
Abstract
Mitochondria contribute to various diseases and aging phenotypes. Reactive oxygen species (ROS), mainly formed by the respiratory chain, were long thought to cause these effects by damaging proteins, DNA, and lipids. The emerging understanding that ROS act not only destructively but also as dedicated signaling molecules, and that aging processes are regulated by specific signaling networks has stimulated research on mitochondrial signaling systems and the regulation of mitochondrial ROS metabolism. p66Shc is a lifespan-regulating protein contributing to mitochondrial ROS metabolism and regulating the mitochondrial apoptosis pathway. It was found to participate in aging processes and has been implicated in several pathologies. Considerable progress has been made recently concerning the molecular function of p66Shc. It appears that p66Shc responds to a variety of proapoptotic stimuli by increasing ROS levels in the mitochondrial intermembrane space through an inherent ROS-producing activity, and that this ROS formation might trigger initiation of the mitochondrial apoptosis pathway. In this review, we will discuss the current knowledge on the molecular architecture of the p66Shc protein, its role in ROS metabolism and apoptosis regulation in the mitochondrial intermembrane space, the regulation of its mitochondrial transport, and the molecular mechanisms and interactions involved in these processes. Antioxid. Redox Signal. 13, 1417–1428.
Mitochondrial Signaling Mechanisms in Aging and Disease
The long-known importance of mitochondria in apoptosis, aging, and diseases (6, 24, 77), the connection between energy metabolism and lifespan regulation (6), and the emerging understanding that defined signaling systems regulate lifespan (9) are stimuli for strong research efforts on signaling mechanisms in mitochondria. From this work, it appears that the role of mitochondria in cellular regulation and aging is more complex than merely damaging proteins, DNA, and lipids through ROS formation. In addition to this destructive effect (26), ROS are more and more recognized also to act as mediators of a network of redox signaling mechanisms (24, 73). An important mechanism in doing so seems to be the reversible oxidation of cysteine residues in signaling proteins, resulting in modulation of the proteins' activities (7). For example, a redox-dependent regulation of kinases, such as cGMP-dependent protein kinase (PKG), and of transcription factors, such as heat shock factor 1 (Hsf1), has been reported (4, 11, 73). And even though additional cellular ROS sources, such as NADPH oxidases (36), are known, the mitochondrial respiratory chain [electron transfer chain (ETC)] constitutes the major ROS source and appears to play a dominant role in ROS signaling (43). The ETC can convert as much as 0.15% of cellular oxygen to ROS (68), and various factors, such as uncoupling or changes in NADH/NAD+ ratios, can influence the ROS formation rate (43). The ETC mainly forms superoxide radicals that can further be converted by superoxide dismutase (SOD) to H2O2, which can then react to yield highly reactive hydroxyl radicals. Additional proteins, such as Erv1 (essential for respiration and vegetative growth 1) and p66Shc, appear to be able to connect the ETC to other processes, in particular protein translocation and the inducible formation of ROS for signaling purposes (see below), respectively. Due to their toxicity, but likely also for making ROS available as signaling molecules, cells have a variety of antioxidant mechanisms for scavenging ROS, such as SOD and glutathione peroxidase. The complex interplay of all these mechanisms determines the level of ROS formation (73). Consequently, the relationship between metabolic rate and ROS formation was found not to be straightforward (6), consistent with ROS contributing to aging processes as signaling molecules whose generation is highly regulated.
Confirming an important role of the ETC and ROS formation in aging mechanisms, a number of Caenorhabditis elegans longevity mutants show changes in mitochondrial energy metabolism (27, 37). A defect in Complex III or downregulation of the ETC via RNAi leads to a decrease in oxygen consumption and an increase in lifespan (15, 18). Similarly, the mitochondrial protein deacetylase Sirt3 appears to contribute to longevity regulation (63), likely through its influence on the ETC (3) or its possible influence on antioxidants regeneration (65). Other mechanisms might also exist, but influencing mitochondrial ETC and ROS metabolism seems to be a common feature and an integrating mechanism for many lifespan influencing proteins (6).
p66Shc—A Longevity Regulator Influencing Mitochondrial Metabolism and Apoptosis
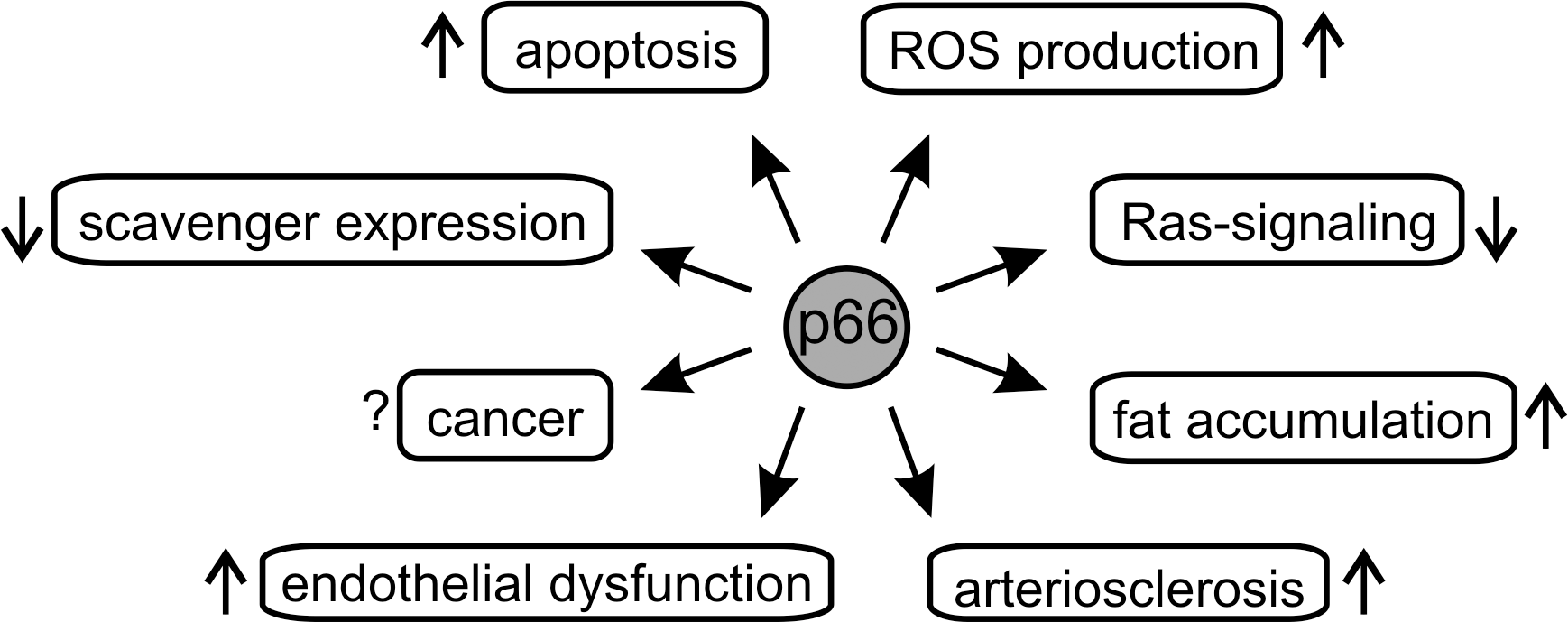
The protein p66Shc was identified in 1999 as a negative lifespan regulator (41). Knocking out part of the corresponding ShcA gene prolonged lifetime of mice about 30% (41), and its expression rises in the elderly (55). Like many other proteins regulating lifespan, p66Shc was found to influence energy metabolism (45), but it was also shown to contribute directly to ROS formation (21, 23) (see below). It seems to act as a sensor for cellular stress, such as UV or oxidants. The activated protein induces the mitochondrial apoptosis pathway, and its “deadly” activity could serve in cell homeostasis as a “suicide switch” if cellular damage exceeds stress response capacities (20, 21, 23). Consistent with its function in regulating this crucial cell fate switch, several pathologies have been associated with disturbed p66Shc signaling (Fig. 1). For example, a variety of human cancers are associated with elevated p66Shc levels (30, 74), possibly a cellular attempt to compensate for other blocked apoptotic pathways to antagonize uncontrolled cell proliferation. However, p66Shc was also reported to promote growth of cancer cells (74), and its exact role in carcinogenesis and tumor growth remains to be established. Furthermore, roles for p66Shc in arteriosclerosis (44) and endothelial dysfunctions (12) have been reported. The role of p66Shc in these physiological systems and associated diseases has been reviewed elsewhere (see e.g., (14, 57)). In this review, we will focus on molecular mechanisms employed by p66Shc, in particular its ability to form ROS, assisted by the mitochondrial respiratory chain, which was proposed to trigger apoptosis initiation (23) and most other effects of p66Shc (70). We will thus discuss in detail the molecular architecture of p66Shc, its molecular function in ROS metabolism and in the mitochondrial intermembrane space (IMS), the regulation of its mitochondrial transport, and the molecular mechanisms and interactions involved in these processes.
