
Abstract
Protein sulfenic acids (SOHs) are the principal oxidation products formed when redox active proteins interact with peroxide molecules. We have developed a new antibody reagent that detects protein SOHs derivatized with dimedone. Using this new antibody, we found that glyceraldehyde 3-phosphate dehydrogenase (GAPDH) is the predominant protein sulfenate present in isolated rat ventricular myocytes under basal conditions. During oxidative stress with hydrogen peroxide (H2O2), GAPDH SOH labeling is lost, but a number of secondary dimedone-reactive protein sulfenates then appear. As the sulfenate labeling is lost, the Cys-149 sulfinic/sulfonic acid oxidation states of GAPDH appear. This hyperoxidized GAPDH is associated with both the inhibition of glycolysis and its ability to reduce H2O2. We examined whether inactivation of GAPDH was causative in the generation of secondary protein sulfenates that coincide with its hyperoxidation. The selective GAPDH inhibitor koningic acid (which functions by forming a covalent adduct at Cys-149) fully prevented basal SOH labeling, as well as subsequent peroxide-induced hyperoxidation. However, koningic acid–mediated inhibition of GAPDH alone did not induce the formation of intracellular H2O2 or secondary protein sulfenates and also failed to potentiate their peroxide-induced formation. Overall, GAPDH appears to have peroxidase-like properties, but its inhibition failed to impact on downstream oxidant signaling involving secondary protein sulfenation. Antioxid. Redox Signal. 14, 49–60.
Introduction
Once oxidants have been produced in the cell, they have to be sensed to manifest signaling and ultimately be transduced into a functional response by individual enzymes, as part of an integrated cellular homeostatic control. A principal mechanism by which this occurs involves the oxidative alteration of selected protein thiols (P-SHs) with the correct structure (16). These posttranslational structural modifications are coupled to alterations in protein function, providing a mechanism of regulation.
In terms of H2O2 and P-SH signaling chemistry, the initial principal reaction product is the sulfenic acid (SOH) (28). Most protein sulfenates are unstable and consequently short-lived. In most scenarios, SOHs are redox intermediates that are rapidly recycled back to the free SH via reaction with reducing equivalents, although other more stable oxidation states such as disulfides can accumulate. Some stable protein sulfenates have been identified in bacterial proteins (26) and also in mammalian albumin and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (4, 11). Protein sulfenation may therefore also directly regulate protein function (15, 28), although lack of analytical methods has slowed progress in this area.
4-Chloro-7-nitrobenzo-2-oxa-1,3-diazole forms specific products depending upon whether it reacts with either a reduced or a sulfenated cysteine (Cys) SH, each product having a specific ultraviolet–visible spectrum, allowing protein sulfenation to be detected (18). 5,5-Dimethyl-1,3-cyclo-hexanedione (dimedone) selectively adducts with SOHs, and radiolabeled derivatives have allowed the selective detection of this oxidation state (4). However, both these methods are only applicable to in vitro analysis of SOHs with purified proteins. To allow the study of protein SOHs in a cellular setting, we previously developed an arsenite-dependent biotin switch method (35). However, this method is limited by the use of sodium dodecyl sulfate (SDS) denaturing conditions (which are likely to destabilize many sulfenates) and the labor-intensive nature of the preparation. The development of biotinylated (13) or fluorescent (29) derivatives of dimedone partly addressed these problems, but these compounds have limited availability because of the organic synthesis expertise required and the expense of production.
We reasoned that an antibody to dimedone that pan-specifically detected protein sulfenates derivatized with this generic, and inexpensive, reagent would be of value to the community, complementing the other approaches and potentially providing novel insights into peroxide signaling. Further, as antibody-based approaches are widely utilized, such a reagent could broaden the study of protein sulfenation because of the potential simplicity and ease of access of such an approach. Here we describe the generation and testing of an anti-dimedone antibody. Initial studies with this novel reagent have allowed us to identify a key role for GAPDH in peroxide-induced redox signaling in ventricular cardiac myocytes.
Materials and Methods
This investigation was performed in accordance with the Home Office “Guidance on the Operation of the Animals (Scientific Procedures) Act 1986,” published by Her Majesty's Stationery Office, London, United Kingdom.
Chemicals
The chemicals were obtained from Sigma Chemical, unless stated otherwise, and were of AnalaR grade or above.
Preparation of a pan-specific antibody to dimedone-modified proteins
A schematic outlining the strategy used to generate this novel dimedone SOH antibody is shown in Figure 1. Keyhole limpet hemocyanin (KLH) (Calbiochem) was dissolved in phosphate-buffered saline (PBS) and subsequently denatured and reduced at high pH as follows: this dissolved KLH (10 mg/ml) was denatured and reduced, generating a final concentration of 4 mg/ml in a buffer containing 100 mM Tris (pH 8.0), 100 mM dithiothreitol, and 4 M guanidinum–HCl. The mixture was incubated at 23°C for 3 h before dithiothreitol was almost entirely removed using PD10 desalting columns (GE Healthcare) equilibrated in 100 mM Tris (pH 8.0) and 4 M guanidinum–HCl. Crystalline dimedone (Sigma) was added to saturation to derivatize all Cys SOHs, as a result of partial oxidation by naturally occurring traces of reactive oxygen species or molecular oxygen. Owing to the low solubility of dimedone in aqueous solutions, the final dimedone concentration was measured empirically by high-performance liquid chromatography as follows: a standard curve was obtained from known dimedone standards (0–100 mM) prepared in dimethyl sulfoxide and injected onto a C18 column (Jones Chromatography) and eluted using a 0%–90% gradient of acetonitrile in 0.1% trifluoroacetic acid. Peaks were quantified by measuring the area under the curve from the absorbance profile at 312 nm. Against these standards, the saturated dimedone concentration typically reached ∼85 mM. After incubation of the denatured, reduced KLH with dimedone overnight at 4°C, the protein was dialyzed extensively against 6 M urea at 23°C, followed by repeated dialysis against PBS at 4°C, using Slide-A-Lyzers (Thermo Scientific). Dialysis into urea was necessary to ensure that the modified KLH remained soluble, once dialyzed into PBS. At this stage, the extent of the KLH Cys oxidation was analyzed colorimetrically at 412 nm using 5,5′-dithiobis(2-nitrobenzoic acid) (Ellman's reagent), comparing the amount of SHs present in the dimedone-reacted KLH against unmodified control KLH. In this way, 13% of the total KLH Cys were consistently found to be modified by dimedone. The dialyzed KLH was dried down under vacuum and the antigen was stored for further use. The dimedone-reacted KLH antigen was subsequently resuspended in Freund's adjuvant and injected into two rabbits (3 mg antigen per rabbit) twice over 120 days. Total plasma bleeds were collected and a plasma fraction was prepared by centrifugation, which was stored in aliquots at −70°C. This plasma was utilized in immunoblotting studies to investigate protein dimedone derivatization as an index of SOH formation. This antiserum is now available from Millipore (catalog no. 07-2139).
