
Abstract
Disulfide bond formation is a crucial step for oxidative folding and necessary for the acquisition of a protein's native conformation. Introduction of disulfide bonds is catalyzed in specialized subcellular compartments and requires the coordinated action of specific enzymes. The intermembrane space of mitochondria has recently been found to harbor a dedicated machinery that promotes the oxidative folding of substrate proteins by shuttling disulfide bonds. The newly identified oxidative pathway consists of the redox-regulated receptor Mia40 and the sulfhydryl oxidase Erv1. Proteins destined to the intermembrane space are trapped by a disulfide relay mechanism that involves an electron cascade from the incoming substrate to Mia40, then on to Erv1, and finally to molecular oxygen via cytochrome c. This thiol–disulfide exchange mechanism is essential for the import and for maintaining the structural stability of the incoming precursors. In this review we describe the mechanistic parameters that define the interaction and oxidation of the substrate proteins in light of the recent publications in the mitochondrial oxidative folding field. Antioxid. Redox Signal. 13, 1189–1204.
Introduction
Disulfide bonds constitute a post-translational covalent modification that occurs between cysteine residues during protein folding and favor the attainment of the protein native conformation by decreasing the entropy of the polypeptide chain. The impact of disulfide bonds on folding kinetics is dependent on whether the disulfides are introduced close to the folding core. Disulfide bonds introduced close to the folding core accelerate protein folding, and conversely protein folding can be slowed down two to three times when they are introduced outside the folding core (1). The formation of disulfide bonds, depicted below, occurs by oxidizing two sulfhydryl groups (-SH), a reaction which is coupled to the release of two electrons.
This reaction can be completed spontaneously in vitro since the electron acceptor can be molecular oxygen. However, these kinds of spontaneous, random oxidation reactions are kinetically very slow and therefore are difficult to occur in the cell where fast reactions are required (89, 123). This observation has lead to the discovery of the first cellular oxidase PDI (protein disulfide isomerase), by the laboratory of Anfinsen (4).
It is now well established that dedicated cellular machineries wired by specific protein networks are responsible for the coordinated catalysis of disulfide bonds in substrate proteins. In this case, disulfide bonds are introduced by thiol–disulfide exchange reactions depicted below.
These involve the exchange of electrons from a molecule that contains free sulfhydryl groups (R'SH) to one which is already disulfide bonded (RS-SR). Thiol disulfide exchange reactions are fundamental in protein disulfide bond formation and result in the concomitant oxidation and reduction of the proteins involved. The reaction is conducted in four consecutive steps (Fig. 1). In the first step, a thiolate anion (S–), which has occurred from the deprotonation of a free sulfhydryl group (SH) of the substrate, performs a nucleophilic attack on the disulfide bond of the oxidase. During this reaction the two proteins form a transient mixed disulfide intermediate. The mixed disulfide intermediate is then attacked by the second thiolate anion of the substrate, hence releasing the substrate in an oxidized state, concomitantly with the reduction of the oxidase. Similarly, disulfide bonds can be formed intramolecularly where a thiolate anion can attack a pre-existing disulfide bond of the same protein. Disulfide bond reshuffling is essential as it allows the protein to acquire its native conformation and is mediated by enzymes called isomerases (Fig. 1).

In this review we will describe one of the major pathways for disulfide bond formation in the cell, the recently identified pathway of protein oxidation in the intermembrane space of mitochondria.
Oxidative Pathways in the Cell
The formation of disulfide bonds is favored in an oxidative environment. Due to the reducing nature of the cytosol, they are usually catalyzed in specialized subcellular compartments both in prokaryotic and eukaryotic cells. In prokaryotic cells, protein disulfide bond formation occurs in the periplasm, while in eukaryotic cells oxidation pathways are found predominantly in the lumen of the rough endoplasmic reticulum (ER) and in the intermembrane space (IMS) of mitochondria. The core pathways that promote oxidation in the ER, the IMS of mitochondria, and the bacterial periplasm have been identified and share many similarities in the mechanism and the proteins involved (Fig. 2).The oxidative pathways in the bacterial periplasm and in the ER will be briefly described in the following section, as a mechanistic basis for analyzing the properties of oxidation in the mitochondrial intermembrane space.
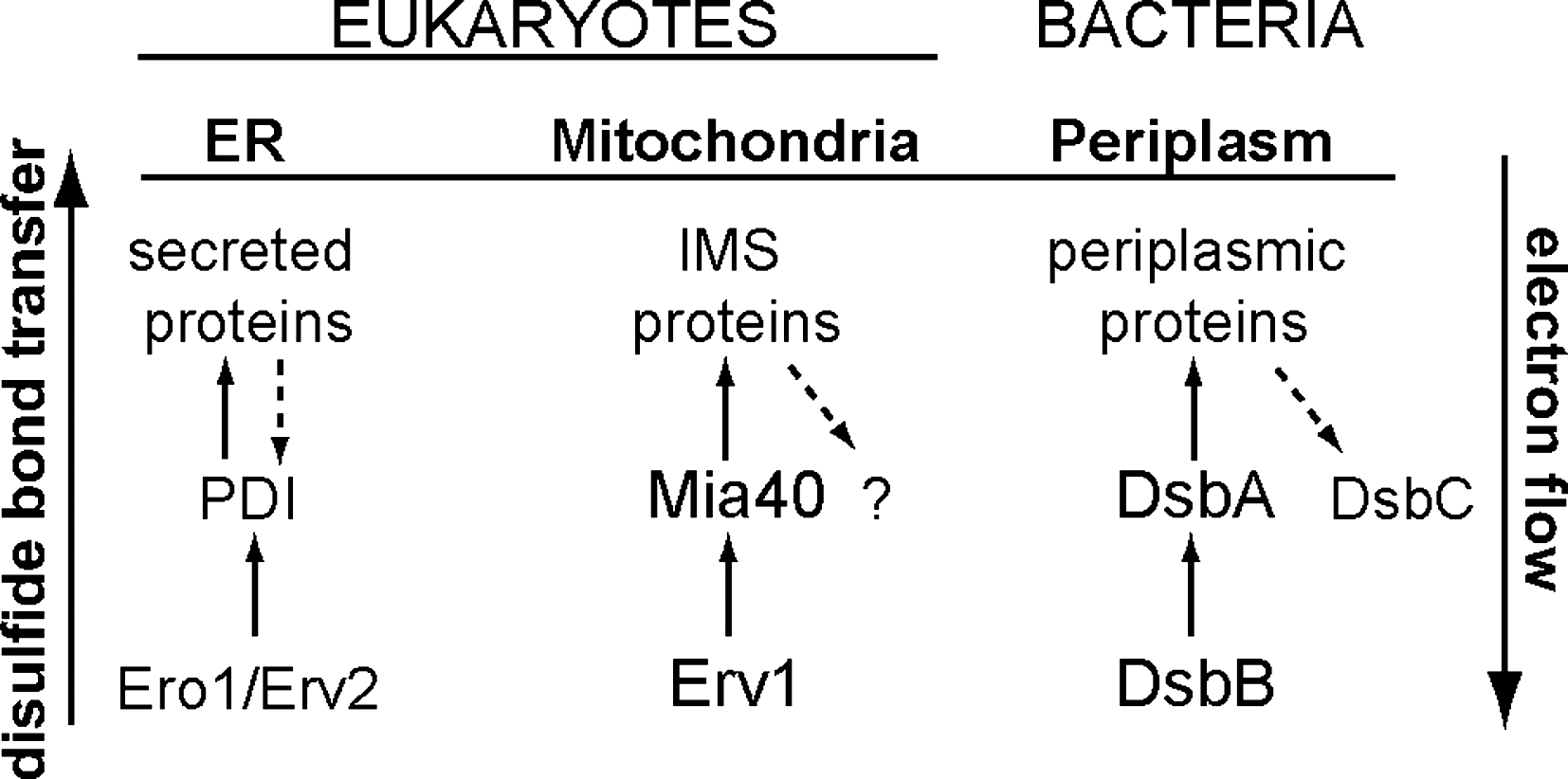
Bacterial periplasm