The Multiple Faces of Shc Proteins
p66Shc is a member of the Shc protein family, whose size apparently increased during evolution, from one locus in Drosophila (dShc) to at least three loci in mammals (Rai, Shc, Sli) (38). The three mammalian loci encode at least six Shc-type proteins, based on alternative initiation codons and splicing. The isoforms encoded by the mammalian ShcA locus are called p46Shc, p52Shc, and p66Shc (Fig. 2), based on their molecular weight, and they promote opposite cellular fates, growth (p46Shc/p52Shc) or apoptosis (p66Shc) (38). p46Shc and p52Shc appear to act as “adaptor proteins” that bind to phosphorylated tyrosines in cytoplasmic motifs of growth factor receptors (70). p46Shc/p52Shc can be tyrosine-phosphorylated by various tyrosine kinase receptors, leading to recruitment of the Grb2/SOS (growth factor receptors bound protein 2, son of sevenless) complex, Ras (rat sacroma) activation, and finally activation of the mitogen-activated protein kinase (MAPK) cascade (70).

p66Shc appears to be a vertebrate protein, it is present in Xenopus and mammals, but absent in lower organisms, such as Saccharomyces, Drosophila, and Caenorhabditis (70). As the largest of the three proteins expressed from the ShcA gene, it carries all domains present in the shorter isoforms p46Shc and p52Shc (Fig. 2): An N-terminal phosphor-tyrosine-binding domain (PTB) is fused to a central collagen homology region (CH1) and a C-terminal Src homology 2 domain (SH2). These domain names were assigned based on sequence similarity, and in the case of the PTB and SH2 domain functional analogies have been shown (17, 56, 79). Compared to the shortest isoform p46Shc, the longer isoforms carry N-terminal extensions. A short domain, termed cytochrome c (Cyt c)-binding (CB) domain (23), common to p52Shc and p66Shc, and an N-terminal extension comprising a second collagen homology domain (CH2) unique to p66Shc. This N-terminus was shown to be responsible for an apoptotic function of p66Shc (21), and despite having the other domains in common with the shorter isoforms, there is no indication that p66Shc activates the Ras signaling pathway. Although p66Shc, like p52Shc/p46Shc, can be phosphorylated by receptor tyrosine kinases and can interact with the Grb2/SOS complex, p66Shc appears to have a negative effect on the Ras-MAPK and Erk pathways (42, 70). This effect was proposed to be due to competition with p52Shc for Grb2 binding (49) or p66Shc-induced displacement of SOS from Grb2 (33). However, p66Shc knockdown studies showed no role for p66Shc in growth factor response or Ras signaling, but revealed a function in regulating intracellular ROS levels (41). Analyzing the oxidation of redox sensitive probes as well as the accumulation of endogenous oxidative stress markers (23, 46, 71) showed decreased ROS levels in p66Shc-depleted cultured cells compared to wildtype (wt) cells. Furthermore, p66Shc k.o. mice showed reduced oxidative stress (44, 71), indicating p66Shc as a positive regulator of ROS generation. In addition, p66Shc was found to be a regulator of mitochondria-mediated apoptosis (23, 71). Both activities might be linked, as a direct ROS-forming activity of p66Shc was proposed to be the mechanism inducing apoptosis, but the mechanism remains to be fully understood (see below). However, p66Shc seems to sense various stress signals and to respond by inducing apoptosis. p66Shc k.o. mice are resistant to apoptosis induced by stresses, such as paraquat (oxidative stress), hypercholesterolemia, or ethanol (70). In addition to its direct apoptosis-inducing activity, p66Shc seems also to amplify other apoptotic pathways. In pharmacological studies, p66Shc was shown to sensitize T cells to Ca2+-dependent apoptosis by inducing mitochondrial dysfunction and by impairing Ca2+ homeostasis (58). Similarly, p66Shc expression in T cells causes imbalance between expression of proapoptotic and antiapoptotic genes, including Bcl-2 (b-cell lymphoma 2) family members, thereby promoting apoptosis initiation (53). Also, other physiological functions for p66Shc and its activity in ROS metabolism are still emerging, such as a role in promoting fat accumulation (8).
It is thus apparent that p66Shc is implicated in a variety of physiological functions and pathologies (Fig. 1), but a unifying molecular framework for its functions remains to be further refined. Likewise, our understanding of the molecular details of the reported interactions and functions of p66Shc is incomplete. However, considerable progress has been made in recent years concerning the role of p66Shc in the mitochondrial redox system, and we will now focus on the emerging molecular mechanisms of this activity and its regulation.
p66Shc Is a General and a Mitochondrial Redox Signaling Protein
p66Shc appears to be a general regulator of ROS metabolism and to increase cellular ROS levels through several mechanisms (Fig. 3). p66Shc was reported to decrease expression of several ROS scavenging enzymes through the inhibition of Forkhead transcription factors (46), although another study found no effect on the scavenging capacity (23). Further, p66Shc was proposed to promote Rac1 activation, thereby triggering ROS production by NADPH-dependent membrane oxidases (33, 70). Finally, p66Shc was reported to localize to the mitochondrial IMS, where it can upregulate O2 consumption and NADH metabolism. It was thus proposed to influence lifespan by acting as a switch that can route energy metabolism toward aerobic glycolysis (45). Although this mechanism, through a general increase of oxidative metabolism and thus formation of the associated side product could explain the influence of p66Shc on ROS levels, a more direct mechanism has been reported recently. In this study, p66Shc was shown to be able to act as a redox enzyme, forming ROS by using electrons from the respiratory chain (23) (see below). Although p46Shc was also reported to localize to mitochondria (76), neither for p46Shc nor for p52Shc a ROS-forming oxidoreductase activity comparable to p66Shc has been described, indicating that p66Shc is the only ROS-producing Shc-isoform in mitochondria. Various functions have been proposed for p66Shc-produced ROS. H2O2 produced by mitochondrial p66Shc was suggested to induce opening of a mitochondrial megachannel, the permeability transition pore (PTP), leading to rupture of the organelle's outer membrane and apoptosis initiation. This mechanism would indeed explain why p66Shc k.o. cells are resistant to apoptosis induced by a variety of signals (70). Another role proposed for p66Shc-mediated ROS is the regulation of gene expression and fat accumulation (8, 46). It remains to be seen, however, whether and how p66Shc generates ROS outside mitochondria, or whether these effects can be explained by an extended model with p66Shc as mitochondrial effector of respiratory chain and local redox metabolism.