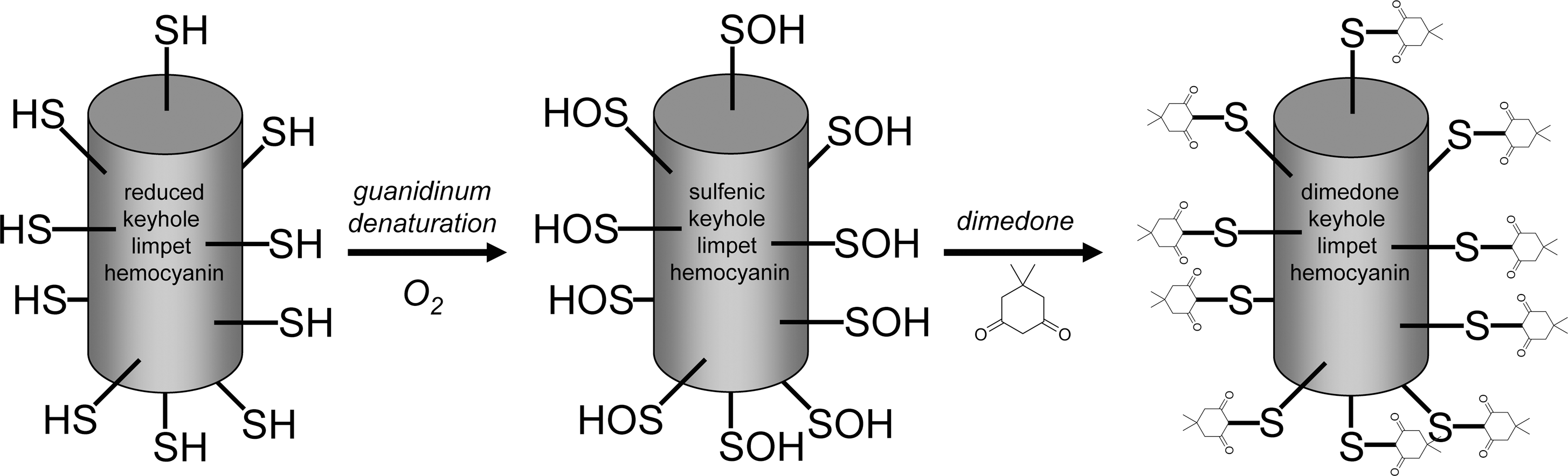
Studying protein sulfenation via dimedone incorporation in isolated rat myocytes
Ventricular myocytes were isolated from male Wistar rats (250–300 g) using a standard collagenase digestion protocol as described previously (9). The cell suspension was maintained in modified Tyrode's solution (8) at room temperature for ∼1 h, after which the pellet of cells was resuspended in fresh modified Tyrode's solution. The cells were treated with H2O2 (0.05–10 mM) for 10 or 20 min. Subsequently, the cells were pelleted (30 s at 1000 g) and resuspended in dimedone lysis buffer (1 mM dimedone, 0.1% Triton X-100, 0.012 M Na2HPO4/0.003 M citric acid, pH 6) for 20 min. Nonreducing SDS sample buffer containing 100 mM maleimide was subsequently added. Koningic acid (TMS) was used in some experiments as an inhibitor of GAPDH. Cells were pretreated with koningic acid or dimethyl sulfoxide as the vehicle control for 30 min at room temperature before being treated with H2O2 as described earlier.
Western blotting and immunodetection
Protein samples were analyzed by 12% SDS–polyacrylamide gel electrophoresis (PAGE) using the Bio-Rad mini-Protean III system (Bio-Rad). Following electrophoresis, samples were transferred onto polyvinylidene fluoride membrane (GE Healthcare) using a Bio-Rad semidry blotter. Proteins were detected using 1:10,000 antidimedone antisera and 1:2000 anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibody (GE Healthcare) and visualized with electrochemiluminescence reagent (GE Healthcare). Additionally, in some studies, polyclonal rabbit antibodies (Lab Frontiers) that detect either peroxiredoxin (Prx) or GAPDH only in the sulfinic/sulfonic forms were used (both at 1:2000), as were antibodies against unmodified Prx1/Prx2 (mouse, 1:1000) (Abcam) and GAPDH (goat, 1:1000) (Santa Cruz Biotechnology). These were detected using either 1:1000 anti-mouse HRP-conjugated secondary antibody (GE Healthcare) or 1:1000 anti-goat HRP-conjugated secondary antibody (Dako), respectively. Quantification was performed using Gel Pro Analyzer 3.1 (MediaCybernetics).
GAPDH peroxidase assays
The peroxidase activity of GAPDH was assessed by a spectrophotometric assay (27). Briefly, GAPDH was prereduced with dithiothreitol and then desalted using desalting columns (Pierce). Prereduced recombinant rabbit GAPDH (20 μM) was incubated with a variety of H2O2 concentrations in the presence and absence of 1 mM sodium arsenite at 37°C for 30 min. The amount of H2O2 remaining was assessed using Phenol Red and HRP, with the samples being read at 610 nm. A standard curve of H2O2 concentrations alone was also determined. Samples were taken at the end of the incubation period into nonreducing sample buffer and subjected to SDS-PAGE and immunoblotting.
