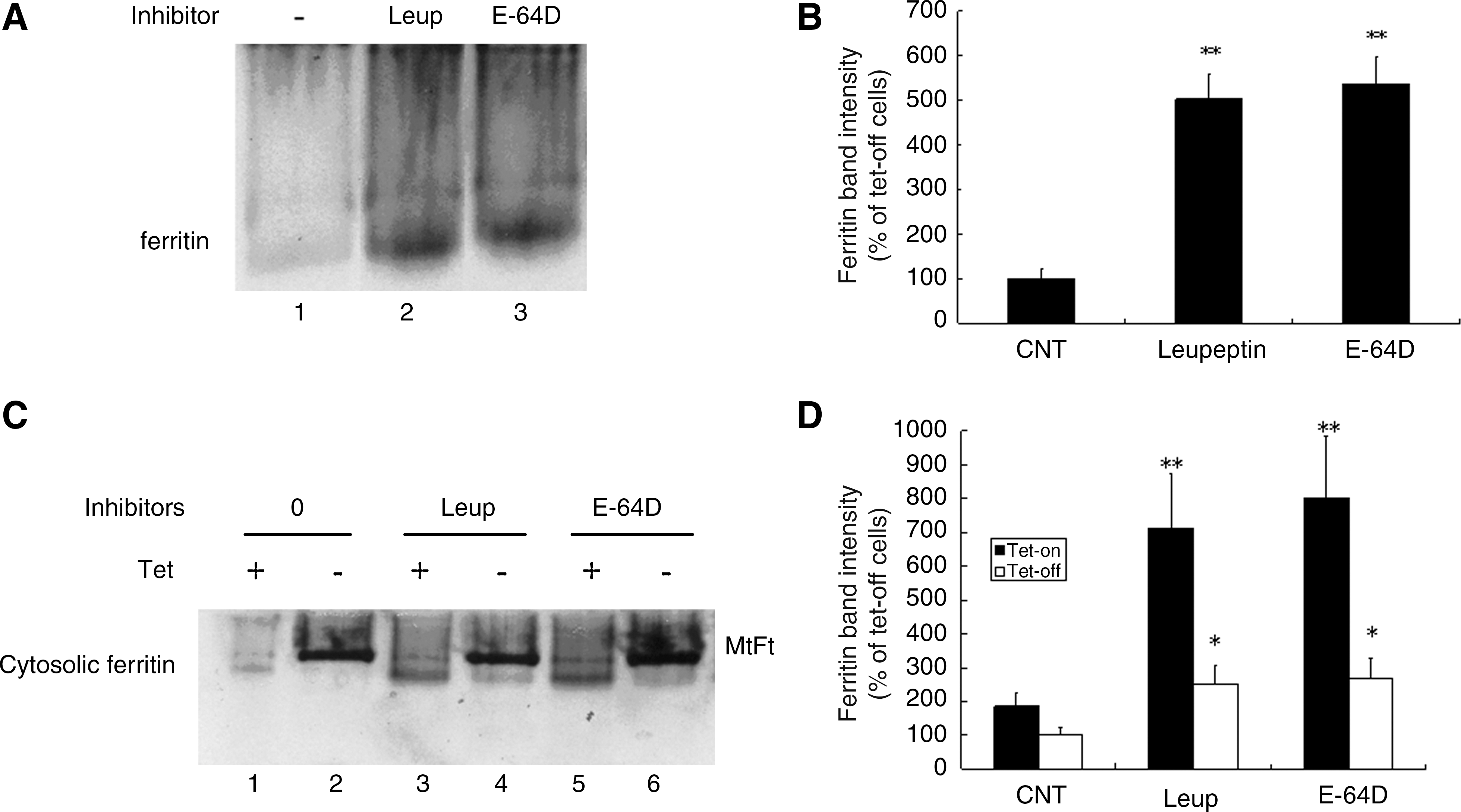
The results presented in Figures 1 –3 show that inhibitors of lysosomal proteases significantly prevented cytosolic ferritin degradation and decreased protein synthesis. Based on these observations, we speculated that if degradation of cytosolic ferritin protein precedes iron release, then stored iron release will be dependent on this step. To test this hypothesis, uninduced B9 cells were labeled with 59Fe-transferrin for 48 h, followed by treatment with leupeptin and E-64D for 24 h. Native, nondenaturing PAGE combined with 59Fe autoradiography was then used to evaluate 59Fe release from cytosolic ferritins. As shown in Figure 4A, both leupeptin and E-64D prevented iron release from cytosolic ferritins. There was about 5 times increase in radio-iron stored in cytosolic ferritin bands in both leupeptin and E-64D treated cells (Fig. 4B). This observation is a strong indication that cytosolic ferritin protein shell degradation occurs prior to iron release from the protein cage.