Mechanism and Regulation of p66Shc-Dependent Apoptosis
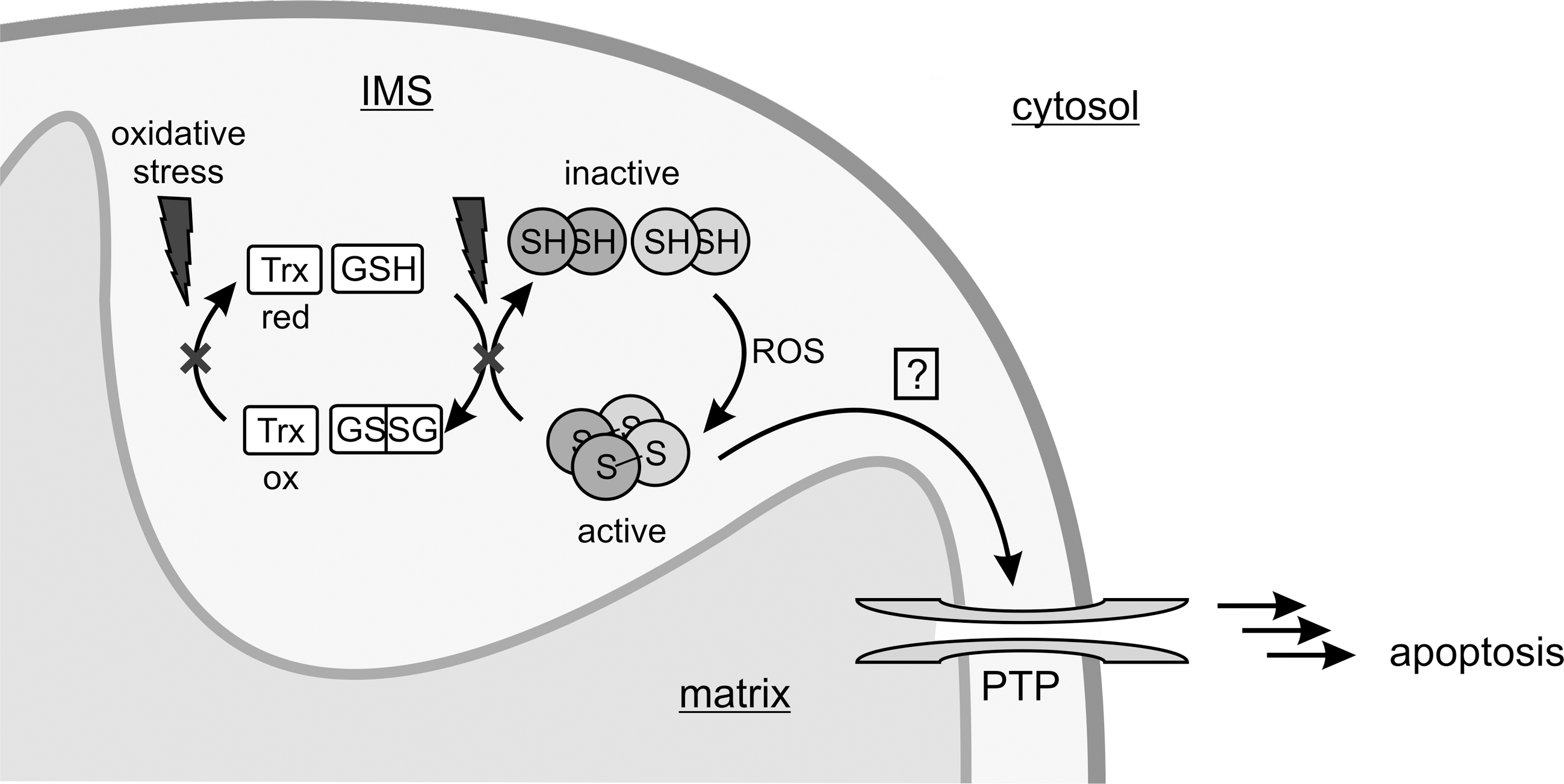
In the mitochondrial apoptotic pathway, outer membrane permeabilization leads to release of Cyt c, AIF (apoptosis-inducing factor) and other IMS proteins for relocation to cytosol and nucleus, where they trigger subsequent apoptotic processes, such as caspase activation and DNA degradation (25). Membrane permeabilization is controlled by interactions of different Bcl-2 family proteins, and by the opening of a complex regulated membrane megachannel, the mitochondrial PTP (80). The PTP is a dynamic multi-protein complex, and the basal proteins forming this pore and the precise mechanisms regulating pore opening are still not known (80). However, PTP opening can be induced by elevated Ca2+ influx into mitochondria and several other triggers, such as excessive ROS production. Further, a decline in electron transfer chain activity as well as changes in transmembrane potential can promote apoptosis initiation, leading to a model with the PTP as a site where mitochondria can integrate multiple cellular signals and metabolic responses (25).
p66Shc is assumed to induce PTP opening in order to induce mitochondria-mediated apoptosis. Consistently, inhibition of PTP opening with cyclosporin A blocked the protein's proapoptotic function (71). Furthermore, addition of recombinant p66Shc to isolated and digitonin-permeabilized rat mitochondria (digitonin makes the outer membrane permeable for proteins) resulted in rupture of the organelles (21, 23), a typical phenomenon of mitochondrial permeability transition (PT) in vitro. Again, mitochondrial rupture was prevented by the presence of PTP inhibitors (23), which strongly hints at PT as a likely key step of p66Shc-induced apoptosis. Consistently, a fraction of cellular p66Shc locates to the mitochondrial IMS (23), where it can regulate the action of the PTP.
The tool proposed to mediate the proapoptotic function of p66Shc is the generation of ROS, which are known as potent PT inducers. In response to proapoptotic stimuli, such as carbon tetrachloride (CCl4), cellular ROS levels increased significantly in wt but not in p66Shc k.o. mouse embryonic fibroblasts (MEFs) (23). Furthermore, addition of the ROS quencher dimethyl-thiourea (DMTU) or catalase, respectively, to isolated mitochondria completely abolished p66Shc’s proapoptotic function (21). Fluorescence-based ROS-assays and electrochemical analyses finally confirmed that p66Shc can act as a redox-enzyme and generate ROS (23). p66Shc thus appears to translate and integrate cellular stresses by converting proapoptotic signals into an oxidative burst.
The role of the p66Shc-specific N-terminus in apoptosis initiation
The N-terminal part is unique for p66Shc and should be responsible for its unique activity. Indeed, experiments using a protein construct covering the CH2 and the neighboring CB domain clearly identified the N-terminus as the ROS-generating and apoptosis-inducing part of p66Shc (20, 21). The N-terminal expansion of p66Shc thus does not only modify given Shc functions, such as regulation of the Ras-signaling pathway, but it creates a completely new function, the induction of mitochondria-mediated apoptosis presumably through ROS generation.