Dichlorofluorescein assay for H2O2 abundance in ventricular myocytes
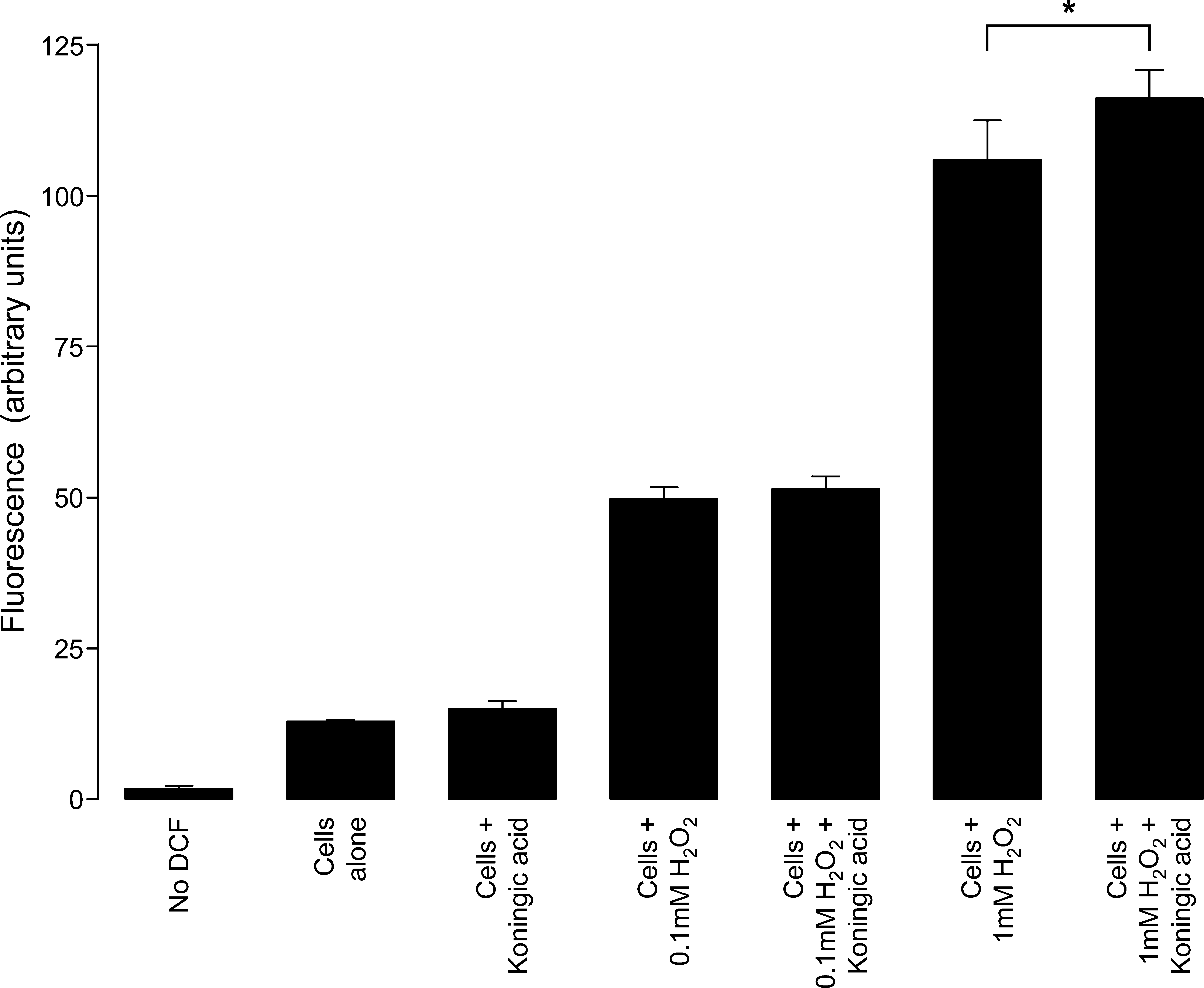
Generation of peroxide was measured using the dichlorofluorescein (DCF) assay (24), with minor modifications. Isolated ventricular myocytes were incubated with 100 μM DCF diacetate in modified Tyrode's solution for 30 min at 37°C. Then, DCF diacetate was removed and the cells were washed and resuspended in modified Tyrode's solution containing either 50 μM koningic acid or vehicle control for 30 min at room temperature. For the final 10 min of the incubation, some samples were spiked with either 0.1 or 1 mM H2O2. The cells were transferred to a black 96-well culture plate (Nunc) and the fluorescence in each cell was measured using a SpectraMax Gemini XS (Molecular Devices). The excitation and emission wavelengths were 485 and 538 nm, respectively.
Statistical methods
Data for all relevant figures are presented as the mean ± standard error. Figures 2B and C, 4A, 5E and F, and 6 were analyzed by one-way analysis of variance followed by Newman–Keuls posttest. Figure 4B was analyzed by one-way analysis of variance followed by Bonferroni posttest. All calculations were performed using Graphpad Prism (Graphpad Software).