Iron redistribution upon leupeptin treatment and MtFt induction
To further confirm that cytosolic ferritin iron release is dependent on protein degradation, we examined iron redistribution following treatment with lysosomal enzyme inhibitors and MtFt induction (Figs. 4C and 4D). The results shown in Figure 4C (lanes 1 and 2) confirmed our previous observations that MtFt migrated more slowly than cytosolic ferritin, and that the induction of MtFt expression blocked iron from being incorporated into cytosolic ferritin, whereas in uninduced cells the majority of the 59Fe was taken up by cytosolic ferritin (37). Treatment with leupeptin and E-64D led to the reappearance of the cytosolic ferritin band (Fig. 4C, lanes 4 and 6). Importantly, treatment with inhibitors of lysosomal proteases to block degradation of the cytosolic ferritin protein shell prevented iron release from cytosolic ferritins in both uninduced and induced cells (compare Fig. 4C, lane 3 vs. 1, lane 5 vs. 1, lane 4 vs. 2, and lane 6 vs. 2). These results show that blocking cytosolic ferritin protein degradation prevented iron mobilization from cytosolic ferritins. The accumulated cytosolic ferritins are functional and incorporate 59Fe into the protein cage.
Effect of proteolysis inhibitors on cellular iron uptake and TfR expression
We examined the role of treatment with proteolysis inhibitors on the uptake of 59Fe via 59Fe-Tf and on 59Fe release from cells. Total iron uptake experiments demonstrated that although MtFt expression significantly induced iron uptake as expected (37), treatment with proteolysis inhibitors did not significantly affect cellular iron uptake capacity (Fig. 5A).
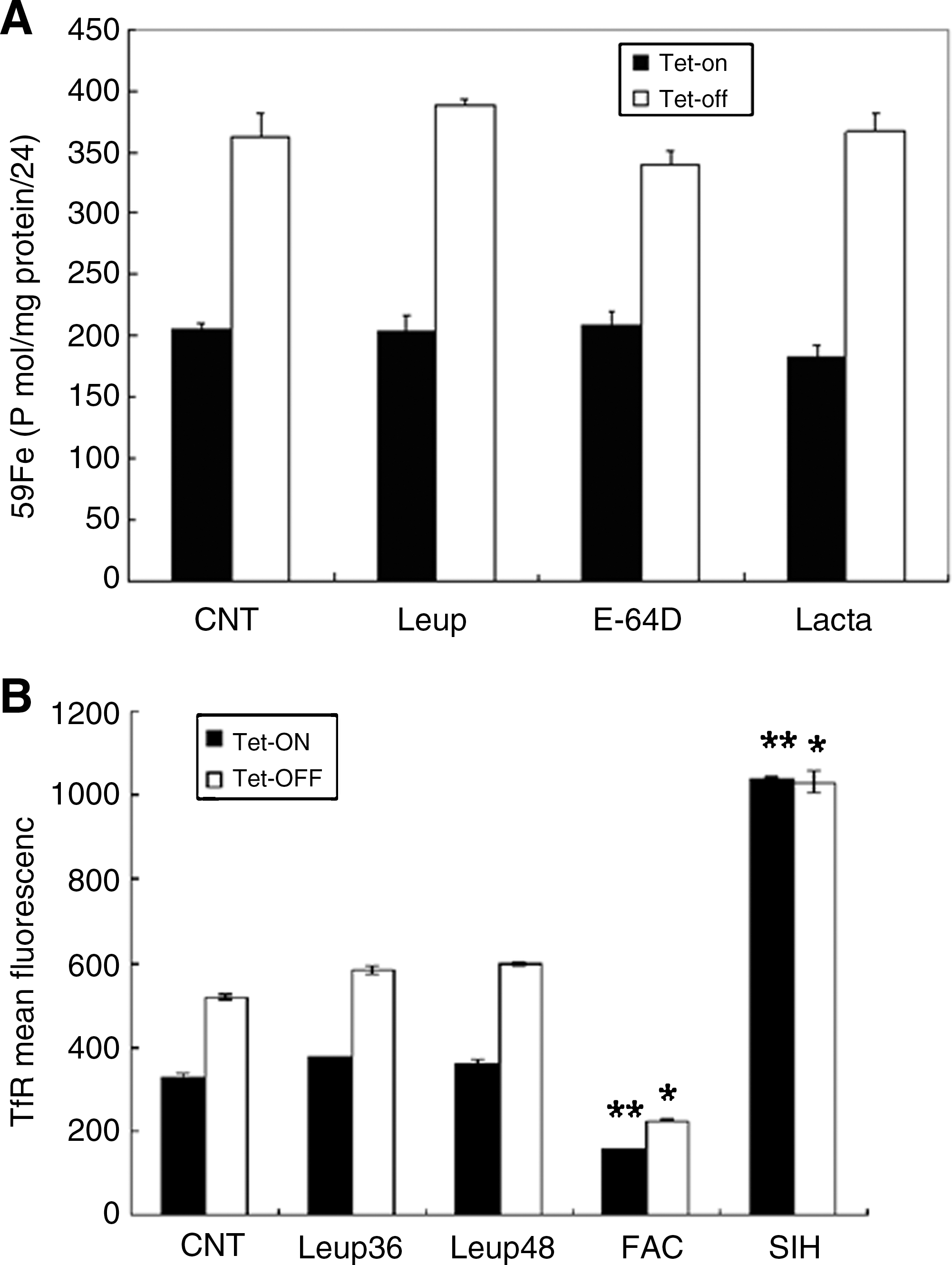
Next, we examined the effect of leupeptin treatment on the expression of membrane associated TfR in both uninduced and induced cells. B9 cells were grown in the presence or absence of tet for 48 h and membrane-associated TfR levels were analyzed by FACS. MtFt expression induced elevated TfR expression as shown previously (37). Leupeptin treatment did not change the levels of TfR, relative to control cells, in either uninduced or induced cells (Fig. 5B).
Proteasome involvement in cytosolic ferritin degradation in iron overload cells