The small CB domain C-terminal to the p66Shc CH2 domain is also part of p52Shc. However, two findings indicate that in p66Shc it might function together with the CH2 domain instead of being a discrete domain. First, the CB domain was disordered in the solution structure of a protein construct covering the N-terminal CB and PTB domain of p52Shc (17, 79). Possibly, the CB domain needs its N-terminal neighbor, the CH2 domain, to fold properly, or, like the PTB domain, its interaction partner (17). Second, an alignment with the Shc family member RalP, which exhibits the typical Shc domain architecture, showed pronounced homology to p66Shc, except for the CH2 and the CB domain (21). We therefore assume a functional, and possibly a structural, CH2CB unit in p66Shc, which is supported by the suggested role of the CB domain/Cyt c interaction in ROS generation (see below).
The ROS-forming oxidoreductase function of p66Shc
For its proapoptotic activity p66Shc seems to exploit the mitochondrial ETC. Mitochondrial PT induced by p66Shc or its isolated N-terminal CH2CB-fragment required the presence of respiratory substrates (e.g., succinate or malonate) (21, 23). Furthermore, rotenone and antimycin A, inhibitors of respiratory complexes I and III, respectively, prevented the organelles from rupture (21, 23), which indicates that the ETC redox equivalent downstream of complex III, Cyt c, might be the electron donor in the ROS-forming and apoptosis-inducing reaction(s). Consistently, the redox pair ascorbate/N,N′-tetramethyl-p-phenyldiamine (TMPD), a reducing system for Cyt c, enabled p66Shc action even in nonenergized mitochondria (23). Cyclic voltammetry experiments indeed revealed an electron transfer reaction between p66Shc and Cyt c, and ROS formation by p66Shc dependent on the presence of Cyt c was detected using a ROS-sensitive fluorescence dye (23). For Cyt c binding, the region was narrowed down to the 52 amino acids located between CH2 and PTB domain, now referred to as CB domain (23). However, using the CH2CB construct, no ROS-generating interaction with Cyt c was observed, but antimycin A still prevented mitochondrial rupture in a mitochondria swelling assay (21). At least in the in vitro fluorescence-based system, the p66Shc N-terminus (CH2 plus CB) alone seems not to be sufficient for proper Cyt c binding, and other domains C-terminal to the CH2CB domain might have to contribute to this interaction. Mutagenesis within the CB domain further identified residues (Glu132, Glu133, Trp134) which are essential for the ROS-forming electron transfer reaction and for apoptosis induction (23).
Copper was identified as essential and specific cofactor for the ROS-forming activity of p66Shc in vitro (21, 23). The metal might function as a redox mediator between the p66Shc active center and oxygen, which would indicate a copper-specific metal site in p66Shc. p66Shc does not bear a typical copper site sequence, but this is also the case in several other copper-dependent proteins (1). However, addition of copper is not needed in experiments in vivo and with purified organelles. Thus, in a physiological environment, p66Shc appears to be able to acquire a copper ion, possibly introduced in the IMS by metallo-chaperones, or another yet unknown copper protein might be involved in the electron transfer reaction. On the other hand, the need for copper might be an artifact of the fluorescence-based ROS assay. Copper is known to convert H2O2 to the much more reactive hydroxyl radical (Fenton reaction), which could amplify the fluorescence signal due to higher sensitivity of the dye to hydroxyl radicals. Also, the exact nature of the ROS species formed by p66Shc remains to be proven. Partial reduction of oxygen (1 electron transfer) generates the superoxide radical, whereas full reduction (2 electron transfer) forms H2O2. In vitro the CH2CB domain forms oligomers (see below), and we speculate that p66Shc might be able to use two active centers for a two electron transfer reaction. Experiments using fluorescence dyes with different specificities in isolated mitochondria and cyclic voltammetry analyses indeed indicated H2O2 to be the product of the p66Shc-catalyzed electron transfer reaction (23). However, measurement of, and discrimination between ROS in complex systems, such as mitochondria, is challenging due to the high scavenging capacity and the high reactivity of these species, which can lead to formation of secondary ROS (43, 72). Furthermore, some evidence suggests that ROS formation and apoptosis induction by p66Shc might not be directly coupled and that the apoptosis-inducing ROS might be produced downstream from p66Shc (see below). Thus, the identity of the p66-derived ROS, the molecular mechanisms of p66Shc-dependent ROS-production, and the exact ROS target(s) for apoptosis initiation remain fascinating questions for future studies.