Results
During pilot experiments in which isolated myocytes were exposed to extracellular dimedone, we failed to detect cellular labeling. This is consistent with the poor membrane permeability of dimedone, as a result of its charge. Consequently, we prepared the cells in a Triton X-100 lysis buffer containing a millimolar concentration of dimedone, to trap any protein sulfenates. Samples were then reconstituted in SDS sample buffer for analysis by Western immunoblotting. Figure 2A shows that a single major band at 37 kDa (representing a protein SOH) was present in control cardiac ventricular myocytes prepared in the presence of dimedone. This band was eventually confirmed as GAPDH through co-migration immunoblots and koningic acid (a selective GAPDH inhibitor) inhibition studies (see below). Further, labeling was wholly dependent on the presence of dimedone, as shown when dimedone was omitted from the preparation and the labeled protein was no longer present. This basal SOH was progressively lost as the cells were treated with increasing concentrations of H2O2. This loss of the dimedone SOH was highly reproducible and observed with both 10 min (Fig. 2B) and 20 min (Fig. 2C) of treatment. The longer treatment time potentiated the H2O2-induced loss of SOH labeling and was more obvious at the lower oxidant concentrations. Indeed, 0.05 and 0.1 mM H2O2 treatment slightly enhanced SOH formation at 10 min, consistent with a proportion of GAPDH being in the SH state, reacting with H2O2 and leading to an elevation in the abundance of the SOH. However, these treatments for 20 min induced a small loss of SOH and are likely explained by the cell not being able to supply adequate reducing equivalents to prevent the SOH being further oxidized during this prolonged H2O2 exposure (as discussed below). Figure 2D shows myocytes that have been treated with various concentrations of H2O2 and then washed and imaged. The myocytes are still healthy, which rules out the loss of labeling being due to necrotic cells.
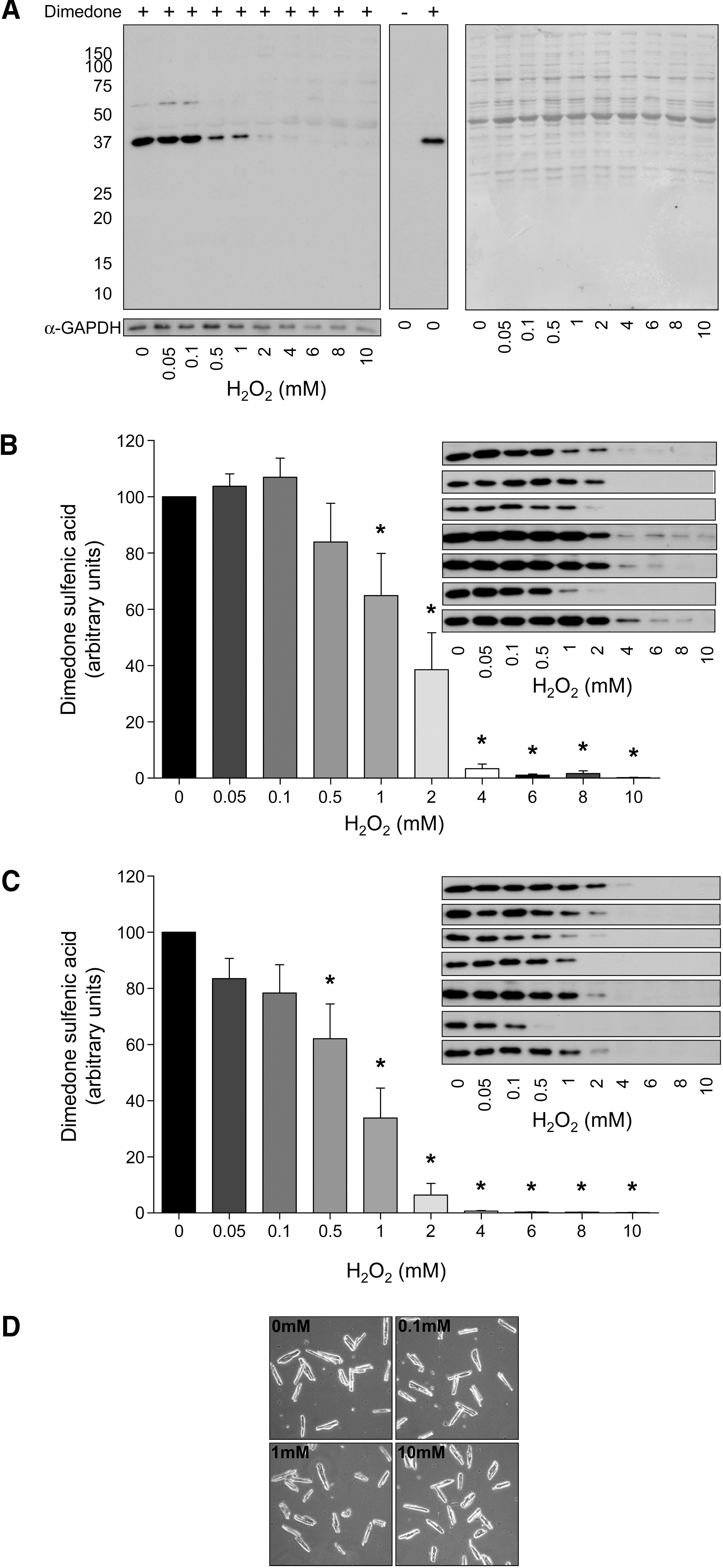
Figure 3A shows myocyte samples prepared for SOH analysis, with the immunoblots being probed more robustly to enhance the sensitivity of detection for oxidized proteins. Again, the GAPDH band dominates in control, untreated cells, but as this band is lost during H2O2 treatment, it is clear that a number of new, secondary protein sulfenates become visible. A longer exposure of different samples (Fig. 3B) further highlights the appearance of these oxidized proteins. The new proteins that appear with H2O2 treatment are notably less intense than the GAPDH sulfenate. It is perhaps to be anticipated that the new bands generated a lower signal than GAPDH, as the dehydrogenase is a highly abundant protein, present at an order of magnitude or more over many proteins in the cell. This is typical of proteomic approaches where a “global picture” of cellular protein status is indexed. When the signal intensity is amplified, so that low or medium abundance components can be visualized, the signal from abundant proteins (and also nonspecific labeling) adds noise. Regardless of this issue, which culminates from differential relative abundance, it is clear that new components do appear as the oxidative stress is elevated. Even with these stronger signals, it is evident that these signals are still wholly dependent on the presence of dimedone (Fig. 3C). As the dimedone SOH labeling is progressively lost with H2O2 treatment, the GAPDH sulfinic/sulfonic acid (SO2/SO3) appears at the corresponding mass on the blot (Fig. 3D). At the same time, the 2-Cys Prxs also progressively hyperoxidize with H2O2 treatment.