As proteasomes have been shown to be involved in cytosolic ferritin degradation in cells with oxidatively damaged and inappropriate protein folding (8, 33), and in HEK293T cells (human embryonic kidney cells) and FM3A cells (mouse mammary carcinoma) (9), we investigated the roles of lysosomal and proteasomal inhibitors on cytosolic ferritin degradation in cells that were pretreated with the iron donor, FAC. Uninduced B9 cells were pretreated with 10 μM FAC for 24 h, followed by incubation with various inhibitors for an additional 16 h. Cytosolic ferritin levels were measured by ELISA. As shown in Figure 6, both tested inhibitors significantly inhibited cytosolic ferritin loss in FAC pretreated cells. These observations indicate that proteasomes are involved in cytosolic ferritin degradation, preferentially in cells with iron overload.

Discussion
Cytosolic ferritins are the major cellular iron storage proteins and their regulation and function have been extensively studied (3). However, some of the fundamental functions of this iron protein are still unclear. For example, it is not known how cytosolic ferritin is degraded, how stored iron is released, and whether cytosolic ferritin iron can be metabolically used. In erythroid cells, transferrin is the major iron donor. It has been proposed that iron acquired from transferrin is targeted to mitochondria and bypasses the cytosol (49). Although it is unlikely that cytosolic ferritins are the source of iron for heme synthesis in reticulocytes (41), in neoplastic and differentiated cardiomyocytes in cell culture, the majority of 59Fe acquired from transferrin was incorporated into cytosolic ferritins, and was subsequently mobilized from cytosolic ferritins into other compartments (24, 25).
Ferritin genes are ubiquitously expressed and the mRNA for L-ferritin is one of the ten most abundant transcripts in the human lens library (54). It has been proposed that age-related cytosolic ferritin accumulation significantly contributes to lens turbidity and cataract formation (17). Accumulation of L-ferritin in lenticular tissue has been found in patients with hereditary hyperferritinemia cataract syndrome who lack IRP dependent regulation as a result of mutations or deletions in the IRE region of L-ferritin mRNA (5, 16, 20). In order to devise therapeutic strategies for treating diseases caused by dysregulated cytosolic ferritin expression, it is important to elucidate the detailed mechanisms of cytosolic ferritin degradation. For example, a strategy aimed at promoting degradation of misfolded proteins by using small compounds to enhance autophagy has been proposed as a therapeutic approach in aggregate-prone protein related diseases (proteinopathies) (46, 55).