Regulation of the proapoptotic function of p66Shc
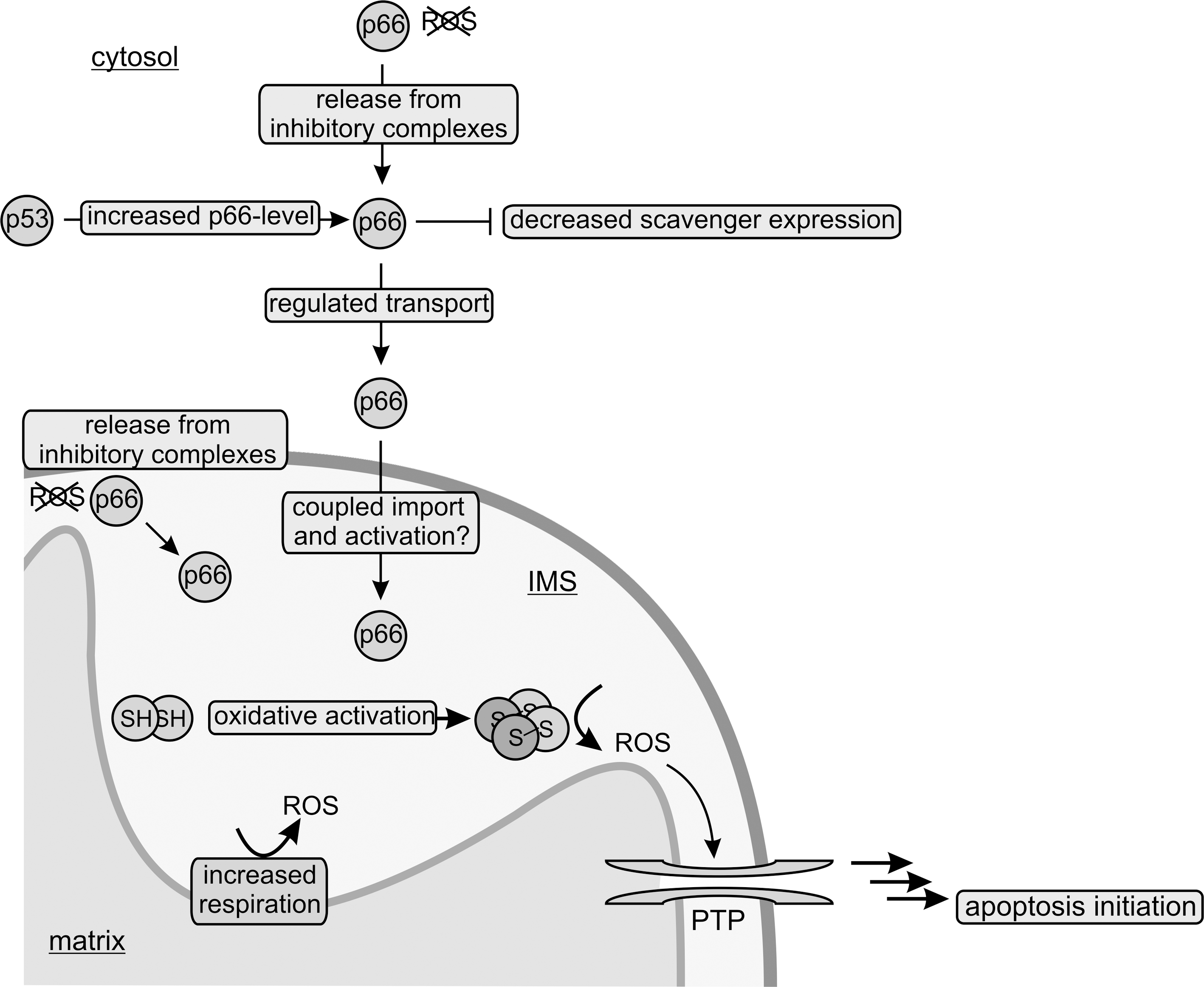
Although the regulation of the potentially cytotoxic activity of p66Shc is not fully understood, it is already clear that it comprises several levels in order to guarantee tight silencing during the normal cell cycle but at the same time effective activation in response to proapoptotic stimuli. This complex regulation of p66Shc includes control of its expression, posttranslational modifications, protein–protein interactions, and its oxidative oligomerization. p66Shc shows a tissue-specific expression as a result of promoter methylation (75), and its expression increases with age (32, 55). Additionally, the transcription factor p53 was reported to upregulate p66Shc protein levels in response to apoptotic stimuli, such as UV or H2O2, through transcription regulation (34), but apparently also through stabilization of the p66Shc protein through an as yet unclear post-translational mechanism (71). p66Shc's proapoptotic function is further regulated by serine phosphorylation (Ser36 of mouse p66Shc) (41), which appears to provoke translocation of cytosolic p66Shc into the mitochondrial IMS (59) (see below). In the IMS, p66Shc was reported to associate into a high molecular weight complex containing subunits of the mitochondrial import machineries TOM and TIM (translocase of the outer/inner membrane) and the mitochondrial heat shock protein 70 (mtHsp70) (51). The authors proposed an inactivating function for this complex until a proapoptotic stimulus induces its dissociation. However, due to their function as chaperone or import complexes, respectively, mtHsp70 and the TOM/TIM machineries bind proteins rather nonspecifically. Additional proteins appear necessary for the specific regulation of this interaction, and thus further investigations seem necessary to reveal the mechanistic basis of such a regulation. Another parameter presumably influencing the level of the p66Shc-mediated electron transfer reaction should be the amount of reduced Cyt c, which could adjust p66Shc-dependent apoptosis to the metabolic status of the cell.
An oxidative tetramerization links p66Shc's proapoptotic function to oxidative stress
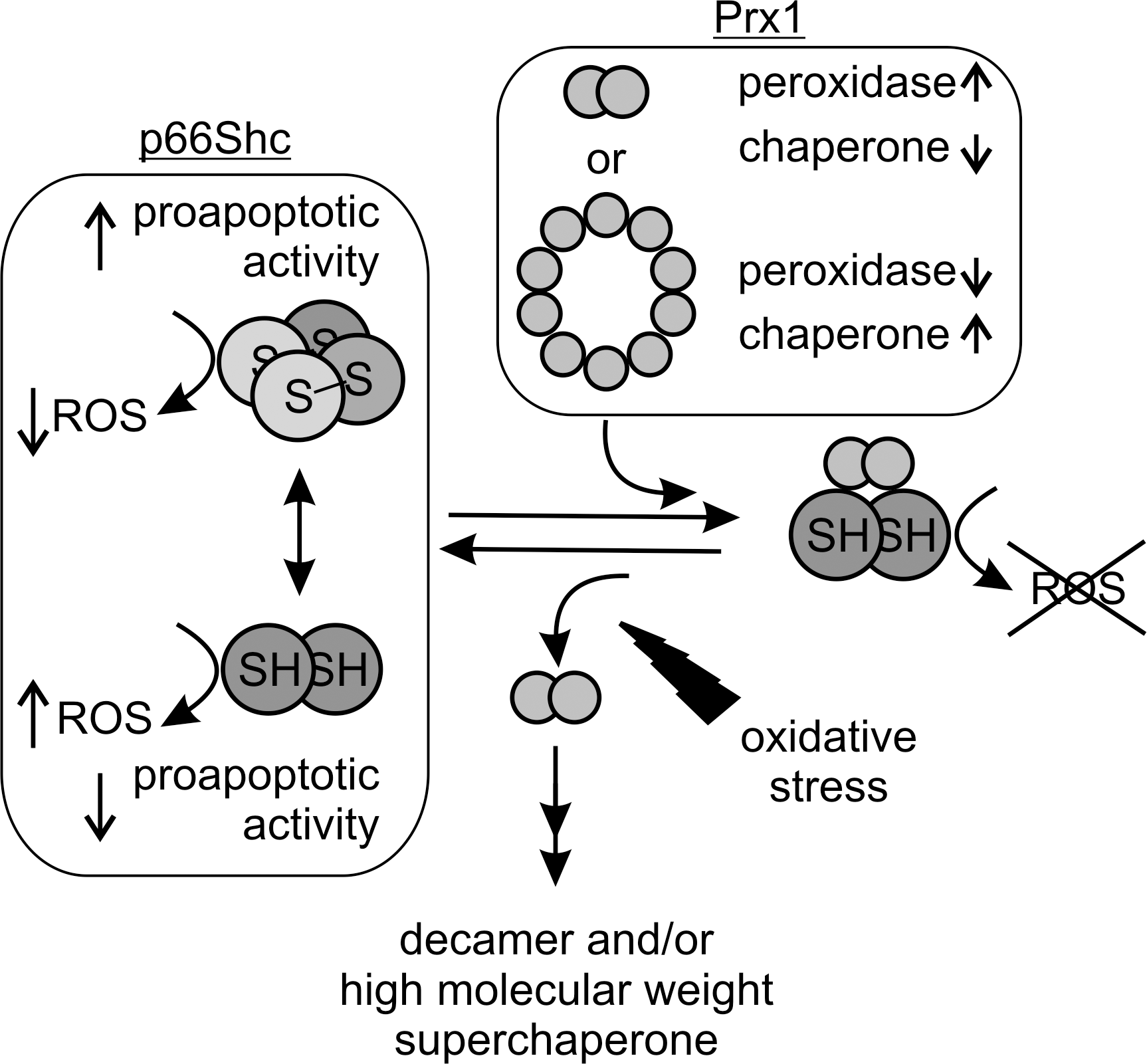
We recently described a mechanism which directly links the proapoptotic function of p66Shc to oxidative stress (21). The proapoptotic activity of the p66Shc CH2CB protein is influenced by a redox-dependent oligomerization. The protein forms non-covalent dimers that can be cross-linked to a dimer of dimers (i.e., to form a tetramer) by disulfide bridge formation through oxidation of a conserved cysteine residue (Cys59 in mouse p66Shc, Fig. 2). The redox-dependent oligomerization remains to be shown for full-length p66Shc in vivo, but this mechanism dramatically activated the protein's proapoptotic activity in isolated mitochondria. The oxidative tetramerization thus directly links p66Shc's proapoptotic activity to the redox status. Redox-dependent structural or functional changes in proteins, such as the transcription factor OxyR or peroxiredoxins (Prx), are indeed more and more emerging as regulatory switches (7, 19). Consistent with a putative physiological function of the dimer/tetramer equilibrium, and the associated change in apoptotic activity, it is controlled antagonistically by the cellular redox systems glutathione, thioredoxin1/2, and Prx1 (see below) and oxidative stress (20, 21). p66Shc thus appears not only to be a redox-enzyme, but also to act as a thiol-based redox-sensor. In our current model (Fig. 4), p66Shc is silenced by the cellular scavenger systems until oxidative stress exceeds a critical threshold, which leads to an overload of the scavengers and oxidative activation of p66Shc's proapoptotic function.