The mechanism of disulfide bond formation in the bacterial periplasm is one of the best characterized systems and is mediated by the Dsb proteins (DiSulfide Bond). The bacterial redox pathway is made up by the proteins DsbA and DsbB that are involved in the oxidation of periplasmic substrate proteins (19, 59, 84), and by the proteins DsbC and DsbD that are involved in isomerization of the substrates (11, 93, 127). The substrate proteins acquire their disulfide bonds through the oxidase action of DsbA. The catalytic center of DsbA consists of a CX2C motif that is placed in a thioredoxin fold and constitutes the most oxidizing protein identified so far amongst the different cellular oxidative pathways (77). This is reflected in the high redox potential of the active cysteine C30 (−120 mV) and in the very low pKa (∼3) (41, 126). The active site of DsbA is recycled by the membrane protein DsbB through a reaction driven by the respiratory chain (46, 61). Under aerobic conditions, DsbB transfers electrons through an intramolecular disulfide cascade to quinone, then to cytochrome oxidase bd and bo, and finally to oxygen being the final electron acceptor (10, 11, 58, 63). Under anaerobic conditions, DsbB transfers the electrons to menaquione. Due to the strong oxidizing capacity of DsbA, substrate proteins are sometimes misoxidized, acquiring non-native disulfide bonds. An isomerization system has been developed in bacteria that promotes disulfide bond reshuffling. Specific structural properties of the different components ensure insulation of the oxidizing and isomerization pathways. The latter is composed of the proteins DsbC and DsbD whose reduction capacity is maintained by the cytoplasmic thioredoxin and NADPH (29, 97).
The endoplasmic reticulum
The endoplasmic reticulum of eukaryotic cells harbors a dedicated oxidative folding pathway composed of the proteins PDI and Ero1 (102). PDI is an essential protein that is organized structurally in four thioredoxin domains and catalyzes the transfer of disulfide bonds to substrate proteins via a CX2C active center (37, 53, 115). The redox state of PDI is determined by the essential protein Ero1, a flavin bound membrane associated protein (36, 43, 92). Ero1 recycles PDI for another round of substrate oxidation by shuttling electrons to FAD (103). Under aerobic conditions, oxygen serves as the final electron acceptor by generating hydrogen peroxide. In fungi, a second pathway for disulfide bond formation in the ER has been identified and involves the flavoenzyme Erv2 (44, 101). This protein has been shown to functionally complement a yeast strain that lacks Ero1 when overexpressed. Erv2 belongs to the Erv/ALR family of flavin-dependent thiol oxidases and can serve as a disulfide bond donor to PDI by consuming oxygen (35, 39). In contrast to the bacterial isomerization pathway, in the endoplasmic reticulum disulfide bond reshuffling is catalyzed by PDI (47, 67). Glutathione, that is transported in the ER by an unknown mechanism, has been shown to mediate PDI reduction, which is required for isomerization to occur (27).
Import in Mitochondria
According to the endosymbiotic theory, the majority of the mitochondrial genes were transferred to the “host genome’, the cell nucleus (42, 125). More specifically, in an effort to delineate the yeast mitochondrial proteome, 800 proteins were identified of which only eight are encoded by the mitochondrial DNA (105). Since the majority of the mitochondrial proteins are encoded in the nucleus, sophisticated import machineries were developed that mediate translocation of mitochondrial precursors to the appropriate submitochondrial compartment. Mitochondria are defined by four subcompartments. The outer membrane (OM) that represents the first point of contact with the newly synthesised precursors, two aqueous compartments the intermembrane space (IMS) and the matrix, and the inner membrane (IM) that separates the two. The basic principles that underlie translocation and sorting of proteins in the different mitochondrial compartments will be briefly discussed, and emphasis will be given to the proteins that reside in the intermembrane space. A more comprehensive description of the mitochondrial import pathways is reviewed in reference (25).
General import properties
The first point of entry for all the mitochondrial precursors is the TOM channel (translocase of the outer membrane). The sorting pathways then segregate according to the nature of the imported protein (Fig. 3). The classical paradigm of mitochondrial protein import is defined by the presence of an amino terminal amphipathic signal, termed a presequence, which contains targeting information. This presequence is typically found on proteins destined for the matrix and is cleaved off by processing peptidases in the matrix. This class of proteins utilize the TIM23 (translocase of the inner membrane) channel for sorting to the matrix (90). The majority of the proteins destined for the inner membrane are polytopic and contain internal targeting information on the polypeptide chain. Those are guided through the intermembrane space by the chaperone complex of the small Tim proteins and are delivered to the carrier translocase TIM22 that mediates insertion in the inner membrane (82). Outer membrane proteins typically contain β-barrels and utilize a separate pathway for insertion. Those first translocate across the TOM channel and via the chaperone Tim complex of the intermembrane space are delivered to the SAM complex (sorting and assembly machinery) that mediates their integration into the outer membrane (66). In all cases the major driving force for mitochondrial import and translocation is provided by the hydrolysis of ATP and by the membrane potential ΔΨ of the inner membrane (94, 98).