The evidence regarding the mechanism of cytosolic ferritin degradation and the relationship between ferritin protein cage degradation and iron mobilization is controversial (3, 30). It has been proposed that cytosolic ferritin-iron release requires degradation of cytosolic ferritins within lysosomes (43, 45). Lysosomes contain large amounts of labile iron, which make them sensitive to oxidative stress. Enrichment of lysosomes with ferritin via autophagocytosis stabilizes autophagosomes and cells by binding lysosomal low mass iron, thereby reducing the concentration of lysosomal redox-active iron and the formation of hydroxyl radicals (14, 15, 23, 39). Ascorbate greatly retards the autophagic uptake of cytosolic ferritin clusters into lysosomes (4). Starvation of NIT insulinoma cells stimulates rapid synthesis of cytosolic ferritins, which has been found to be autophagocytosed and stabilize lysosomes (38). However, entry of the protein into lysosomes may reflect a response to cellular stresses, rather than a physiological route for iron release (9). Most recent studies show that ferroportin-mediated mobilization of cytosolic ferritin iron precedes the protein degradation by the proteasomal pathway (9), and ferroportin-mediated mobilization of cytosolic ferritin iron precedes its degradation by the proteasome in HEK cells and yeast (9). Oxidized cytosolic ferritins can be degraded by the proteasome (33, 47). The route of cytosolic ferritin degradation differs depending on the specific iron chelator that is used (10). In aggregate, these observations suggest that there are multiple degradation pathways which are involved in the response to complex physiologic and pathologic conditions.
Levi et al. (28) recently identified a new intronless gene which encodes an H-ferritin-like protein, mitochondrial ferritin (MtFt), which is expressed only in mitochondria. Most cells and tissues, including normal erythroblasts, express very low levels of MtFt; testes have the highest expression of MtFt in mice (28). However, MtFt levels increase dramatically in ring sideroblasts from patients with sideroblastic anemia (6, 28). Ring sideroblasts are pathologic erythroid precursors containing excessive deposits of non-heme iron with a characteristic perinuclear distribution accounting for the ring appearance (2, 13, 32). Previous work has documented that the overexpression of MtFt in mammalian cells leads to a decrease in cytosolic ferritin levels and enhanced TfR expression (28, 37). Interestingly, when MtFt expression has been induced, direct competition for iron has been observed among MtFt and other iron binding molecules (primarily cytosolic ferritin), and in turn, iron is mobilized from cytosolic ferritin and resulting in decreased cytosolic ferritin levels.
We exploited inducible MtFt expression in stable transfectants in combination with the use of specific proteolytic inhibitors, to investigate cytosolic ferritin degradation mechanisms. The rapid mobilization of iron from cytosolic ferritin to MtFt in this inducible system provides a useful tool for investigating the nature of an enduring dilemma regarding iron metabolism—the release of iron from cytosolic ferritins in situ. The results demonstrated that lysosomes play a more important role than do proteasomes in cytosolic ferritin degradation (Figures 1 –3). Although cytosolic ferritin degradation proceeded at a reduced rate, leupeptin treatment did not completely prevent the loss of cytosolic ferritins induced by MtFt expression (Figs. 1A and 1B). This result can be explained by a decreased protein synthesis rate in MtFt-expressing cells (see Fig. 3A) and an incomplete prevention of cytosolic ferritin proteolytic degradation by leupeptin. Leupeptin inhibits several, but not all, lysosomal proteases (4). MtFt-expressing cells, as compared to their wild-type counterparts, had less cytosolic ferritin content, as shown previously (37). The decreased protein levels in MtFt-expressing cells may represent the combined effects of decreased protein synthesis and increased protein degradation. To address this possibility, we measured cytosolic ferritin stability and synthesis. As shown in Figure 3A, leupeptin treatment led to decreased cytosolic ferritin synthesis in both uninduced and induced cells. As shown in Figure 3B, leupeptin treatment increased protein stability, while MtFt expression decreased cytosolic ferritin stability over the 24 h treatment. This experiment confirmed that leupeptin blocked cytosolic ferritin degradation and prolonged protein half life. Therefore, MtFt expression decreased both cytosolic ferritin stability and synthesis; leupeptin treatment decreased the protein synthesis, but increased stability. This observation suggests that when cytosolic ferritin protein degradation is blocked, the proportion of iron normally recycled from cytosolic ferritins to the LIP is reduced. In the resulting iron-deficient cellular environment, IRPs downregulate cytosolic ferritin synthesis. Based on this reasoning, we speculate that cytosolic ferritin iron release is dependent on degradation of the protein cage. Both lysosomes and proteasomes are involved in cytosolic ferritin degradation in cells pretreated with FAC (Fig. 6), indicating that multiple proteolytic pathways exist for protein degradation under different metabolic and pathophysiological conditions.