It is noteworthy to mention that the critical cysteine residue has exclusively a regulatory function and does not participate catalytically in ROS formation. A cysteine to serine mutant (Cys59Ser) was still fully active in the fluorescence-based ROS assay (21). Moreover, the mutant and also the dimeric wt protein showed significantly increased ROS production rates compared to the tetrameric form. This finding is a clear discrepancy to the pronounced proapoptotic activity of the tetramer and the only basal activity of the dimer and the Cys59Ser mutant. It questions if indeed p66Shc-dependent ROS formation, or at least formation of the ROS detected by the fluorescence-based assay, can be the mediator of p66Shc-induced apoptosis. There are further conflicts that need to be resolved in the current model of p66Shc-induced apoptosis by ROS formation. For example, serine phosphorylation of p66Shc appears to be necessary for the proapoptotic function of the protein (41). Consistently, expression of the responsible protein kinase C β (PKCβ) significantly increased cellular ROS production in MEFs (59), and a significantly increased ROS-generating activity was observed for a Ser36Asp mutant of the CH2CB domain, which mimics the phosphorylation (20). However, the mitochondrial p66Shc pool was reported to be dephosphorylated (23), and the Ser36Asp mutant showed a drastically decreased apoptotic activity when tested on isolated mitochondria (20). A role of this phosphorylation in triggering mitochondrial transport (see below) can explain some of these findings, but the reciprocal effects on ROS formation and apoptotic activity make a direct connection between them unlikely. However, addition of scavengers (catalase, DMTU) to isolated mitochondria abolished p66Shc's proapoptotic function (21), which is consistent with ROS participating in the p66Shc-initiated apoptosis pathway, although not necessarily in the step directly mediated by p66Shc. Possibly, tetramerization does not result in a general increase in ROS-production but increases the affinity to an interaction partner, such as the PTP, which would increase the ROS level locally. Alternatively, ROS generation by p66Shc might only be a side reaction or a waste product of a different p66Shc redox activity, and the true ROS-dependent apoptosis step could be downstream from p66Shc. Interestingly, the NADPH-dependent oxidase Nox4 was recently found in mitochondria (10), and p66Shc could act by regulating this or another ROS-forming enzyme. Yet another possibility is that a different p66Shc activity in ROS metabolism than H2O2 formation is responsible for apoptosis induction. Indeed, we recently found that p66Shc might use H2O2 as a substrate for the Fenton reaction to form the highly reactive hydroxyl radical (20). In this case, p66Shc would not only be able to generate oxidative stress, but it could also amplify and convert oxidative signals. Such a function would be consistent with its role as redox-sensing apoptosis inducer, and the previously proposed physiological importance of so-called ROS-induced ROS release (80).
Despite the wealth of evidence for a physiological importance of p66Shc-derived ROS, the molecular details of p66Shc's action in mitochondria remain to be fully understood. Although ROS definitely play a role in p66Shc-dependent apoptosis initiation, it remains to be shown whether indeed p66Shc-derived ROS induce PTP opening. Further studies on the molecular basis of the p66Shc redox activity will help to define the exact molecular mechanism of p66Shc-mediated apoptosis initiation.
Silencing of p66Shc's proapoptotic function through complex formation with Prx1
Using a combination of pull-down experiments and mass spectrometrical analysis, we identified Prx1 as a new interaction partner of p66Shc participating in its complex regulation network (20). Prxs are a family of thiol-based H2O2-degrading enzymes, so-called peroxidases, which contribute to the cellular redox equilibrium and to ROS-mediated signaling (e.g., in apoptosis regulation) (19, 48). They are weak scavengers, but due to their abundance they effectively degrade H2O2 at lower concentrations. Increasing oxidative stress, instead, inactivates Prxs by overoxidation, making them ideal sensors for H2O2-mediated signaling (19). Prx1 exists in different oligomeric forms, a homodimer, a decameric ring composed of five dimers, and a stress-induced, high molecular weight (HMW) multimer (31, 39). Formation of the decameric ring and of the HMW multimer, which are not yet fully understood mechanistically, appear to be accompanied by a functional switch (31). As a dimer, Prx1 degrades H2O2 (peroxidase activity). The decameric ring, in contrast, has decreased peroxidase activity, but at the same time shows an additional chaperone activity. Finally, oxidative stress or heat stress was shown to induce formation of the HMW multimer, which then acts as a superchaperone without peroxidase activity. Interestingly, when testing the interaction of dimeric and decameric Prx1 with the p66Shc CH2CB domain we observed a destabilization of the decameric Prx1 arrangement and stabilization of dimeric Prx1, the form with higher peroxidase activity (20). In turn, complex formation with Prx1 forced the p66Shc CH2CB protein in its dimeric, apoptosis-inactive form due to a disulfide exchange between the two proteins within the complex. The p66Shc/Prx1 interaction complex therefore appears to be a dual regulatory mechanism to silence p66Shc's proapoptotic function: Prx1 in its dimeric form can immediately degrade the p66Shc-generated H2O2, and it can reduce tetrameric p66Shc, thereby inactivating its proapoptotic function. Increasing oxidative stress, however, should lead to dissociation of the p66Shc/Prx1 complex due to overoxidation of Prx1, which favors the decameric arrangement (19, 66) and also formation of the HMW superchaperone (31). Release of p66Shc from the inhibitory complex would then enable apoptosis induction by p66Shc. We therefore assume that the redox-sensitive p66Shc/Prx1 module can sense cellular ROS levels to keep p66Shc inactive under conditions of moderate stress but to switch on its proapoptotic function in response to overwhelming oxidative stress (Fig. 5). Two Prxs, Prx3 and Prx5, are known to localize to mitochondria; Prx5 additionally localizes to cytosol, peroxisomes, and nucleus (19, 67). Prx1 is mainly found in the cytosol. However, a recent proteomic study identified Prx1 also in the mitochondrial IMS (54). The IMS is indeed strongly connected to the cytosol and also in other scavenging systems the cytosolic isoform is also present in the IMS [e.g., Cu/ZnSOD (SOD1)] (50). Thus, complex formation of p66Shc with Prx1 might be a general mechanism controlling p66Shc activity in the cytosol before its mitochondrial import, but also in the mitochondrial IMS.
Mechanism and Regulation of p66Shc Import in the IMS
The translocation of p66Shc into the mitochondrial IMS appears to participate in the complex regulation of its proapoptotic function. Mitochondria have their own genome, but it encodes only few of the mitochondrial proteins (13 of more than 1000 mitochondrial proteins in mammalia). All other mitochondrial proteins are encoded by the nuclear genome and synthesized by the cytosolic translation machinery. Although a wealth of information has been collected on mitochondrial translocation signals and on at least four different translocation pathways (13, 47), for a substantial number of proteins it is still not fully understood how and when they are translocated to mitochondrial compartments. Especially many IMS proteins are targeted by as yet unknown import signals and mechanisms (28).