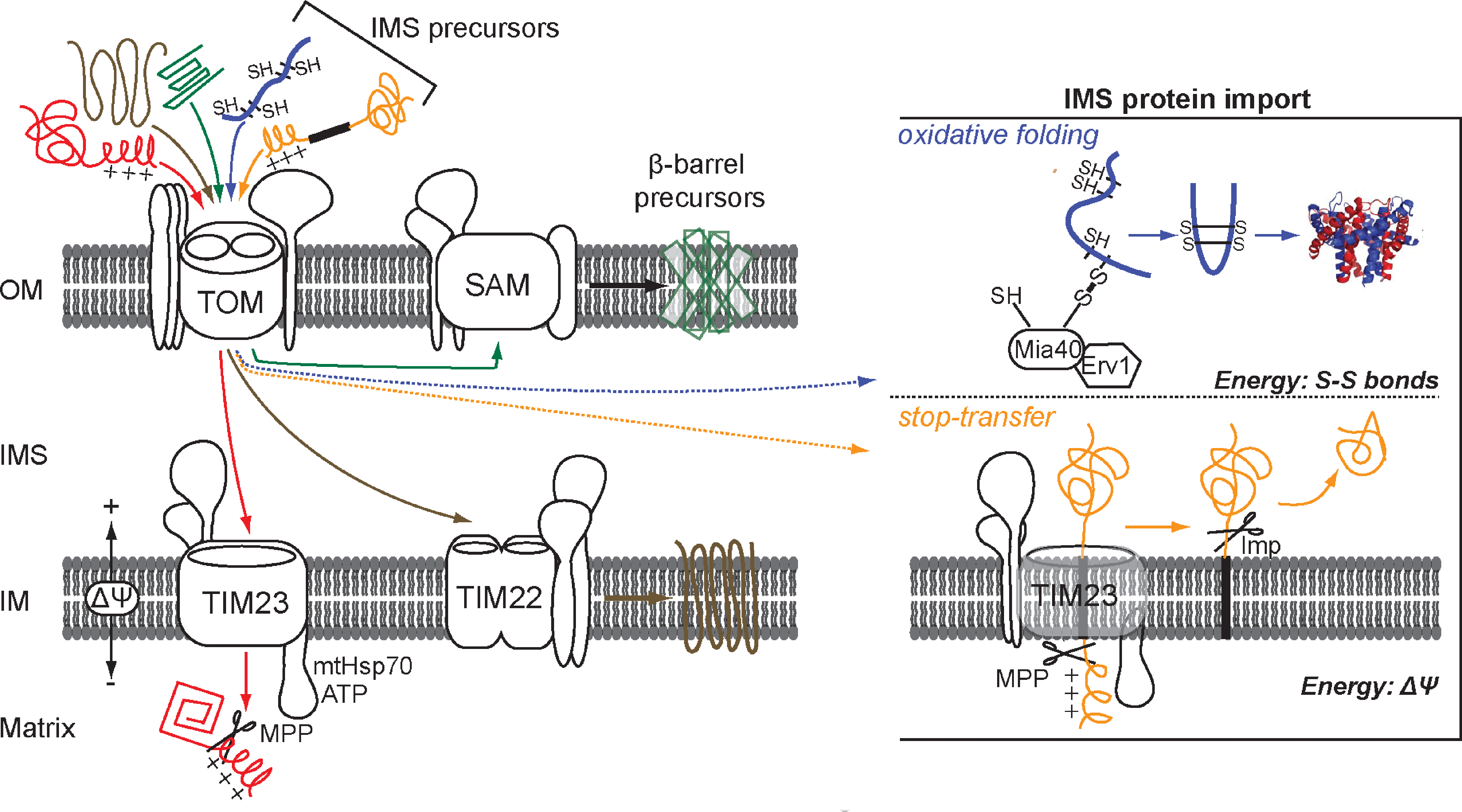
Import in the intermembrane space
Proteins in the intermembrane space of mitochondria participate in vital cellular functions such as transport of metabolites, of heme, and metal ions, induction of apoptosis, and are components of the electron transport chain. Two broad categories of intermembrane space proteins can be assigned depending on their import mechanism. One class of proteins is characterized by the presence of a bipartite signal, namely a presequence followed by a hydrophobic sorting domain. According to the “stop-transfer” model the presequence targets the protein to the TIM23 channel where it is cleaved off by the matrix mitochondrial peptidase (MPP) similarly to the matrix targeted proteins. A second consecutive proteolytic event that occurs in the intermembrane space releases the mature protein in the IMS (49). Different peptidases of the intermembrane space participate in the second cleavage event depending on the substrate protein. Cytochrome b2 and Mgm1 are proteins of the intermembrane space which, during their translocation, are cleaved by Imp1 and Pcp1, respectively. In the stop-transfer pathway, ATP hydrolysis is not necessary and the energy for translocation is provided by the electrochemical membrane potential (Δψ).
The second major class of intermembrane space proteins is comprised of the ones that acquire disulfide bonds during their biogenesis as a mechanism of retention in the IMS. Until very recently the import mechanism by which these proteins are sorted to the intermembrane space was not known. It is now apparent that a dedicated machinery that oxidizes these proteins is directly coupled to their import (26, 79). The identification of this pathway came as a surprise due to the fact that the intermembrane space is thought to be in redox equilibrium with the cytosol. Outer membrane residing porins allow the passage and diffusion of small molecules such as glutathione, which in principle would maintain the IMS proteins in a thiol state much like it does in the cytosol. However, it is now well established that the environment of the intermembrane space of mitochondria allows the oxidation of substrate proteins to occur in a manner analogous to the periplasm, its bacterial counterpart.
Several observations have led to this conclusion. In nature there are examples of thiol–disulfide exchange reactions that occur even in reducing environments, as in the case of poxviruses or eukaryotes. Indeed, proteins that contain disulfide bonds have been found in the capsid of vaccinia virus (99, 100) and in the cytoplasm of eukaryotes (e.g., Cu,Zn-SOD1). Furthermore, it is likely that the intermembrane space of mitochondria has maintained the oxidizing environment of its corresponding bacterial compartment, the periplasm, during the endosymbiotic process. As described earlier, the generation of disulfide bonds and oxidation of substrate proteins in the periplasm is mediated by specific proteins of the Dsb family. In addition, the redox potential of the intermembrane space was measured at −225 mV, which is more oxidizing than that of the cytosol at −290 mV (57), and is closer to the periplasmic one at −165 mV (80), thus providing further evidence that the IMS environment can accommodate disulfide bond formation. Finally, the identification of the enzymes that allow the regulated transfer of disulfide bonds to substrate proteins was the cornerstone for the intermembrane space oxidative pathway. In this pathway, namely the MIA40 pathway, neither ATP nor the membrane potential Δψ are needed, and the energy for translocation is mainly provided by the covalent formation of disulfide bonds in the substrate.
Substrate Proteins of the MIA40 Pathway
An increasing number of disulfide-bonded proteins is found in the intermembrane space of mitochondria. Their biogenesis depends on the MIA40 oxidative pathway. The import of these proteins relies on the “folding-trap” model according to which the proteins are trapped in the intermembrane space due to thiol exchange reactions that lock them in their native folded conformation. The substrates of the MIA40 pathway share some common features, namely that they do not possess a presequence, they are of low molecular mass (6–18 kDa) and that they contain characteristic cysteine motifs. Based on their cysteine motifs they can be assigned in two major categories that are discussed in the following sections.
Substrate proteins with twin CX3C motif
Small Tim proteins belong in a family of homologous proteins that reside in the intermembrane space of mitochondria. Members of this family are the proteins Tim8, Tim9, Tim10, Tim12, and Tim13 that are ubiquitously expressed and highly conserved in eukaryotic organisms (20). They function as chaperones of the intermembrane space (108, 116) operating as two separate soluble 70 kDa complexes, namely the TIM10 complex and the TIM8-13 (87, 91). The two complexes are made by the association of three Tim9 and three Tim10 subunits for the TIM10 complex, whilst three Tim8 and Tim13 associate to make the TIM8–13 complex. The TIM10 complex interacts with the components of the TIM22 channel (Tim12, Tim18, Tim22, and Tim54), forming a 300 kDa complex in the inner membrane that mediates insertion of polytopic hydrophobic proteins (70, 76, 121). In addition to their first described role in facilitating import of inner membrane proteins (64, 65), the small Tim chaperones also mediate the import pathway of proteins destined for the outer membrane by associating with the SAM complex (124).