We also found that the intracellular distribution of cytosolic ferritins largely overlaps with autophagosomes and autolysosomes, as was demonstrated by merged immunofluorescence of cytosolic ferritins and LC3 (Fig. 2). This finding provides direct evidence that cytosolic ferritin is physically localized in lysosomes. In another set of experiments, we documented that stronger intracellular cytosolic ferritin immunofluorescent signals were observed in leupeptin-treated cells, as compared to untreated cells (Fig. 2B), indicating higher levels of cytosolic ferritins after inhibition of lysosomal protease activity. Taking together, these observations strongly suggest that lysosomes are responsible for cytosolic ferritin turnover. Together, these results show that blocking the protein degradation prevented iron mobilization from cytosolic ferritins. The accumulated cytosolic ferritins are functional and incorporate 59Fe into the protein cage. When cytosolic ferritin turnover is blocked by inhibitors of lysosomal proteases, more functional cytosolic ferritin is available to store more iron in the protein cage. At the same time, less new cytosolic ferritin is synthesized because of reduced iron availability. These data imply that cytosolic ferritin protein degradation is necessary for iron mobilization, and suggest that protein degradation precedes iron exit.
To provide direct evidence that cytosolic ferritin degradation precedes iron release, we radiolabeled cytosolic ferritins and treated cells with leupeptin, followed by measurement of radiolabeled cytosolic ferritin band intensity. Treatment with leupeptin prevented significant cytosolic ferritin iron mobilization (Fig. 4). As well, MtFt expression significantly decreased cytosolic ferritin band intensity as shown previously (37). In contrast to uninduced cells in which the majority of the radio-iron was in cytosolic ferritin, induced cells had much less radio-iron in cytosolic ferritin and contained virtually all their radio-iron in MtFt. Leupeptin treatment dramatically increased the cytosolic ferritin band intensity in uninduced cells. In addition, leupeptin treatment resulted in reappearance of the cytosolic ferritin band in induced cells.
We also investigated whether treatment with leupeptin or E-64D affected cellular iron uptake and release, or levels of TfR and actin. The data demonstrated that, despite the fact that induction of MtFt significantly increased cellular iron uptake, lysosomal enzymatic inhibitors did not significantly affect cellular iron uptake or levels of TfR (Fig. 5). The data suggest that in the presence of leupeptin or E-64D, the cells exhibited normal iron acquisition ability.