Figure 4A shows data from in vitro studies assessing the ability of GAPDH to function as a peroxidase and decompose H2O2. The GAPDH peroxidase activity was indexed from 1 to 100 μM H2O2 and the graph shows the percentage of the total available H2O2 that was consumed at each of the concentrations. Of course, the total amount of H2O2 available to the putative peroxidase increases with the H2O2 concentration. In the absence of GAPDH or arsenite, the H2O2 remained stable and did not spontaneously decompose over the duration of these activity assays (data not shown). GAPDH clearly catalyzed the decomposition of H2O2, and it is evident that arsenite alone could also significantly achieve this. This severely hindered our assessment of the peroxidase events, which were further complicated by the fact that the GAPDH hyperoxidized at higher H2O2 concentrations (Fig. 4B). Overall, although GAPDH can clearly reduce H2O2, it is difficult to determine the efficiency of this process and there was not substantially enough H2O2 decomposition to indicate the catalytic turnover of this peroxidase activity driven by arsenite-induced GAPDH SOH reduction.
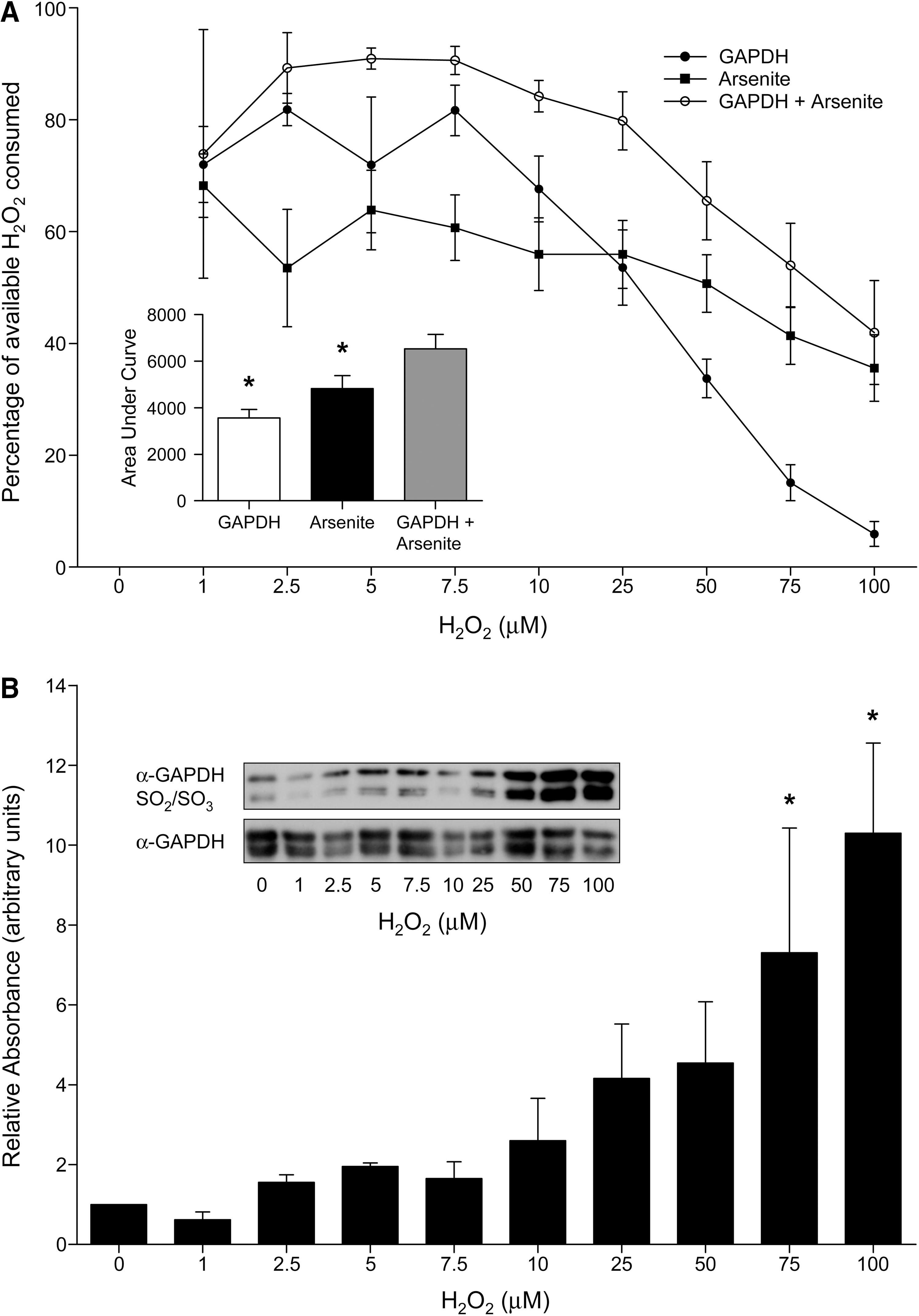
Figure 5A shows that the GAPDH inhibitor, koningic acid (19, 23, 32 –34), prevents the dimedone-dependent basal labeling of GAPDH SOH. The mechanism of action of koningic acid involves it being covalently adducted to the catalytic SH of the dehydrogenase, Cys-149, rendering it catalytically inactive. The action of this selective inhibitor helps verify that this protein is indeed GAPDH. Consistent with GAPDH Cys-149 being the target of koningic acid, this antagonist prevented the hyperoxidation of this residue to the sulfinic/sulfonic state (Fig. 5B, C). Indeed, this hyperoxidation event is known to occur in GAPDH at Cys-149 (6), further substantiating the dehydrogenase as this protein, which is SH redox modulated by H2O2. The 2-Cys Prxs are well known to form SO2/SO3 in response to H2O2, including in ventricular cardiac myocytes (37). We observed this Prx hyperoxidation event (Fig. 5D), which was not prevented by koningic acid, substantiating the selectivity of this antagonist for GAPDH. Further, the inhibition of GAPDH by koningic acid did not modulate the H2O2-induced hyperoxidation of the 2-Cys Prxs (Fig. 5D, E). However, it is interesting to note that there was a trend for koningic acid treatment to marginally increase Prx hyperoxidation over the range of 0–0.1 mM H2O2, whereas, at oxidant concentrations beyond this, the trend was for the inhibitor to decrease it (Fig. 5F). This could reflect a subtle, moderate role for GAPDH in metabolizing cellular peroxide.