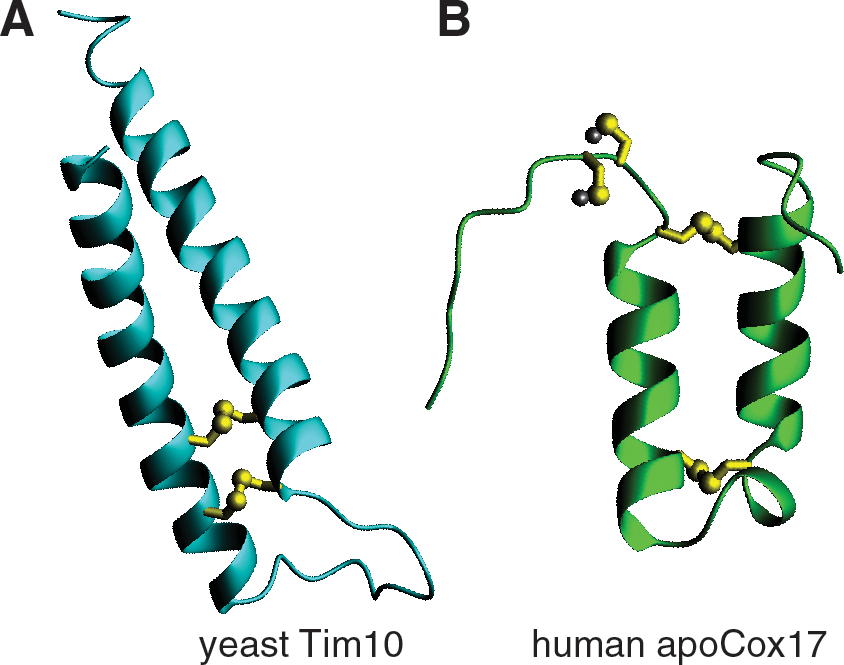
The small Tims possess a characteristic twin CX3C motif. The cysteines were originally proposed to coordinate zinc, and in fact it has been shown that the purified proteins can bind zinc ions in a 1:1 molar ratio (74, 75). Furthermore, it has been proposed that zinc binding in the cytosol retains the proteins in an import competent state by preventing spontaneous oxidation of the precursors (85). However, the role of zinc binding in the biogenesis of small Tim proteins has not been confirmed in vivo. On the other hand, limited trypsinolysis and mass spectrometry have shown that the cysteines are covalently linked and juxtaposed in antiparallel α-helices (Fig. 4) (3). The organization of the cysteines in two intramolecular disulfide bridges was later confirmed in the crystal structure of the TIM10 and the TIM8–13 complex (13, 122). Based on the crystal structure of the complex, each of the subunits is folded in a helix-coil-helix that is stabilized by the disulfide bonds. The complex assumes an alpha-propeller topology with helical tentacles that radiate from a narrow central pore stabilized by salt bridges.
A different functional role has been attributed to the two disulfide bridges. By site directed mutagenesis, it has been shown that the inner disulfide bridge is more important for the stabilization of the protein structure and for the interaction of Tim10 with AAC, whilst mutations of the outer disulfide pair have weaker functional defects and are important for the release of the protein from Mia40 (3, 106). Furthermore, in vitro studies have shown that distinct amino acid domains in Tim10 and Tim9 affect the formation of the TIM10 complex and the interaction with the hydrophobic substrate proteins (119). Conserved charged residues that are found in the loop regions of the Tim proteins are crucial for the interaction of the two partner proteins (13, 118). Furthermore, it has been demonstrated that Tim9 is responsible for maintaining the structural stability of the complex while Tim10 interacts via its amino-terminal domain with the substrate during translocation across the aqueous environment (119).
The in vivo and in vitro studies suggest that there are two main requirements for the import and sorting of the Tim proteins in the intermembrane space. First, it is crucial that the Tim proteins are in a reduced unfolded state in order to get efficiently imported in mitochondria. Finally, the correct intramolecular disulfide bond formation is needed for retaining the proteins in the intermembrane space and is essential for adopting their native conformation and forming the chaperone complex. The importance of the cysteine residues in the biogenesis and protein folding of the small Tims is also evident in the development of neurodegenerative disorders. Mutation of the last cysteine in the human homologue of Tim8 is the causal agent of the deafness-dystonia syndrome (52, 96).
Substrate proteins with twin CX9C motif
The second class of MIA40 substrate proteins contains a twin CX9C motif. Members of this class are proteins of the Cox family that participate in the formation of the cytochrome c oxidase complex (CcO). Bioinformatic searches have also revealed proteins that contain the twin CX9C motif but their functional role is not yet known. Some of them (e.g., Mic14 and Mic17) have been confirmed to be bonafide MIA40 substrates by in organello import experiments (38).
More than 30 proteins are required for the assembly of the CcO complex. These proteins participate in essential processes such as the formation and binding of heme, transfer and metal binding, and the maturation of the CcO complex (23). Among them, Cox12, Cox17, Cox19, and Cox23 contain a signature twin CX9C motif. Cox17 is the best characterized substrate of the oxidative pathway. Its functional role in the intermembrane space is to chaperone copper to the proteins Cox11 and Sco1 (16, 56).
Cox17 contains in total six cysteines, four of which make part of the twin CX9C motif, while the other two are consecutively placed close to the amino terminus of the protein and coordinate a copper(I) ion. The cysteines in the CX9C motif are juxtaposed in antiparallel α-helices and form two disulfide bridges, reminiscent of the small Tim family (Fig. 4). The role of these cysteines is structural as they are required for maintaining the protein in a folded conformation (17). The other two cysteines are either found in an oxidized state, or bound to copper. These constitute the first example of metal binding by two adjacent cysteines (8, 18).
The structure of Cox17 has been solved by NMR and demonstrated that two disulfide bridges stabilize the protein in an α-hairpin structure, much like the small Tims (17). The disulfide bonds that are part of the twin cysteine motif are stable and break open in the presence of 20 mM DTT. This could suggest that the protein is found unfolded in the reducing environment of the cytoplasm, a major prerequisite for efficient import in mitochondria, and once oxidized in the intermembrane space are stable against reduction. On the other hand, the other two cysteines are very sensitive to low concentrations of reducing agents and they have been proposed to be involved in CcO maturation through a disulphide exchange reaction coupled to copper transfer (16). Site-directed mutagenesis of the cysteines involved in metal biding is not tolerated in yeast cells that grow on nonfermentable carbon source (48). Furthermore, it has been proposed that a conserved lysine at position 25, which is close to the amino-terminal cysteines stabilizes the copper ion by maintaining the overall negative charge of the Cu(I)S2 – binding site (17). A similar example has been shown for the cytoplasmic copper chaperone Atx1 and the mitochondrial copper chaperone Cox11 where a conserved lysine is also close to the copper (I) binding site which involves two cysteine residues. Reduction of the protein affects its capacity to bind copper, suggesting that the binding and release of the copper cofactor is regulated by the redox state of the protein (9, 14).
Disulfide Relay in the Intermembrane Space of Mitochondria
A recently identified pathway in the mitochondrial intermembrane space ensures oxidation of the proteins that require disulfide bonds in order to acquire their native conformation. The proteins that constitute the MIA40 pathway and are dedicated in substrate oxidation are Mia40 and Erv1. The structural and functional properties for each protein are analyzed in the following section as the basis for understanding the mechanistic parameters of the pathway.
Erv1, a FAD dependent sulfhydryl oxidase of the intermembrane space
The essential 22 kDa protein Erv1 (essential for respiration and vegetative growth) was originally identified in Saccharomyces cerevisiae and is localized in the intermembrane space of mitochondria (72). The role of Erv1 in various cellular pathways is exemplified by a number of defects observed in cells that lack the endogenous protein. Fluorescence microscopy experiments have shown that an Erv1 thermosensitive mutant when grown at the restrictive temperature has a severe defect in the formation of cristae at the inner membrane. Lack of Erv1 in mitochondria leads to loss of mitochondrial DNA and also causes mitochondrial morphology defects (21, 73). A role of Erv1 in the maturation of the cytosolic iron–sulfur cluster containing proteins Leu1p and Rli1p has been found, although lack of Erv1 has no effect on the Fe/S proteins in the mitochondrial matrix (68). It has been suggested that Erv1 participates in the maturation of Fe/S clusters after they get exported from the mitochondrial matrix by the Atm1 inner membrane transporter (62). In Erv1-lacking cells, the maturation of heme is affected possibly due to a defect in the binding of heme to cytochrome c and cytochrome c peroxidase (Ccp1) (32). The endogenous levels of disulfide bond containing intermembrane space proteins have been shown to be significantly decreased (2, 79, 95). Finally, the human Erv1 homologue (ALR) is linked to liver regeneration by an unknown mechanism (40).