p66Shc was reported to be an ubiquitous cellular protein locating to the cytosol, the endoplasmic reticulum, and mitochondria (23, 45, 51), which indicates further functions for p66Shc independent from the mitochondrial ETC. In mitochondria, p66Shc was reported in one study to mainly associate with the inner mitochondrial membrane (56% of mitochondrial p66Shc), besides being a soluble IMS protein (35%), or locating to the mitochondrial matrix (9%) (23). In contrast to p52Shc and p66Shc, the shortest Shc-isoform p46Shc was found in a single study to mainly localize to mitochondria, and a 32-amino acid motif in p46Shc was identified to be essential for mitochondrial translocation (76). However, the wealth of functional studies available hints at interactions with cytosolic proteins as the main mechanism of p46Shc action, but localization of the protein could also be dynamically regulated. p66Shc of course also contains the translocation sequence described in p46Shc; however, mutagenesis within this region suggests that p66Shc import into mitochondria is independent of this motif (45), possibly because the mitochondrial targeting sequence is masked by the N-terminal extension in p66Shc (76). Similar to the localization signal, the machinery and mechanism employed for p66Shc import remain to be characterized. p66Shc was found to associate into a high molecular weight complex including subunits of TOM and TIM (52), which indicates that import of the matrix fraction of p66Shc is mediated by this well characterized transport system (Fig. 6). For IMS import, a system that couples import and oxidative folding has been described (29, 40, 62), and we speculate that this mitochondrial import and assembly 40 (Mia40) relay system might be responsible for p66Shc import into the IMS. Mia40 is a conserved IMS protein that oxidizes incoming proteins, thereby facilitating disulfide bond formation and protein import (62). Subsequently, Mia40 is reoxidized by Erv1, a FAD-containing sulfhydryl oxidase that routes the electrons through Cyt c to the mitochondrial ETC. Mainly small proteins (7–15 kDa) with a certain cysteine motif were found to be imported by the Mia40/Erv1 system. However, also other proteins, such as SOD1 and its copper chaperone, were identified as substrates for this oxidizing import machinery (61), indicating a broader substrate range for Mia40-dependent mitochondrial import, possibly including p66Shc.
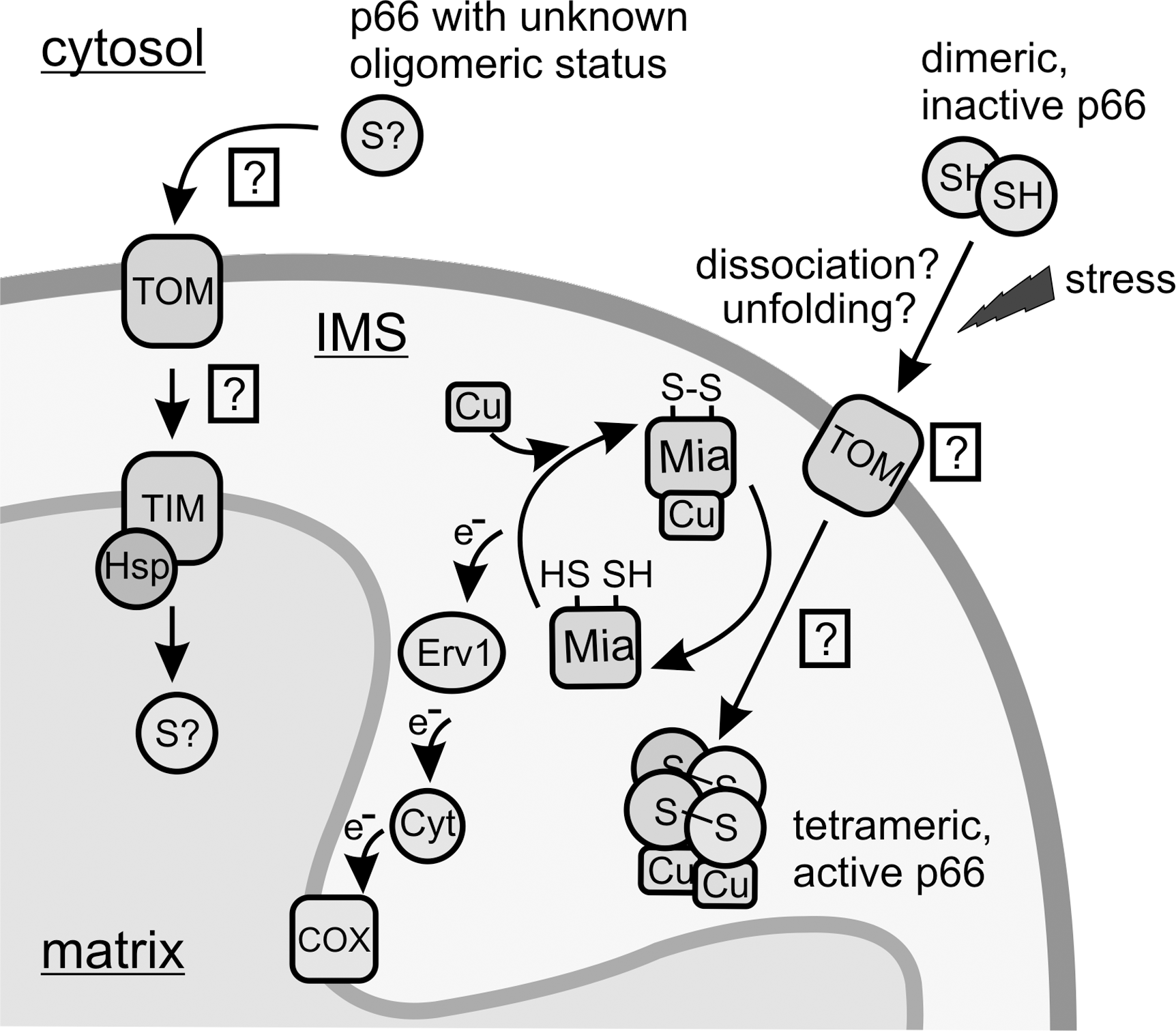
Proapoptotic stimuli, such as UVc or H2O2, result in net translocation of cytosolic p66Shc into the mitochondrial IMS (51, 59). It is tempting to speculate that import of p66Shc by Mia40 further activates its proapoptotic function by oxidative formation of the more active p66Shc tetramer (Fig. 6). Interestingly, Mia40 can bind copper and zinc ions (69) and was suggested to equip incoming proteins with their metal cofactors (40). Import of p66Shc by Mia40 might thus result in oxidatively activated and copper-constituted p66Shc protein. Experimental verification of the machinery mediating p66Shc import and of the potential concurrent activation steps, however, remains an interesting topic for future studies.