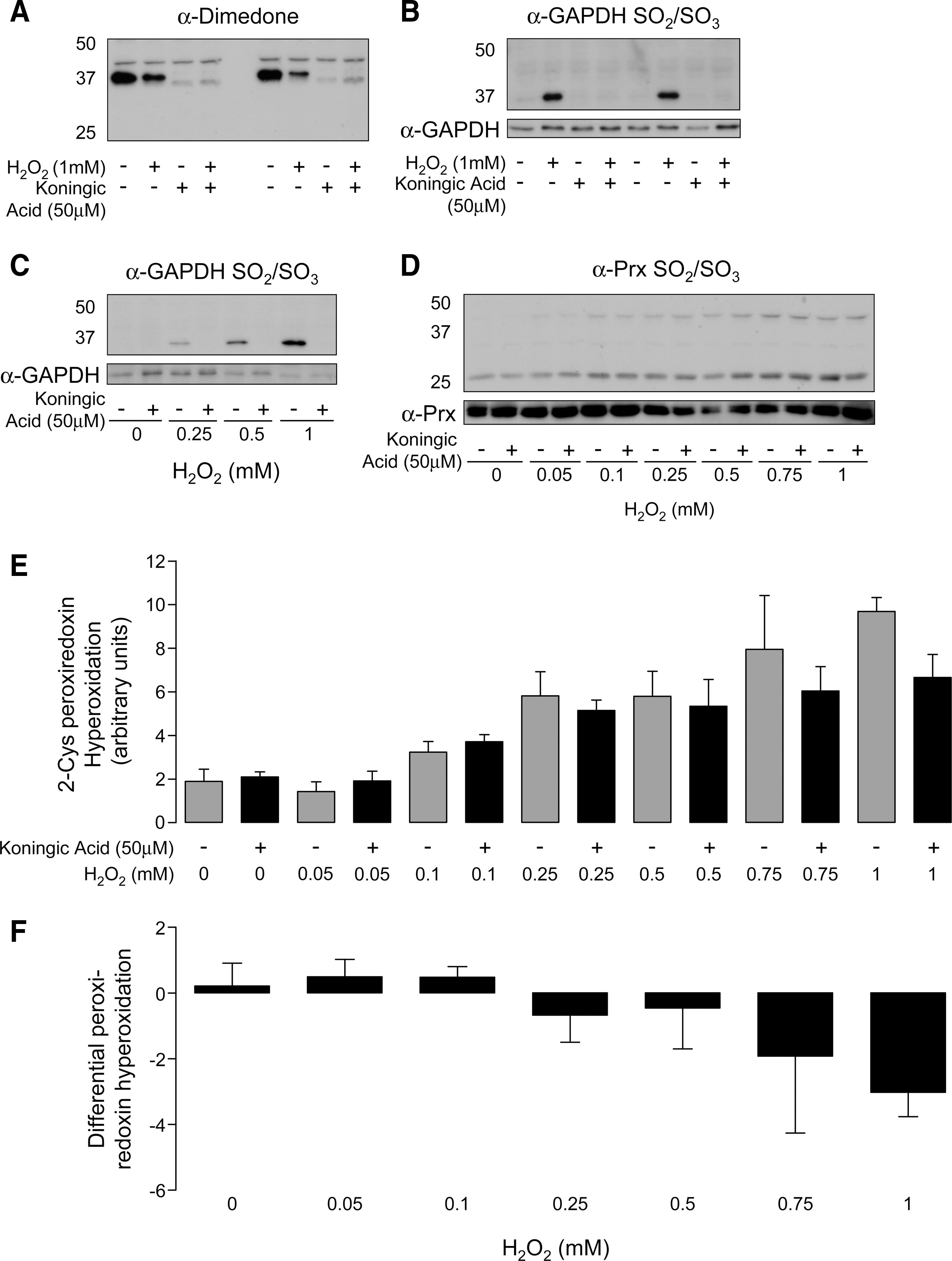
Figure 6 shows the intracellular H2O2 abundance in myocytes loaded with the reporter dye DCF. Although it is clear that exogenous oxidant treatment increased intracellular H2O2, treatment with the GAPDH inhibitor, koningic acid, failed to do this. Further, koningic acid did not modulate the extent to which exogenous H2O2 accumulated intracellularly following exogenous application except at high concentrations of H2O2.
Discussion
Oxidants such as H2O2 and lipid hydroperoxides are produced in healthy tissues in a regulated way, playing crucial roles in the homeostatic regulation of fundamental cellular functions. A key event in the sensing and subsequent transduction of these signals is the chemical reaction of the oxidant molecules with P-SHs, leading to the posttranslational oxidative modification of Cys residues (16). Indeed, key events in the life history of cells, such as cell growth, proliferation, and apoptosis, are associated with precise alterations in the SH reduction potential, as a result of alterations in oxidant production during these specific scenarios.
A primary redox couple involves an equilibrium between nucleophilic reduced P-SHs and peroxides, resulting in the formation of a reversible SOH. Proteins known to form SOHs include nitrile hydratase, the reduced nicotinamide adenine dinucleotide phosphate disulfide reductase family, various peroxidases, tyrosine phosphatases, OxyR, OhrR, and methionine sulfoxide reductases (15). Most protein sulfenates are labile, reacting rapidly with reducing equivalents such as secondary SHs to generate more stable disulfides. However, if a protein sulfenate encounters more oxidizing molecules, as may occur during heightened or chronic oxidant stress, it will be further oxidized, first to the SO2 and then to the SO3 state. Some protein sulfenates are stable enough to be long-lived, for example, the bacterial transcription factor OxyR and, the focus here, GAPDH. GAPDH is primarily known as a glycolytic enzyme whose activity is regulated by Cys modification. However, GAPDH is now also well established as a multifunctional protein, rather than just a “simple” metabolic enzyme. Mammalian GAPDH is a homotetramer, with a well-defined active site and mechanism based on Cys-149 and His-176. Cys-149 switches redox state between the reduced (SH), thiolate (S−), and covalent (S-C) hemithioacetal states during glycolysis.