Erv1 belongs to the Erv/ALR sufhydryl oxidase family and homologous proteins are found in the endoplasmic reticulum (Erv2) (39, 101, 109), in the extracellular environment (Quiescin sulfhydryl oxidase) (54, 55) and in the poxvirus family (E10R) (99, 100). All the protein members of this family can bind FAD that they use during substrate oxidation to transfer electrons to oxygen, a reaction which results in hydrogen peroxide (H2O2) generation (30, 113).
The crystal structures of yeast Erv2 and Erv1 from Arabidopsis thaliana have shown that the Erv/ALR family is characterized by a catalytic core of about 100 amino acids that has noncovalently bound FAD stabilized by a four helix bundle (35). Proximal to the isoalloxazine ring of FAD is situated the conserved redox active CX2C motif which allows the efficient electron transfer from the cysteine pair to the FAD cofactor. The core domain is fused to a tail segment that contains a conserved cysteine pair called a “shuttle pair.” This pair can be found either in the amino or the carboxy terminus and the amino acid spacing from the catalytic pair is different amongst the Erv/ALR family members. The yeast and the human homologue have the shuttle pair positioned near the N-terminus in a CX2C motif, while in Arabidopsis thaliana the pair it is found in the carboxy terminus in a CX4C motif (71).
Biochemical evidence and in particular the crystal structure of Erv2 (the Erv1 yeast paralogue localized in the endoplasmic reticulum) point to a well-orchestrated mechanism of substrate oxidation (28, 44). The “shuttle cysteine pair” is found in an unstructured flexible domain and as such it is directly involved in disulfide bond transfer to substrate proteins. The tail segment of the Erv/ALR family members has been suggested to confer substrate specificity due to the differences in the primary sequence and in the cysteine spacing of the shuttle pair among the homologues. In line with this hypothesis are the results by the study of Thorpe in 2009 (33) who showed that the full length ALR isoform can oxidize the authentic substrate Mia40, but not the ALR isoform that lacks this tail segment. After substrate oxidation, the electrons are shuttled to the redox active pair and then to oxygen via the FAD cofactor. In the crystal structure of Erv2, the CGC shuttle pair found on the carboxy terminus has been captured in two conformations (44). In one case the CGC motif was found close to the catalytic pair near the FAD cofactor. In another case the CGC motif was found 10 Å away from the redox active pair which hinted that the CGC motif can transfer disulfide bonds inter- and intramolecularly from the catalytic pair to the substrates (28).
Early work on Erv1 suggested that it participates in a thiol–disulfide exchange pathway. The Erv1 protein contains six conserved cysteines, four of which are in a CX2C motif. A role for the carboxy terminal cysteines C159–C176 in stabilizing FAD binding has been proposed. Recombinant Erv1 mutated in those two cysteines is unable to bind the FAD cofactor (50). The catalytic pair C130–C133 transfers electrons to FAD while the N-terminus pair C30–C33 has been suggested to shuttle electrons from the substrate to the catalytic pair (109). The redox potential for each of the cysteine pairs has been measured and is in agreement with a putative intramolecular disulfide bond exchange that leads to an electron cascade from the substrate to the FAD cofactor. More specifically the shuttle pair is measured at −320 mV and the catalytic pair at −150 mV (32).
Erv1 has been suggested to function as a dimer in the intermembrane space of mitochondria. In fact, recombinant Erv1 can form covalent dimers as monitored by nonreducing SDS–PAGE. The Erv1 variant that has the shuttle cysteines mutated into serines is unable to form covalent dimers, suggesting a direct involvement of the particular cysteine pair in dimerization (50). Furthermore, the endoplasmic reticulum paralogue Erv2 has been crystallized in a noncovalent dimeric form, where the two monomers interact in opposite orientation (28, 44). Consistently, the dimeric form of the Arabidopsis thaliana Erv1 homologue is necessary for the protein function (71). In agreement with these observations, it was recently shown that simultaneous incubation of an Erv1 mutant lacking the shuttle cysteines with one that has the catalytic pair mutated can oxidize Mia40, which indicates that electrons can be transferred from one monomer to another (6). Taken together, this evidence points to a possibly crucial role of the Erv1 dimer, however direct evidence showing the dimeric state and the nature of the dimer in vivo is still lacking.
The functional role of Erv1 in vitro was originally studied with artificial substrates such as lysozyme and DTT because authentic substrates had not been identified (50, 69, 119). The first physiological substrate of Erv1 was found to be the mitochondrial protein Mia40. The key experimental evidence in support of this finding are (i) the fact that in Erv1-lacking mitochondria Mia40 is predominantly reduced, and (ii) the finding that a measurable fraction, albeit a small one, of the endogenous Erv1 can be isolated in a covalent complex with Mia40 under physiological steady-state conditions (79).
Mia40, a redox-regulated receptor of the intermembrane space
Mia40 is an essential protein that was originally identified when the yeast mitochondrial proteome was delineated. The protein with the systematic name YKL195w was first characterized by the group of N. Pfanner (26). The theoretical molecular weight of the precursor protein is 44 kDa (and 40 kDa for the mature form), but migrates as a 64 kDa protein on SDS-PAGE. The fact that 23.5% of its primary amino acid sequence is composed of negatively charged residues could account for the aberrant electrophoresis migration. The oligomeric state of the endogenous protein is not yet clearly defined. Glycerol density gradient centrifugation experiments have showed that the human homologue of Mia40 forms dimers and tetramers, but static multiangle light scattering and NMR data showed that it is monomeric in vitro (15, 51). Moreover, the yeast Mia40 that migrates as 140 kDa protein under native electrophoresis that is resistant to reducing agents and to strong detergents (26) indicating a monomeric native state.
Homologous proteins to Mia40 are found only in eukaryotes. The protein is found either soluble in the intermembrane space of mitochondria or anchored to the inner membrane. Members of the Mia40 protein family vary substantially in the length of the primary sequence. Yeast Mia40 contains an amphipathic presequence in the amino terminus that targets the protein to the inner membrane via the TIM23 pathway (26, 88). The protein is imported via the “stop-transfer” mechanism where the signal is degraded by the MPP protease of the matrix which results in the lateral insertion of the protein in the inner membrane. However, anchoring of Mia40 on the inner membrane is not a prerequisite for the function of the protein and deletion of the hydrophobic segment does not compromise yeast viability (24, 88). In higher eukaryotes, the targeting signal of Mia40 is absent and the protein is found soluble in the intermembrane space which indicates that the functional segment of the protein is found in the conserved domain of Mia40. Indeed, the highest degree of sequence identity (∼60%) is found near the carboxy terminus and includes the conserved CPC-CX9C-CX9C motif. Mutagenesis of the cysteines is not tolerated in vivo suggesting an important structural and/or functional role (88, 112).