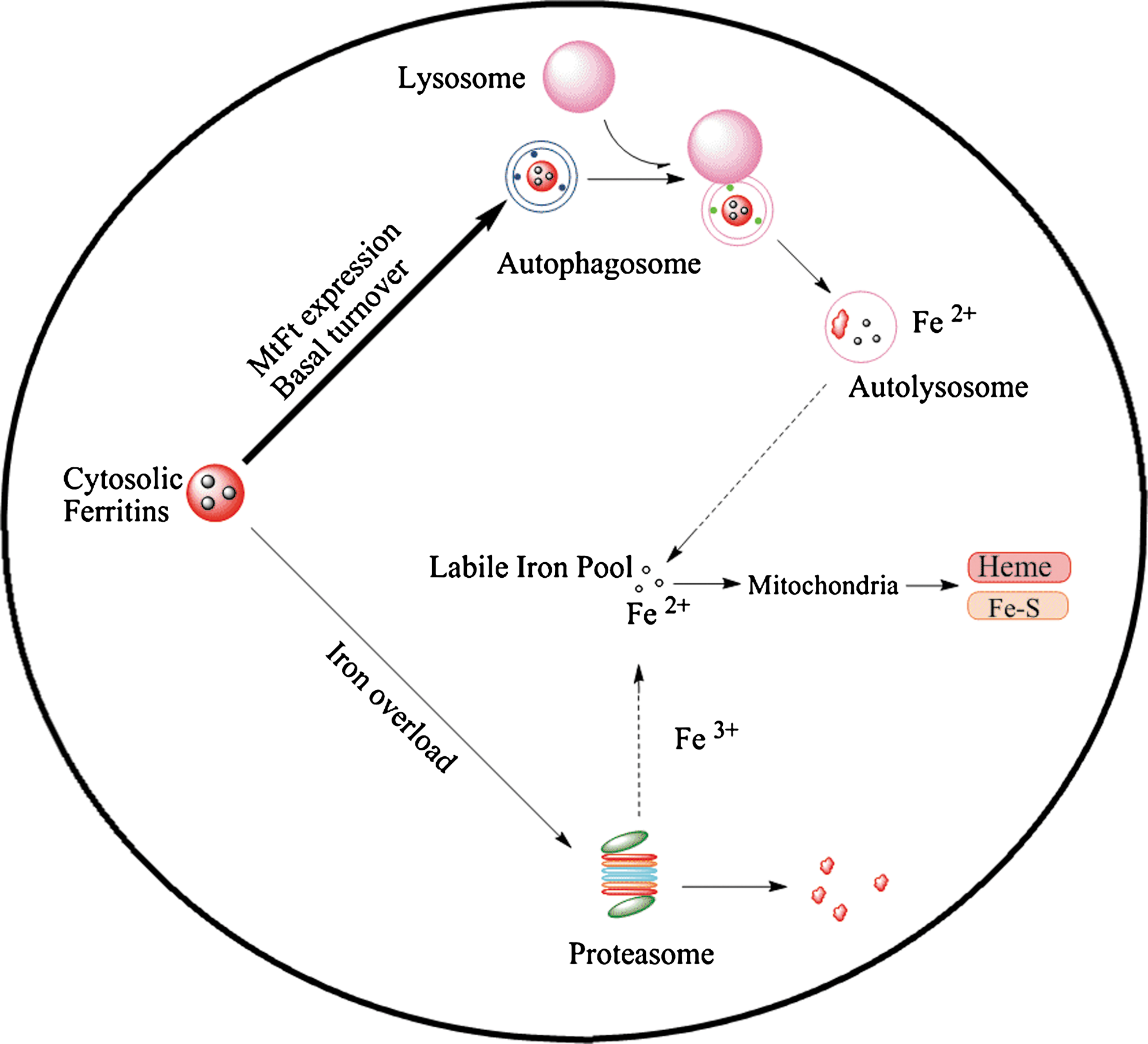
In summary, the current study has demonstrated that lysosomal enzyme inhibitors, leupeptin and E-64D, are capable of preventing cytosolic ferritin loss and iron release. Moreover, treatment with leupeptin stabilized cytosolic ferritin and decreased cytosolic ferritin synthesis. The proteasomal inhibitor, lactacystin, also prevented cytosolic ferritin degradation in cells under conditions of iron overload. Leupeptin treatment did not compromise cellular iron uptake. Collectively, the studies presented here suggest the following mechanisms of ferritin degradation and iron release (Fig. 7). (a) Cytosolic ferritin is degraded primarily in lysosomes in both MtFt expressing cells and wild type cells; (b) multiple degradation mechanisms exist that operate under metabolic conditions, such as iron overload condition; (c) cytosolic ferritin degradation precedes the release of iron and that cytosolic ferritin turnover may be a necessary step for iron release and utilization; and (d) MtFt-induced cytosolic ferritin decrease was prevented by lysosomal protease inhibitors. Elucidation of these mechanisms of cytosolic ferritin degradation could be potentially important for deciphering pathological cytosolic ferritin accumulation, such as occurs in aging or as a result of genetic defects related to development of cataracts (5, 16, 17, 20). Therapeutic strategies aimed at promoting protein degradation might provide a feasible means of treating conditions related to pathologic cytosolic ferritin accumulation.
Footnotes
Acknowledgments
This work was supported by grants from Chinese Academy of Sciences (Hundred Talents Program) (GN), National High Technology Research and Development Program of China (2009AA03Z335), National Basic Research Program of China (973; No.2010CB933600), National Natural Science Foundation of China (30900278, 10979011) and Canadian Institutes of Health Research (PP).
Y. Zhang designed and performed research, analyzed, and interpreted data, performed statistical analysis; M. Mikhael performed research, and analyzed the data; D. Xu and Y. Li performed research, analyzed, and interpreted data; S. Soe–Lin designed research, analyzed, and interpreted data, and helped to edit the manuscript; B. Ning performed research; G. Nie conceived, designed, and performed research, analyzed, and interpreted data, and wrote the manuscript; Y. Zhao conceived and designed research and edited the manuscript; P. Ponka designed research and edited the manuscript.
Author Disclosure Statement
The authors declare no competing financial interests.