Regulation of p66Shc import in the IMS
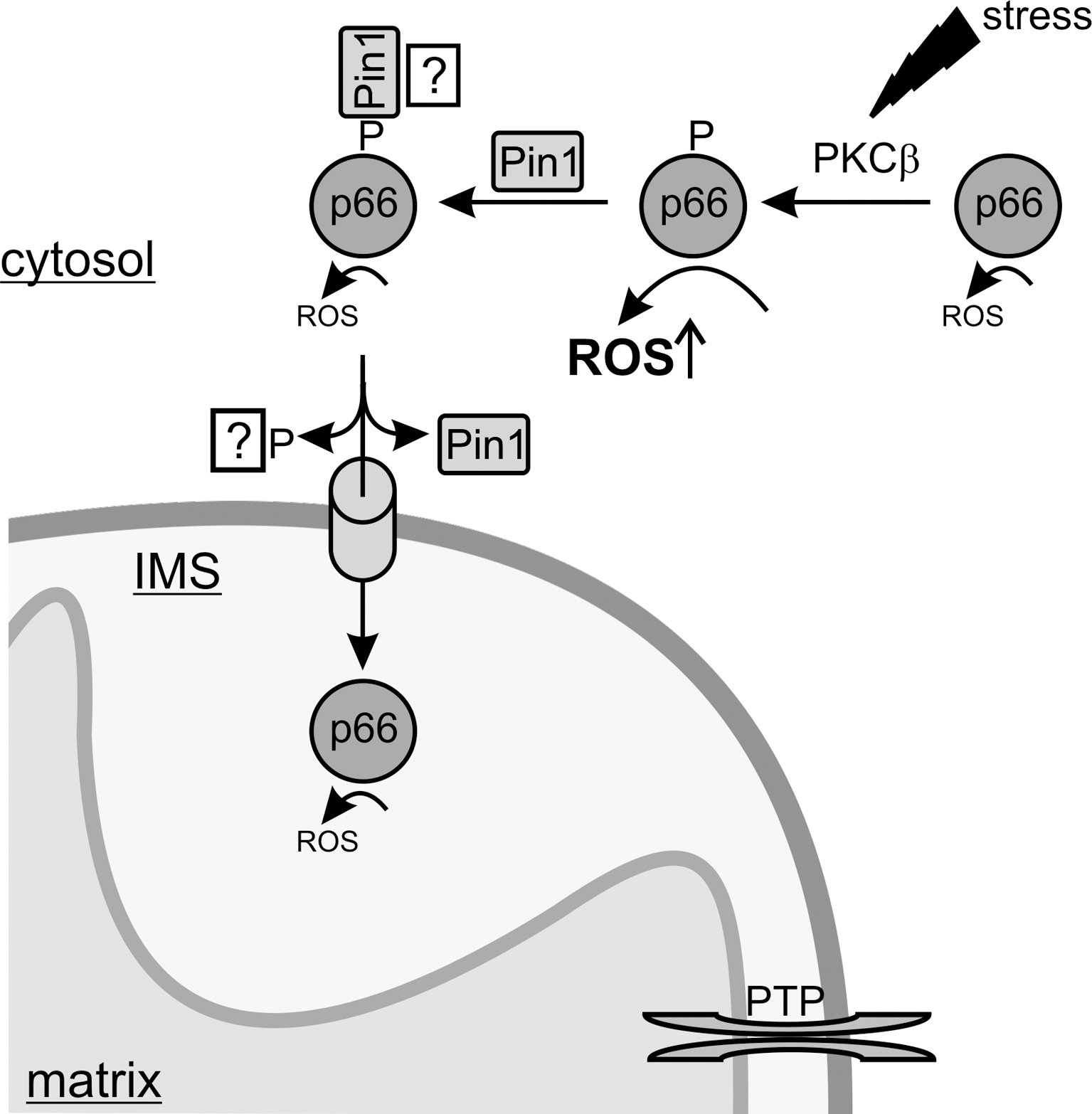
Similar to the mechanism of mitochondrial translocation, the regulation of this event is little understood. Only recently, a first mechanism regulating p66Shc translocation depending on p66Shc phosphorylation by stress kinases like JNK-1 (just another kinase 1) and PKCβ, followed by prolyl-isomerization by Pin1 (protein interacting with nima 1), in response to stress signals has been described. Serine phosphorylation of p66Shc occurs in response to proapoptotic stimuli (59) and is a prerequisite for its proapoptotic function (41). The phosphorylation was proposed to act as signal for translocation of cytosolic p66Shc into the mitochondrial IMS, as inhibition of PKCβ prevented p66Shc translocation (59). Beside PKCβ-dependent serine phosphorylation, an interaction with the prolyl isomerase Pin1 was shown to control p66Shc translocation. In Pin1 k.o. MEFs the mitochondrial fraction of p66Shc was markedly decreased (59). Pull-down experiments indeed revealed that Pin1 physically interacts with p66Shc, and that this interaction required proapoptotic stimuli, such as UVc exposure (59). As mentioned, these stimuli result in phosphorylation of Ser36, leading to formation of a typical Pin1 binding motif (phosphoSer36-Pro37), which was indeed shown to be a prerequisite for this interaction. Phosphorylation and interaction with Pin1 thus seem to compose a signal cascade for translocation of p66Shc into the mitochondrial IMS where it can fulfill its proapoptotic function (Fig. 7). However, more recent experimental evidence suggests that the interaction with Pin1 does not only lead to prolyl-isomerization and mitochondrial import, but that it additionally silences p66Shc's toxic oxidoreductase function until p66Shc is delivered into the IMS. Phosphorylation of p66Shc results in a stimulation of its oxidoreductase activity (see above), although it is as yet unclear which cellular component would act as electron donor outside the IMS. Interestingly, a stoichiometric interaction with Pin1 was shown to antagonize this increased ROS-generating activity (20) (Fig. 7). Consistently, competitive inhibition of Pin1-dependent isomerization activity did not weaken the inhibitory function of Pin1, indicating a silencing mechanism independent of its isomerization activity. It is indeed a matter of debate whether Pin1 regulates target proteins solely through isomerization or possibly also by stabilization through complex formation, as shown for its interaction with the transcription factor p53 (78). Thus, some questions remain about the details of the PKCβ/Pin1 regulation of p66Shc, especially the mechanistic role of serine phosphorylation in regulating the interaction with Pin1 and in regulating p66Shc's ROS-producing and apoptosis initiating activity.
Perspective
p66Shc appears to be a critical determinant of cell fate: Activated by proapoptotic stimuli, it responds by inducing ROS formation and by initiating PTP opening, the first step in mitochondria-mediated apoptosis (Fig. 8). The finding that the proapoptotic function of p66Shc is regulated in a redox-dependent manner (21) suggests that p66Shc is not simply a redox enzyme, but also a ROS sensor and possibly a ROS amplifier. Redox regulation through disulfide bond formation is indeed emerging as a general principle for adaptation of protein function to oxidants levels (7). For example, a recent study described the actin-binding protein cofilin to be regulated by disulfide bond formation (35). Surprisingly, cofilin showed further similarities to p66Shc: Oxidative stress induces its dissociation from actin and its translocation to mitochondria where it promotes apoptosis through PTP opening. It thus appears that other proteins carry similar proapoptotic activities as p66Shc, and that similar mechanisms, such as protein/protein complex formation and disulfide bridging, regulate their activity. It will be exciting to characterize further members and regulation mechanisms for this putative network of cellular stress sensors, which together appear to regulate apoptosis through the PTP as a signaling integration point.
The lack of any reported redox activity for cofilin and some recent results for p66Shc might indicate that the latter protein might induce apoptosis through a mechanism different from direct ROS formation. However, ROS definitely play a role in p66Shc's proapoptotic function, and their exact target as well as the mechanism and function of p66Shc's redox activity are interesting questions for future studies. p66Shc has been recognized as an important lifespan determinant and is associated with several diseases, which could make it a target for chronic and aging-related diseases. Specific intervention in the p66Shc-based signaling network is challenging, however, because characterization of the protein at a molecular level has remained limited, in contrast to an abundance of physiological studies. Previous attempts to interfere with cellular redox systems were based on the idea that treatments reducing mitochondrial energy metabolism or treatments with antioxidants could be used for fighting ROS-involving diseases and for lifespan extension through a general reduction of ROS levels. However, in particular, treatments with antioxidants have not provided convincing results in higher organisms, most likely due to the much more complicated role of ROS. A deeper understanding of the structural and mechanistic basis of the redox activity of p66Shc and its function could thus enable a more targeted approach toward interfering with a ROS-based signaling system, making it an exciting system for future mechanistic studies on ROS signaling.
Footnotes
Acknowledgment
Work on p66Shc in our laboratory is supported by Deutsche Forschungsgemeinschaft (grant STE1701/5 to CS).