Mia40 is the central component in the import pathway of a set of proteins destined to the intermembrane space of mitochondria. These proteins are generally small, (6–22 kDa), lack a targeting signal and possess a highly conserved signature cysteine motif. As already mentioned in the previous section, substrate proteins have a twin CX3C or CX9C, like small Tims and proteins of the Cox family, and are trapped in the intermembrane space by oxidative folding mediated by Mia40. Mitochondria lacking functional Mia40 have reduced endogenous levels of substrate proteins (26, 79). Furthermore, newly incoming substrate proteins cannot get imported in mitochondria that lack Mia40, which is suggestive of a receptor role for Mia40. Many studies have now conclusively shown that the newly imported substrates interact with the endogenous Mia40 by forming transient mixed disulfide intermediates in the first step of substrate oxidation (38, 79, 81, 106, 111). Consistently, the endogenous Mia40 is found predominantly in an oxidized form under steady-state conditions and any treatment that inhibits thiol–disulfide exchange between Mia40 and the newly incoming precursor, such as pretreatment of mitochondria with a reducing agent or blocking the free thiols of the substrate with an alkylating agent, abolishes their import in the intermembrane space (79).
The recent conformational characterization of Mia40 revealed the exact structural properties of the protein and provided valuable insight into the functional properties of the protein. The structure was determined first for the human Mia40 by NMR (15). Subsequently, the yeast Mia40 structure was solved by X-ray crystallography (60) displaying essentially the same features as the human Mia40 (Fig. 5). From the solution structure of hMia40 it became apparent that it has minimal secondary structure made up of only two α-helices and unlike other cellular oxidoreductases (DsbA, PDI) does not possess a thioredoxin fold motif. The majority of the protein (∼80%), mainly at the N and C-termini, are unfolded and do not assume any secondary structure. The structured part of the protein is composed of a lid and a core segment. The core part consists of two antiparallel α-helices stabilized by two disulfide bridges made of the juxtaposed cysteines of the CX9C motif, as originally suggested by trypsinolysis/mass spectrometry data (45). The lid segment contains the CPC motif that forms an intramolecular disulfide bond, is structurally rigid, and folds onto the core. Furthermore, the lid contains conserved hydrophobic residues that interact with conserved hydrophobic residues located on the core segment. The hydrophobic interactions between the two domains position the CPC motif in a solvent exposed conformation and create a shallow hydrophobic cleft. This is made of residues Leu60, Met63, Phe72, Phe76, Phe79, Phe95, Met98, and Met102 in the hMia40 (Fig. 5). This shallow cleft was proposed to function as a putative substrate binding domain, a concept that was substantiated by mutagenesis experiments. Interestingly, the second cysteine of the CPC motif (that turns out to be involved in the disulfide exchange reaction with the substrate) lies directly above the hydrophobic cleft. This can rationalize in structural terms the function of this hydrophobic region as the substrate binding domain, physically allowing facile oxidation by the protruding and adjacent CPC motif. In conclusion, the structure of both the human and the yeast homologues provided evidence for the first cellular thioredoxin-unrelated oxidoreductase (Fig.5).
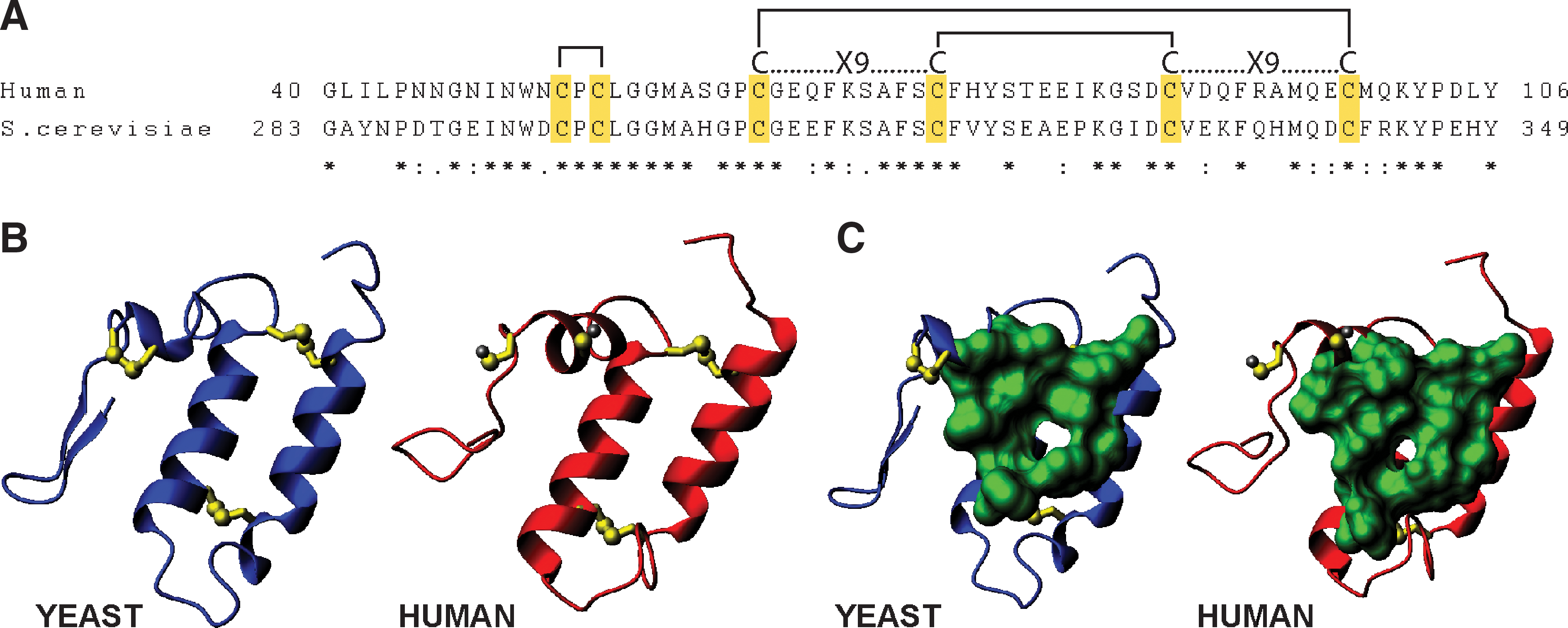
Biochemical experiments substantiated the structural observations. Specifically, it was shown experimentally that the CPC motif acts as the catalytic center for substrate oxidation and in particular the second cysteine is crucial for mixed disulfide formation with the substrate (15, 60, 112, 116). Notably the disulfide bond between the cysteines of the CPC motif is very sensitive to low concentrations of DTT, apparently because it is solvent exposed. On the other hand, the disulfide bridges of the CX9C are extremely stable against high DTT concentrations and require incubation at high temperature for efficient reduction (15, 45). This result is functionally relevant as the catalytic CPC center of an oxidoreductase needs to be recycled quickly between the oxidized and reduced form while the CX9C disulfide bridges ensure the structural stability of the protein. The redox potential of the CPC motif was measured at −200 mV which makes the oxidation of substrate proteins (e.g., small Tims −320 mV) thermodynamically favored (15). Mutagenesis of the hydrophobic cleft had an effect in substrate oxidation which agrees with the putative role of the cleft in substrate recognition (15, 60). It seems that substrate binding in the cleft occurs in such a way that the mixed disulfide intermediate can easily be formed between the protruding second cysteine of the CPC motif and the substrate.