The novel antibody reagent that we have generated has demonstrated that GAPDH exists basally as an SOH in ventricular myocytes and can be trapped by exposure to dimedone. We were a little surprised by the presence of this single, dominant product in control cells and had anticipated the labeling of additional proteins. For example, cardiac cells are replete with Prx proteins, which form SOHs, especially when exposed to H2O2 (37). Indeed, we observed the hyperoxidation of the 2-Cys Prxs to the sulfinite and sulfonite states during H2O2 treatment, and these presumably transition through the sulfenate oxidation state. A likely explanation for the lack of Prx SOH formation is that it rapidly forms a disulfide by reacting with a proximal SH (42), which outcompetes the potential reaction with dimedone. Indeed, the Prxs are known to be dimedone reactive, but this was only observed when a cysteineless resolving mutant was used (30). Thus, the limited number of dimedone-modified SH residues we observed may indicate the slow reactivity of the compound with Cys-SOH, compared with alternate reactions they may undergo in the cell. GAPDH formed a stable SOH in vitro, accumulating in enzyme preparations as reducing agents were depleted, rendering it inactive (4). These studies utilized radiolabeled dimedone and showed that the enzyme could be rejuvenated to its standard glycolytic function by the addition of a fresh reducing agent. However, what is less apparent is why the GAPDH is stably present as an SOH, as this state would be anticipated to inhibit glycolysis.
The fact that glycolysis occurs in ventricular myocytes despite the presence of sulfenated GAPDH is perhaps consistent with the protein cycling back and forth between the reduced and sulfenated redox states. Repetitive conversion to the reduced state would enable hemithioacetal conjugation with glyceraldehyde 3-phosphate and consequently glycolysis. This redox cycling scenario requires a cellular reducing equivalent to catalyze the reduction of the SOH. This reversal of SOHs indicates that GAPDH can, in principle, reduce H2O2 in a cyclical fashion, highlighting a potential peroxidase activity. The decomposition of H2O2 by GAPDH is already acknowledged, although the rate at which this occurs is thought to be slow (41). Although, such in vitro data are potentially misleading, as the kinetic analysis was likely performed without the optimal physiological reducing equivalent, as this remains unidentified. In addition to H2O2, GAPDH is also redox modified by glutathione (GSH) or glutathione disulfide (9), nitric oxide (21), peroxynitrite (10, 40), nitroalkenes (2), 4-hydroxynonenal (22), disulfide S-oxides (20), and by alkylation (36). Of these, nitric oxide and peroxynitrite may potentially give rise to Cys-SOH formation within GAPDH. More credence for a GAPDH peroxidase activity comes from the substitution of Cys-149 with selenocysteine, which turns the enzyme into an effective GSH-dependent peroxidase (7). GAPDH SOH formation and turnover can also be inferred from our earlier studies in which we observed reversible glutathiolation (17) and interprotein disulfide bond formation in response to ischemia and H2O2, respectively (35). Further, GAPDH hosts a Cys-His ion pair in its active site, which is crucial for catalysis in some peroxidases (44).
Exposure of cellular GAPDH beyond ∼0.1 mM H2O2 caused further oxidation of Cys-149 sulfenate to the SO2/SO3 forms, defined as hyperoxidation (5, 45). Hyperoxidation inactivates all glycolytic activity, alters the cellular localization of GAPDH, and is nonreversible through direct reduction (5, 17). Although this hyperoxidation can be considered as damage, it can confer new functions on GAPDH, such as nuclear translocation and proapoptotic signaling, and is also a metabolic stress response that stimulates reduced nicotinamide adenine dinucleotide phosphate production (21). Hyperoxidation is a defining characteristic of the Prx peroxidases (37) and for DJ-1, which was recently identified as a peroxidase (1). Baty et al. (3) identified H2O2-sensitive proteins using an unbiased proteomic strategy and identified three proteins with SHs that were susceptible to oxidation. These included the Prxs and thioredoxin, which would be anticipated, but the oxidation of GAPDH was also notably prominent.
Altogether, SOH formation and turnover at Cys-149 suggests a possible, biologically significant role for GAPDH as a peroxidase or regulator of H2O2-induced cell signaling. An intriguing feature of H2O2 signaling is the hyperoxidation of the 2-Cys Prxs, which inactivates the enzymes, leading to potentiated cellular peroxide concentrations. In this scenario, as outlined in the floodgate hypothesis of H2O2 signaling (43), the oxidant can then reach such a concentration so as to enable reaction with less-reactive proteins, which are not modified at lower H2O2 concentrations. Indeed, when we exposed cells to higher concentrations of H2O2, we observed a concomitant loss of GAPDH dimedone SOH, appearance of hyperoxidized Cys-149 GAPDH, and generation of new secondary dimedone-positive protein sulfenates. Further, exposure of GAPDH in vitro to concentrations of H2O2, which induced hyperoxidation, correlated with loss of GAPDH peroxidase activity. The complete loss of basal GAPDH dimedone labeling required ∼2 mM H2O2, and it is not until this loss occurs that the other targets of sulfenation become labeled. One question is the relevance of this oxidant concentration to health and disease. We have reviewed this complex issue in depth (38) and concluded that the evidence supports this H2O2 concentration as being potentially relevant to pathophysiology. This question of the concentration of oxidants in cells remains a matter of debate and reflects the difficulty in measuring these reactive entities.