Finally, it should be noted that recombinant Mia40 has also been shown to bind to zinc and copper via its cysteines and this metal binding renders the protein more resistant to proteolysis (113). This may indicate a potential role of metal binding in maintaining its structural stability. Consistently, Hot13, an additional factor of the mitochondrial oxidation machinery has been suggested to improve the electron transfer between Mia40 and Erv1 by maintaining Mia40 in a zinc-free state, yet a Mia40 metal bound form still remains to be identified (78).
The pathway
All available data converge to a mechanism for coordinated transfer of disulfides in the oxidative folding of proteins imported into the mitochondrial intermembrane space. According to this, the substrates enter through the OM in a reduced and therefore unfolded state, disulfide bonds are introduced by Mia40, and the relevant pairs of electrons are transferred initially to Mia40, then to oxidized Erv1, and from then on to CytC or Cytochrome C peroxidase. In all cases, the final electron acceptor is oxygen under aerobic conditions (22, 32), but remains unknown in anaerobic conditions. The coordinated transfer of disulfides is supported by the thermodynamic redox potentials of the components involved (Fig. 6).Parts of the disulfide transfer reactions have been reconstituted successfully in vitro using purified components. A dissection of the pathway in subreactions is shown schematically in Figure 7. The first subreaction is the recognition of the substrate by Mia40. Initial studies by Chacinska and collaborators and Tokatlidis and collaborators (81, 106) showed that this is a site-specific event, determined by the N-terminal cysteine in the case of small Tims or one of the inner cysteines for the case Cox17 (15). However, although these studies demonstrated the cysteine specificity of Mia40, at the same time they raised the issue how different substrates can be recognized so efficiently by Mia40 involving different cysteines in each case. The answer to this came from two publications reporting the important discovery of a targeting signal specifically recognized by Mia40. This was initially found in the small Tims, spanning four residues upstream and three residues downstream of the N-terminal cysteines, termed MISS (83). A more recent study (107) identified a similar signal but only partially overlapping with MISS in the case of the small Tims (spanning the nine residues upstream of the N-end cysteine, called ITS, for Intermembrane space Targeting Signal), and also found in CX9C substrates like Cox17. The ITS is characterized by some key properties (107), namely (i) it can function upstream or downstream of the docking cysteine depending on the substrate (upstream of the docking cysteine for Tim10 or downstream for Cox17); (ii) is sufficient for crossing the outer membrane and for targeting nonmitochondrial proteins; (iii) forms an amphipathic helix with crucial hydrophobic residues on the side of the docking cysteine and dispensable charged residues on the other side; (iv) fits complementary to the substrate cleft of Mia40 via hydrophobic interactions measured to be of micromolar affinity (Fig. 8).The MISS/ITS represents a new type of mitochondrial targeting signals. Its propensity to form an amphipathic helix is a common feature with the typical matrix targeting signal (MTS) which may explain why MISS/ITS is sufficient for crossing the OM. The MISS/ITS is much shorter and its function relies on packing of the hydrophobic residues to create a complementary surface to the Mia40 binding cleft and the crucial Cys being positioned in the hydrophobic side of the helix (Fig. 8). This is in contrast to the matrix targeting signal where both hydrophobicity and charge are key for function. The combination of the experimental data on the cysteine specificity and the MISS/ITS signal allows us to rationalize the dual function of Mia40 as a receptor and disulfide donor in the first subreaction indicated in panel A, Figure 7. A two-step mechanism seems to operate: a MISS/ITS-guided ‘sliding’ step orients the substrate noncovalently through weak hydrophobic interactions with the substrate binding cleft of Mia40; this is followed by ‘docking’ of the substrate cysteine now juxtaposed to pair with the Mia40 active cysteine.

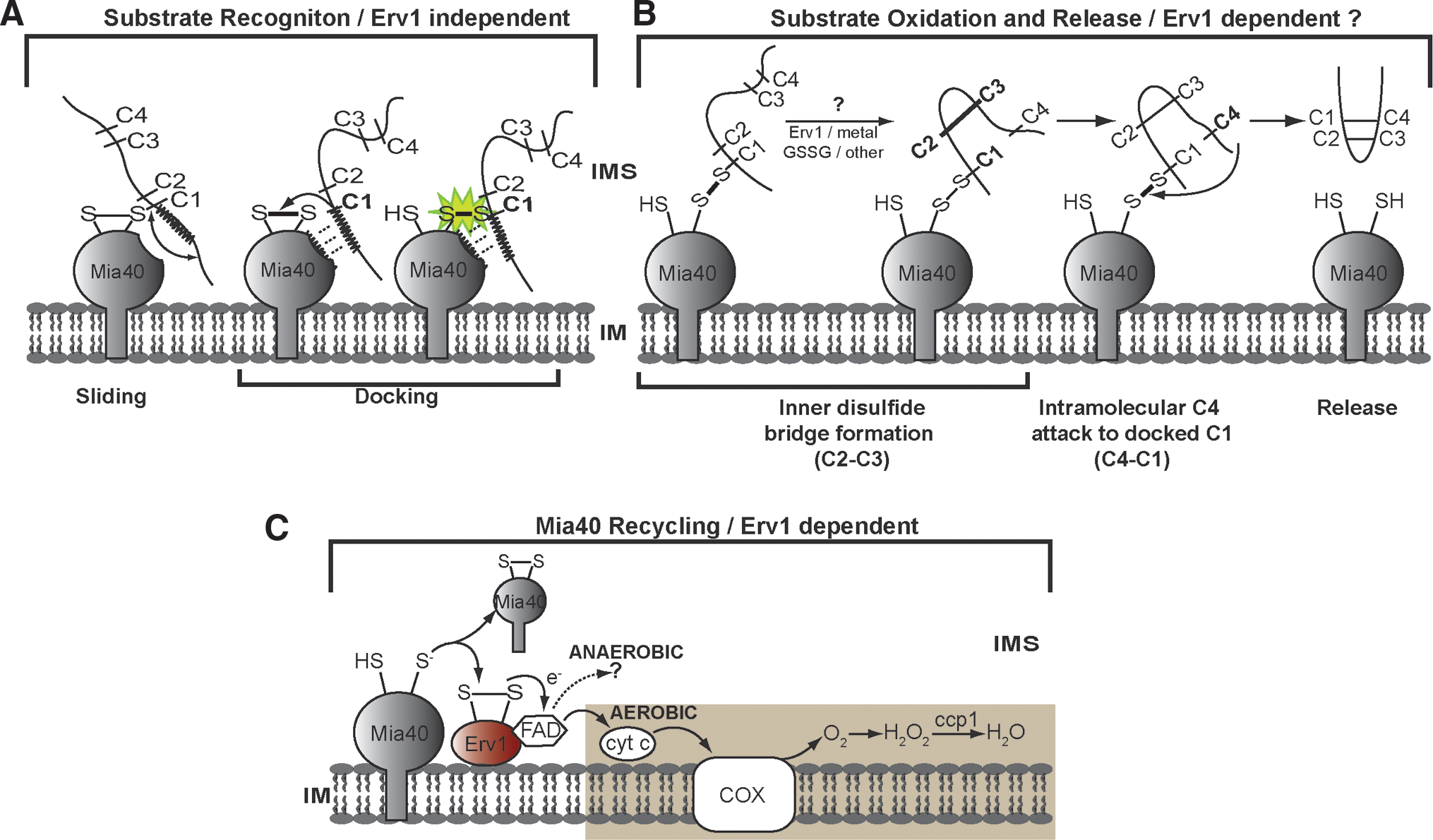

Correct cysteine priming in each substrate as determined by the MISS/ITS is key to this process and provides a logical explanation for the accuracy of the Mia40 substrate recognition. This represents a major difference to the Dsb and PDI oxidative folding systems and may conceivably allow substrate oxidation in mitochondria to proceed in the absence of isomerization. In vitro studies have shown that Mia40 is necessary and sufficient for complete oxidation of the substrate when in stoichiometric amounts and in the presence of oxygen (15). On the other hand, catalytic amounts of Mia40 are sufficient to oxidize the substrates (45, 112), whilst Erv1 alone cannot oxidize the substrate at all (2, 112). By contrast, complete oxidation of the substrate in vitro by catalytic amounts of Mia40 can occur only if Erv1 is present in the assay (6, 45, 112, 116). A clear role of Erv1 in recycling Mia40 to its oxidized state is therefore substantiated by these in vitro studies in agreement with the in vivo data showing a physical and functional interaction between Mia40 and Erv1 (2, 26, 79, 95, 111). In panel C of Figure 7, the recycling reaction of Mia40 by Erv1 and the subsequent transfer of electrons to CytC and Ccp1 (2, 22, 32, 34) is shown.
However, the steps involved in complete oxidation of the substrate and its subsequent release in a folded conformation are less clear. Sideris and Tokatlidis (106) showed that for Tim10 the formation of the second disulfide pair C2–C3 is prerequisite to the final attack of C4 onto C1 to break the mixed disulfide intermediate with Mia40 and release the substrate (panel B, Fig. 7), but how this second disulfide is formed is still not understood. Oxidation via a metal or GSSG is conceivable, but there are no data in support of these. In fact, complete oxidation of the substrate in vitro can proceed even in the presence of chelating factors, arguing against the importance of metal-dependent oxidation (15). Oxygen-dependent oxidation that was suggested to facilitate the process in vitro (15) is less clear in vivo since the process must occur also in anaerobic conditions. A second oxidation round by Mia40 is in principle an alternative possibility, although this is not very likely in view of the strong dependence of Mia40 binding to the targeting signal MISS/ITS (which is occupied on the substrate cleft while the substrate is still engaged to Mia40 via the docking cysteine). If a second oxidation by Mia40 occurred, one would have to assume either that the second Mia40 molecule would bind elsewhere on the substrate, not on the high affinity MISS/ITS region, or that the C4–C1 is made before C2–C3 (which is not supported by existing data). A third possibility is that complete oxidation of the substrate is mediated by Erv1. This possibility is attractive because there is compelling evidence that Erv1 is needed for complete oxidation of the substrates in vivo (2, 79, 86, 109). Despite the important observation of a ternary complex containing Erv1-substrate-Mia40 (110), a direct demonstration of disulfide-bonded species between Erv1 and the substrate is yet missing, in contrast to the clear presence of bimolecular disulfide-bonded species between substrate-Mia40 or Mia-Erv1 in an in vitro reconstituted system (116). The demonstration of the player(s) that are facilitating the second disulfide bond on the substrate remains elusive. This may reflect the fact that this second oxidation event becomes much more rapid (and hence the relevant intermediate much more transient) due to a very favorable positioning in space of the two cysteines involved (C2 and C3 in the case of Tim10, as shown in Fig. 7) after the initial docking to Mia40 occurs.
Finally, the presence of Hot13, a zinc-binding protein that affects the biogenesis of small Tims (31) and physically interacts with Mia40 (78), was proposed to have an indirect/auxiliary role in this process functioning as a buffering system for zinc ions in the IMS (78).
Versatility in substrate recognition
The extended studies on the CX3C and CX9C type of Mia40 substrates revealed important clues as to how these are targeted via the MISS/ITS to Mia40 and how their oxidation is initiated through the docking of selective cysteines. Although the ITS are present in essentially all CXnC substrates, recognition of other substrates that do not belong to this large family (like the copper chaperone Ccs1, and Erv1 itself ) is still unclear. The general features of the sliding-docking model for recognition by Mia40 as presented above may still hold as this depends on conformational properties rather than amino acid sequence features (as exemplified for the case of Cox17 vs. Tim10).
Conclusions
Five years after the original suggestion for the existence of an oxidative folding pathway in the mitochondrial intermembrane space (74), the crucial components have been identified, and some salient features of the mechanism have been elucidated. The development of in vitro reconstituted systems, the demonstration of some of the crucial mixed disulfide species in vitro and in vivo as well as the elucidation of the structure of Mia40 have been pivotal in advancing our knowledge of this mechanism.
Still, some key questions are still open and will no doubt be in the focus of future efforts. First, the precise role of Erv1 in the context of the ternary complex with Mia40 and the substrate needs to be understood (110). Second, it will be of interest to identify other physiological substrates for Erv1/ALR in addition to Mia40. Third, the cascade of oxidizing steps subsequent to initial docking to Mia40 is obviously crucial for the attainment of a folded state and we need to understand how the second disulfide of the substrate is formed. In this respect, the presence of the ITS signal in different positions within the polypeptide depending on the substrate results in substantially different topologies of the substrate while this is docked to Mia40. Further structural studies will need to be performed to dissect the structural basis of recognition in each case. Fourth, as this pathway must operate also in anaerobic conditions, it will be crucial to find out what the final electron acceptors in these conditions may be. Finally, recent reports on the Ero1 system of the endoplasmic reticulum suggested the presence of regulatory disulfides (7, 12, 104). It will be of interest whether similar regulatory functions could be assigned to noncatalytic disulfide pairs of Erv1/ALR.
The oxidative folding pathway of protein import into mitochondria is the only one of the very versatile import pathways for this organelle that involves covalent modification of the imported precursor and is linked to the function of the respiratory chain. Mia40, the key component in this pathway, represents the first cellular oxidoreductase that is unrelated to the thioredoxin family, and seems to operate by a different mechanism ensuring specificity already at the level of the very first protein–protein encounter with the substrate via the sliding-docking mechanism outlined here. This may be one fundamental reason why isomerization may not be needed for this system that operates on the basis of high accuracy and specificity at the very early stages of substrate recognition, in contrast to the ER and periplasmic oxidative folding machineries.
Several proteins of the IMS endowed with a diverse array of functions in crucial cellular processes like respiration, energy metabolism, apoptosis, and neurodegenerative diseases are either known to be or putative substrates of the mitochondrial oxidative folding pathway. It is therefore of paramount importance to understand the important mechanistic and structural basis of this process not only to draw parallels and understand unique features compared to the other cellular oxidoreductase systems, but also to uncover putative functional links to pathological conditions.
Footnotes
Acknowledgments
This work was supported by intramural funds from IMBB-FORTH (Institute of Molecular Biology and Biotechnology-Foundation for Research and Technology Hellas), the University of Crete, and the European Social Fund and national resources to KT. DPS was supported by a PENED grant (Greek General Secretariat for Research and Technology). We are grateful to Lucia Banci (Centre for Magnetic Resonance, University of Florence, Italy), Ivano Bertini (Centre for Magnetic Resonance, University of Florence, Italy) and Carolyn Sevier (Dept of Biology, MIT) for comments and discussions, to Simone Ciofi–Baffoni (Centre for Magnetic Resonance, University of Florence, Italy) for generating the structural figures and for helpful suggestions and to members of our lab for comments.