Overall, the interaction of GAPDH with H2O2 is reminiscent of the floodgate scenario, and it is possible that hyperoxidation of the dehydrogenase is a component of this signaling paradigm. However, as the Prxs hyperoxidize in parallel with GAPDH, it was difficult to dissect their potential relative contributions. To address this issue, we utilized koningic acid, a selective inhibitor of GAPDH that operates by covalently adducting to the catalytically essential Cys-149 (19, 23, 32, 33). The specificity of koningic acid for GAPDH Cys-149 was confirmed in our studies, as it fully prevented hyperoxidation, which is known to occur at this residue. Further, koningic acid treatment also prevented dimedone labeling. This observation highlighted the fact that Cys-149 is redox cycling between the SH and the sulfenate, being trapped by koningic acid or dimedone in each of these respective oxidation states. If GAPDH hyperoxidation and inactivation participates in the floodgate scenario of oxidant signaling, koningic acid treatment would be expected to mimic the effects of H2O2. However, there was no evidence that koningic acid treatment alone induced significant protein sulfenation (i.e., dimedone labeling) of secondary proteins. However, it is interesting that at lower concentrations of H2O2 treatment, koningic acid tended to marginally enhance Prx hyperoxidation. In contrast, at 0.25 mM H2O2 and above, koningic acid progressively decreased this hyperoxidation compared with its respective control. Although the differential effect is subtle, there is a consistent trend.
This lack of an effect of koningic acid may be due to the absence of peroxide or because the Prxs can efficiently cope with any additional oxidative burden resulting from basal GAPDH inhibition. Consequently, we compared the dose–response of cells, with or without koningic acid inhibition of GAPDH, to H2O2. If GAPDH deals with a biologically meaningful turnover of H2O2, koningic acid treatment would be anticipated to “push” this component on to the Prxs. Consequently, we might expect koningic acid to have induced hyperoxidation of Prx at a lower concentration of H2O2 than in corresponding controls without the inhibitor. In addition, there should be a corresponding sensitization of H2O2-induced dimedone SOH labeling by koningic acid. However, despite searching for experimental conditions where koningic acid potentiated H2O2 signaling, we could find no evidence for this. Further, although in vitro experiments showed that GAPDH can indeed decompose H2O2, we found little evidence of efficient catalytic turnover. However, this issue is complicated by the fact that there may be a particular electron donor that can indeed efficiently reduce GAPDH-SOH. Arsenite failed to do this, although it is efficient in this role with GSH-dependent peroxidase assays. Amongst other complications were the facts that arsenite itself directly reduced H2O2, and that GAPDH becomes inactive as a result of hyperoxidation, complicating the interpretation of these data. Clearly, without a molecule that donates electrons rapidly enough to reduce GAPDH-SOH, the enzyme will at some point hyperoxidize as the H2O2 concentration is progressively increased. Given that myocyte GAPDH is in the sulfenate form basally and not hyperoxidized, and that there is glycolytic flux despite constant H2O2 turnover, suggests that there is a cellular molecule that reduces the sulfenated enzyme.
Ascorbate is a candidate reductant in vivo, as it abolishes the acyl phosphatase activity of reversibly oxidized GAPDH (by reducing Cys-149 SOH), restoring the dehydrogenase activity that requires Cys-149 SH. Ascorbate, therefore, appears to reduce Cys-149 SOH back to the SH state. Further, ascorbate is abundant in mammalian tissues (∼mM), enzymatically recycled upon oxidation, and supports the peroxidase activity of the 1-Cys Prx 6 with a catalytic efficiency of ∼105 M −1 s−1 (25). Reduced nicotinamide adenine dinucleotide (NADH) is an another candidate reductant, as GAPDH can utilize it in reducing the SH-ester intermediate to glyceraldehyde 3-phosphate during glycolysis. Hence, under conditions that favor peroxidase activity over catabolic activity, it is plausible that GAPDH's Cys-SOH is reduced by NADH, in a way analogous to peroxide reduction by bacterial NADH peroxidase and NADH oxidase (28). However, we found it impossible to study these compounds with our GAPDH peroxidase assays as they alone rapidly reduced H2O2.
Overall, it is clear that ventricular myocyte GAPDH undergoes intriguing changes in redox state when H2O2 is elevated. Although there is potential for GAPDH-SOH recycling to provide a cellular mechanism for H2O2 decomposition, we have found no strong indication that this occurs or impacts on oxidant signaling in the context of the floodgate model. Further, we found no evidence that inhibiting myocyte GAPDH increases myocyte H2O2. Koningic acid also did not modulate the amount of H2O2 accumulating inside myocytes when the oxidant was applied extracellularly. This further adds to the conundrum regarding the cellular significance of GAPDH forming SOH, and its hyperoxidation response to elevated oxidant stress. As GAPDH's acyl phosphatase activity is dependent on Cys-149 SOH, it is clear that hyperoxidation will result in loss of this activity, as well as inhibition of glycolysis. As highlighted earlier, GAPDH is established as a multifunctional enzyme and its redox state may be crucial in controlling these activities during H2O2 signaling. For example, GAPDH's oxidation state can control the stability of mRNA and so regulate protein abundance (31), and its S-nitrosylation is important in induction of apoptosis (21). On a final note, it is notable that another group has recently generated an antibody that also recognizes protein sulfenates derivatized by dimedone (39). Their study showed increased protein SOH formation in cancer cells, consistent with them being under oxidative stress involving elevated H2O2. Although the two antibodies would both broadly detect protein dimedone SOHs, the antigen used in their production was a little different. We induced SOH formation directly in KLH and trapped it with excess dimedone (outlined in Fig. 1). As such the derivatized sulfenates will be directly adjacent to other amino acid residues, as they would be when we use dimedone to trap cellular protein sulfenates. In contrast, Seo et al. synthesized an amine variant of dimedone Cys SOH and then conjugated this to succinylated KLH. This generated KLH with dimedone coupled via a lengthy spacer arm distant from the secondary or tertiary structure of the protein. Consequently, it is likely that there may be some subtle differences in the performance of these novel antibody reagents.
Footnotes
Acknowledgment
This research was supported by a project grant from the Medical Research Council (G0600785).
Author Disclosure Statement
The novel dimedone SOH antibody described in this manuscript was licensed to Millipore (